the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 29 Nov 2019
| 29 Nov 2019
Isotopic characterization of nitrogen oxides (NOx), nitrous acid (HONO), and nitrate (pNO3−) from laboratory biomass burning during FIREX
David J. Miller
Eric Scheuer
Jack Dibb
Vanessa Selimovic
Robert Yokelson
Kyle J. Zarzana
Steven S. Brown
Abigail R. Koss
Carsten Warneke
Meredith Hastings
New techniques have recently been developed and applied to capture reactive nitrogen species, including nitrogen oxides (), nitrous acid (HONO), nitric acid (HNO3), and particulate nitrate (p), for accurate measurement of their isotopic composition. Here, we report – for the first time – the isotopic composition of HONO from biomass burning (BB) emissions collected during the Fire Influence on Regional to Global Environments Experiment (FIREX, later evolved into FIREX-AQ) at the Missoula Fire Science Laboratory in the fall of 2016. We used our newly developed annular denuder system (ADS), which was verified to completely capture HONO associated with BB in comparison with four other high-time-resolution concentration measurement techniques, including mist chamber–ion chromatography (MC–IC), open-path Fourier transform infrared spectroscopy (OP-FTIR), cavity-enhanced spectroscopy (CES), and proton-transfer-reaction time-of-flight mass spectrometry (PTR-ToF).
In 20 “stack” fires (direct emission within ∼5 s of production by the fire) that burned various biomass materials from the western US, δ15N–NOx ranges from −4.3 ‰ to +7.0 ‰, falling near the middle of the range reported in previous work. The first measurements of δ15N–HONO and δ18O–HONO in biomass burning smoke reveal a range of −5.3 ‰ to +5.8 ‰ and +5.2 ‰ to +15.2 ‰, respectively. Both HONO and NOx are sourced from N in the biomass fuel, and δ15N–HONO and δ15N–NOx are strongly correlated (R2=0.89, p<0.001), suggesting HONO is directly formed via subsequent chain reactions of NOx emitted from biomass combustion. Only 5 of 20 p samples had a sufficient amount for isotopic analysis and showed δ15N and δ18O of p ranging from −10.6 ‰ to −7.4 ‰ and +11.5 ‰ to +14.8 ‰, respectively.
Our δ15N of NOx, HONO, and p ranges can serve as important biomass burning source signatures, useful for constraining emissions of these species in environmental applications. The δ18O of HONO and obtained here verify that our method is capable of determining the oxygen isotopic composition in BB plumes. The δ18O values for both of these species reflect laboratory conditions (i.e., a lack of photochemistry) and would be expected to track with the influence of different oxidation pathways in real environments. The methods used in this study will be further applied in future field studies to quantitatively track reactive nitrogen cycling in fresh and aged western US wildfire plumes.
- Article
(1306 KB) - Full-text XML
-
Supplement
(278 KB) - BibTeX
- EndNote





Biomass burning (BB), which occurs in both anthropogenic processes (e.g., cooking, heating, and prescribed fire that is controlled burning for management purposes) and natural wildfire (lightning-ignited vegetation burning), is a significant source of atmospheric reactive nitrogen species, including nitrogen oxides (), nitrous acid (HONO), nitric acid (HNO3), particulate nitrate (p), organic nitrates, peroxyacyl nitrate (PAN), and ammonia (NH3), that have major impacts on air quality and climate from regional to global scales (Crutzen and Andreae, 1990). Globally, biomass burning emits ∼6 Tg of nitrogen oxides () per year, contributing at least 14 % to total NOx emissions (Jaeglé et al., 2005), with large interannual and seasonal variation due to fire frequency and intensity (Jaffe and Briggs, 2012). Primarily emitted NOx plays an important role in the photooxidation of volatile and semi-volatile organic compounds, which are present in high concentrations in BB plumes, and strongly influences the production of tropospheric ozone (O3) and secondary aerosols (Alvarado et al., 2015). In BB plumes, NOx can be converted to PAN, which can be transported long distances (hundreds to thousands of kilometers) in lofted plumes before rereleasing NOx. Therefore, BB-emitted NOx could widely influence air quality downwind for days to weeks (Val Martín et al., 2006; Ye et al., 2016). In addition, NOx is also the major photochemical precursor of HNO3 and p, which can be transported downwind, mix with anthropogenic emissions, and impact air quality and ecosystem health (Hastings et al., 2013).
HONO has been observed in BB plumes in both laboratory and field experiments, with HONO mixing ratios in the range of ∼5 %–33 % of observed NOx (Akagi et al., 2012, 2013; Burling et al., 2010, 2011; Keene et al., 2006; Liu et al., 2016; Roberts et al., 2010; Selimovic et al., 2018; Yokelson et al., 2007, 2009). The photolysis of HONO is a major OH precursor in the daytime; therefore, HONO plays an important role in the photochemical aging of BB plumes and atmospheric oxidation capacity at regional scales (Alvarado and Prinn, 2009; Liu et al., 2016; Tkacik et al., 2017; Trentmann et al., 2005). HONO has been proposed as a significant OH source in BB plumes, and the inclusion of HONO in photochemical models could explain much of the uncertainty in the modeled O3 (Alvarado et al., 2009; Alvarado and Prinn, 2009; Cook et al., 2007; Travis et al., 2016; Trentmann et al., 2005).
Direct BB emission factor measurements of HONO and NOx exhibit significant uncertainties due to limited observations and the large spatial and temporal variability of burning conditions, making it challenging to build an accurate inventory of BB emissions relative to other major sources (Lapina et al., 2008). Emission factors vary and mainly depend on (1) fuel nitrogen content (0.2 %–4 % by mass), which is a function of vegetation type, and (2) modified combustion efficiency () that is determined by combustion conditions including fuel moisture, fuel load, temperature, relative humidity, wind speed, and other meteorological parameters (Burling et al., 2010; Jaffe and Briggs, 2012; Yokelson et al., 1996). Additionally, the temporal evolution of HONO in BB plumes varies greatly in different fires, and relative contributions from direct emission versus NO2 conversion to HONO remain unclear. For instance, significant concentrations of HONO and correlations between HONO and NO2 have been observed in aged plumes, indicating the importance of the heterogeneous conversion of NO2 to HONO on BB aerosols (Nie et al., 2015). By contrast, no evidence was found for secondary HONO formation in a BB plume during the Southeast Nexus Experiment (Neuman et al., 2016). It is important to constrain HONO directly emitted from BB compared to HONO formed during plume aging. This would reduce uncertainties associated with the total HONO budget and increase our understanding of HONO impacts on O3 and secondary aerosol formation downwind of BB regions.
In an effort to better understand reactive nitrogen emissions and chemistry, especially for HONO, new techniques have been developed to analyze the isotopic composition of various species. Stable isotopes provide a unique approach for characterizing and tracking various sources and chemistry for a species of interest (Hastings et al., 2013). Fibiger et al. (2014) developed a method to quantitatively collect NOx in solution as for isotopic analysis, which has been verified to avoid any isotopic fractionation during collection in both lab and field studies. This allows for high-resolution measurement of δ15N–NOx in minutes to hours depending on ambient NOx concentrations ( ‰, and ‰ where VSMOW is Vienna Standard Mean Ocean Water). δ15N has also been used to track gaseous NOx from a variety of major sources including emissions from biomass burning (Fibiger and Hastings, 2016), vehicles (Miller et al., 2017), and agricultural soils (Miller et al., 2018). Using this method, Fibiger and Hastings (2016) systematically investigated BB δ15N–NOx from different types of biomass from around the world in a controlled environment during the fourth Fire Lab at Missoula Experiment (FLAME-4). NOx emissions collected both immediately from the BB source and 1–2 h after the burn in a closed environment ranged from −7 ‰ to +12 ‰, and primarily depended on the δ15N of the biomass itself. BB-emitted HONO isotopic composition has never been measured before. Our recently developed method for HONO isotopic composition analysis (Chai and Hastings, 2018) enables us to not only characterize δ15N and δ18O of HONO, but also explore the connection between δ15N–NOx and δ15N–HONO.
Fire Influence on Regional to Global Environments and Air Quality (FIREX-AQ) investigates the influence of fires in the western US on climate and air quality via an intensive multi-platform campaign. The first phase of FIREX-AQ took place at the US Forest Service Fire Sciences Laboratory (FSL) in Missoula, Montana, in the fall of 2016, where we measured δ15N–NOx, δ15N–HONO, δ18O–HONO, δ15N–p, δ18O–p, and δ15N–biomass in 20 “stack burns” of a variety of fuels representative of northwestern North America. Here we report on the results and explore relationships between the isotopic composition of these reactive nitrogen species, as well as the corresponding mixing ratios for HONO that were concurrently measured by a variety of techniques. This work offers a characterization and quantification of the BB source signatures of these species, which can be applied in the interpretation of observations in future field studies.
2.1 FIREX Fire Sciences Laboratory design
The room for controlled BB experiments is 12.5 × 12.5 m × 22 m, with a continuously weighed fuel bed at the center of the room. The combustion exhaust was vented at a constant flow rate (∼3.3 m s−1) through a 3.6 m diameter inverted funnel, followed by a 1.6 m diameter stack, and collected at a platform 17 m above the fuel bed via sampling ports that surround the stack, resulting in a transport time of ∼5 s. Further details have been described in the literature (Stockwell et al., 2014). All of our instruments for sampling and online measurements were placed on the platform, which can accommodate up to 1820 kg of equipment and operators. Measurements were focused on the stack burns, for which fires lasted a few minutes up to 40 min.
Table 1Concentration (mean, derived from solution concentration and flow rate) and N isotopic composition for various biomass burning experiments (all δ denotations: ‰). MCE values are extracted from the NOAA FIREX fire archive. Note: fire nos. 1, 7, and 13 were missing due to technical issues; NOx results are only shown when the blank-to-sample ratio is < 70 %. Biomass acronyms are defined in Sect. 2.1; d – duff, c – canopy, l – litter.

For this study, we investigated 20 stack fires of vegetation types abundant in the western US representing coniferous ecosystems, including ponderosa pine (PIPO), lodgepole pine (PICO), Engelmann spruce (PIEN), Douglas fir (PSME), and subalpine fir (ABLA), with replicate burns for most of these types (Table 1). Some of the fires proceeded with the burning of an individual fuel component such as litter, canopy, duff, and rotten logs. Other fires simulated actual biomass in the coniferous ecosystem by mixing various fuel components in realistically recreated ecosystem matrices using the first-order fire effects model (FOFEM) (Reinhardt et al., 1997).
2.2 Instrumentation
2.2.1 Collection of HONO, NOx and nitrate for isotopic analysis
HONO was completely collected for isotopic analysis using an annular denuder system (ADS) (Chai and Hastings, 2018). The ADS deployed in this laboratory experiment consisted of a Teflon particulate filter and a Nylasorb filter to remove HNO3, followed by two annular denuders, each coated with a solution of 10 mL of Na2CO3 (1 % w∕v) + glycerol (1 % v∕v) + methanol–H2O solution (1:1 volume ratio) following a standard US Environmental Protection Agency (EPA) method. Methanol and glycerol are certified American Chemical Society (ACS) plus with a purity of ≥99.8 % and ≥99.5 %, respectively. After coating, the denuders are dried using zero air and capped immediately. Within 6 h after each collection, the coating was extracted in 10 mL of ultrapure water (18.2 MΩ) in two sequential 5 mL extractions. The extracted solution with a pH of ∼10 was transported to Brown University for concentration and isotopic analysis 3–14 d after the sampling. The timescales for sample extraction and isotopic analysis preserve both the solution concentration and isotopic composition of HONO in the form of nitrite (Chai and Hastings, 2018). The two-denuder setup allows us to minimize interference for both concentration and isotopic analysis from other N-containing species that could be trapped and form nitrite in residual amounts on the denuders, especially NO2. Our method development study showed that NO2 tends to absorb in the same amount (difference < 4 %) on the walls of each denuder in a train setup, which is consistent with other studies (Perrino et al., 1990; Zhou et al., 2018). On the basis of this validation, the second denuder extract is used to correct the first denuder extract for both concentration and isotopic composition (Chai and Hastings, 2018). Note that HONO levels were above the minimum detection limit (0.07 µM), and the breakthrough amount of HONO threshold is far from being reached given the concentrations (Table 1), flow rate (∼4 L min−1), and collection times (5–40 min). The necessary minimum amount of nitrite collected for isotopic analysis is 10–20 nmol.
To avoid scrubbing HONO, a flowmeter (Omega) and the NOx collection system for the analysis of δ15N–NOx are placed following the ADS (Fibiger et al., 2014; Fibiger and Hastings, 2016; Wojtal et al., 2016). In brief, NOx is collected in a solution containing 0.25 M KMnO4 and 0.5 M NaOH, which oxidizes NO and NO2 to by pumping sampled air through a gas-washing bottle with a 65 W diaphragm vacuum pump. The flow rate (∼4 L min−1 with ±1 % uncertainty) is controlled with a critical orifice inserted between the pump and gas stream outlet and is monitored and recorded with a flowmeter placed prior to the NOx collector. The NOx trapping solution blanks are also collected every day to quantify background for concentration and isotopic blank corrections. The Omega flowmeter was calibrated with another flowmeter (Dry Cal Pro) by varying flow rates. Within a day after collection, we stabilized the samples in the wet chemistry lab in the Fire Sciences Lab by adding 30 % w∕w H2O2, which reduces to MnO2 precipitate, before shipping them back to Brown University for further processing. This effectively excludes the possible interferences from NH3 that could be oxidized to by after a week (Miller et al., 2017, and references therein). The samples were neutralized with 12.1 N HCl in the Brown lab before concentration measurement and isotopic analyses. on the upstream Millipore filters and HNO3 from the Nylasorb filters, if there was any, were extracted by sonicating the filters in ∼30 mL of ultrapure H2O (18.2 MΩ) for 30 min. Samples with [] > 1 µM were analyzed for isotopic composition (concentration techniques detailed below).
All treated samples from both HONO collection and NOx collection and their corresponding blanks were analyzed offline for concentrations of and with a WestCo SmartChem 200 Discrete Analyzer colorimetric system. The reproducibility of the concentration measurement was ±0.3 µmol L−1 (1σ) for and ±0.4 µmol L−1 for when a sample was repeatedly measured (n=30). A detection limit of 0.07 µmol L−1 for and 0.1 µmol L−1 for was determined, and no detectable nitrite or nitrate was found in the blank denuder coating solution, whereas blank concentrations of ∼5 µM are typical for the NOx collection method (Fibiger et al., 2014; Wojtal et al., 2016). Note that concentration was measured on the ADS solutions to verify whether and to what extent was oxidized to on denuder walls because the denitrifier method converts both and to N2O for isotopic analysis (see below). In addition, samples collected with a mist chamber–ion chromatography system (described in Sect. 2.2.2) were also tested for their concentrations, and only those with sufficient nitrite quantity were further analyzed for isotopic composition.
2.2.2 NOx and HONO online concentration measurement
NO and NOx concentrations were measured with a Thermo Scientific model 42i chemiluminescence NO ∕ NOx analyzer, which is described the Supplement. The NOx measurement verified the concentration of the NOx collected for isotopic analysis, shown in Table S3 and Fig. S1 in the Supplement.
HONO and HNO3 concentrations were measured using the University of New Hampshire's dual mist chamber–ion chromatograph system (Scheuer et al., 2003) with the sampling inlet placed right next to that of the ADS. The dual-channel IC system is custom built using primarily Dionex analytical components. Briefly, automated syringe pumps are used to move samples and standard solutions in a closed system, which minimizes potential contamination. A concentrator column and 5 mL injections were used to improve sensitivity. Eluents are purged and maintained under a pressurized helium atmosphere. The background signal is minimized using electronic suppression (Dionex-ASRS). The chromatography columns and detectors are maintained at 40 ∘C to minimize baseline drifting. A trifluoroacetate tracer spiked into the ultraclean sampling water is used as an internal tracer of sample solution volume, which can decrease due to evaporation in the exhaust flow by 10 %–20 % depending on the ambient conditions and length of the sample integration interval. The spike was analyzed to correct the final mist-chamber-sampled solution volume with an uncertainty of ±3 %. This system has been deployed in various field studies for HONO measurement (Dibb et al., 2002; Stutz et al., 2010) and showed reasonable intercomparison with other HONO measurement techniques (within 16 % uncertainty) during the 2009 SHARP campaign in Houston (Pinto et al., 2014). The detection limit for HNO3 and HONO is 10 ppt for 5 min sample integrations. During the experiments, two mist chambers were operated to collect gas samples in parallel, each with an integration interval of 5 min. One channel of the IC was utilized for concentration measurement; in the other, the mist chamber's solution was transferred into a sample bottle using the syringe pump, and the collected solution was brought to Brown University for isotopic analysis of HNO3 if a sufficient amount (10–20 nmol) was collected for each sample.
In addition to MC–IC, the HONO mixing ratios were also measured using high-time-resolution (∼1 s) measurement techniques, including open-path Fourier transform infrared spectroscopy (OP-FTIR) (Selimovic et al., 2018), a cavity-enhanced spectrometer (CES) (Min et al., 2016; Zarzana et al., 2018), and a proton-transfer-reaction time-of-flight mass spectrometer (PTR-ToF). Inlet ports of CES and PTR-ToF were placed 5′ apart but at the same height on the platform as those for ADS and MC–IC, while the OP-FTIR had an open-path cell at the stack. Smoke has been shown to be well mixed at the sampling platform (Christian et al., 2004), and the mean HONO mixing ratios across each fire obtained from the four techniques were compared with that retrieved from ADS collection. This offers comprehensive verification of the complete capture of HONO by ADS, which is extremely important for conserving the isotopic composition of HONO.
The details of OP-FTIR are described in previous works (Selimovic et al., 2018; Stockwell et al., 2014). The setup included a Bruker MATRIX-M IR cube spectrometer with a mercury cadmium telluride (MCT) liquid-nitrogen-cooled detector interfaced with a 1.6 m base open-path White cell. The White cell was positioned on the platform and its open path spanned the width of the stack. This facilitates direct measurement across the rising emissions. The optical path length was set to 58 m. The IR spectra resolution was 0.67 cm−1 from 600 to 4000 cm−1. Pressure and temperature were continuously recorded with a pressure transducer and two temperature sensors, respectively, which were placed adjacent to the White cell optical path. They were used for spectral analysis. The time resolution for stack burns was approximately 1.37 s. The OP-FTIR measures CO2, CO, CH4, a series of volatile organic compounds, and various reactive nitrogen species (Selimovic et al., 2018). Mixing ratios of HONO were retrieved via multicomponent fitting to a section of the mid-IR transmission spectra with a synthetic calibration nonlinear least-squares method (Griffith, 1996; Yokelson et al., 2007), and both the HITRAN spectral database and reference spectra recorded at the Pacific Northwest National Laboratory (Rothman et al., 2009; Sharpe et al., 2004; Johnson et al., 2010, 2013) were used for the fitting. The uncertainty is ∼10 % for the HONO mixing ratio measurement, and the detection limit is no more than a few parts per billion as reported in previous studies (Stockwell et al., 2014; Veres et al., 2010).
HONO measurements by cavity-enhanced spectroscopy used the airborne cavity-enhanced spectrometer, ACES, recently described by Min et al. (2016). This instrument consists of two channels, one measuring over the spectral range from 438 to 468 nm, at which glyoxal (CHOCHO) and NO2 have structured absorption bands, and one measuring from 361 to 389 nm at which HONO has structured absorption. In the HONO channel, light from an LED centered at 368 nm with an output power of 450 mW and collimated with an off-axis parabolic collector illuminates the input mirror of a 48 cm optical cavity formed from mirrors with a maximum reflectivity R=99.98 % at 375 nm. The effective path length within the optical cavity is > 3 km over the region of greatest reflectivity. The mirror reflectivity (effective path length) was calibrated from the difference in Rayleigh scattering between helium and zero air to provide an absolute calibration of the instrument response. A fiber-optic bundle collects light exiting the optical cavity and transmits it to a grating spectrometer with a charge-coupled device (CCD) detector, where it is spectrally dispersed at a resolution of 0.8 nm. The resulting spectra are fit using DOASIS software (Kraus, 2006) to determine trace gas concentrations, including NO2, HONO, and O4. Mixing ratios of NO2 and HONO are reported at 1 s resolution, although the NO2 precision is higher in the 455 nm channel. The 1 Hz HONO precision is 800 pptv (2σ). (The precision of the HONO instrument in ACES is somewhat degraded by the optimization of the 455 nm channel for glyoxal detection, which reduces the photon count rate on the 368 nm channel.) The accuracy of the HONO measurement is 9 %. Air was sampled directly from the stack at a height of 15 m above the fuel bed through a 1 m length of OD Teflon (FEP) tubing as described in Zarzana et al. (2018). The residence time in the inlet and sample cells was < 1 s. Comparison between the ACES HONO and an open-path FTIR agreed to within 13 % on average, and ACES HONO was well correlated with 1 Hz measurements from a PTR-ToF (r2=0.95) (Koss et al., 2018).
The PTR-ToF instrument used in the FIREX Fire Lab experiment is described in detail in previous studies (Koss et al., 2018; Yuan et al., 2016). The PTR-ToF instrument is a chemical ionization mass spectrometer typically using H3O+ reagent ions, and a wide range of trace gases can be detected in the range of tens to hundreds of parts per trillion (pptv) for a 1 s measurement time. At the Fire Lab, PTR-ToF detected several inorganic species including HONO with an uncertainty of 15 %. HONO is detected at a lower sensitivity than most trace gases in PTR-ToF, but mixing ratios for all fires were well above the detection limit.
2.2.3 Isotopic composition measurements
The denitrifier method was used to perform nitrogen and oxygen isotope analyses (15N∕14N, 18O∕16O) of and/or by complete conversion to N2O by the denitrifying bacteria P. aureofaciens (Casciotti et al., 2002; Sigman et al., 2001). The isotopic composition of N2O is then determined by a Thermo Finnigan Delta V Plus isotope ratio mass spectrometer at m∕z 44, 45, and 46 for 14N14N16O, 14N15N16O, and 14N14N18O, respectively. Sample analyses were corrected against replicate measurements of the isotopic reference materials USGS34, USGS35, and IAEA-NO-3 (Böhlke et al., 2003). Additional correction was performed for δ18O–HONO following previous studies (Casciotti et al., 2002, 2007; Chai and Hastings, 2018). Precisions for δ15N–HONO, δ18O–HONO, and δ15N–NOx isotopic analysis across each of the entire methods are ±0.6 ‰, ±0.5 ‰, and ±1.3 ‰, respectively (Chai and Hastings, 2018; Fibiger et al., 2014). δ18O–N2O from the NOx collection samples was measured but is not reported as δ18O–NOx because it is greatly impacted by oxidation and does not represent the δ18O–NOx in the sample air. The total δ15N of the starting biomass (δ15N–biomass) was measured at the Marine Biological Laboratory Ecosystems Center Stable Isotope Facility. The materials measured for δ15N–biomass (Table S1) cover most but not all the biomass types burned in the experiments depending on the availability of the leftover materials. Analyses were conducted using a Europa ANCA-SL elemental analyzer–gas chromatograph preparation system interfaced with a Europa 20-20 continuous-flow gas source stable isotope ratio mass spectrometer. Analytical precision was ±0.1 ‰ based on replicate analyses of international reference materials.
Collection time spanned the whole fire burning (5 to 40 min) in order to maximize the signal. We chose to report the samples whose concentrations are at least 30 % above the 5 µM present in the blank KMnO4 solution upon purchase (Fibiger et al., 2014) such that the propagated error through the blank correction does not exceed the analytical precision of ±1.5 ‰ for δ15N–NOx. We found identical concentration and isotopic signatures for both Fire Lab and Brown University lab blanks, which ensures that no additional contamination was introduced into the KMnO4 solutions in the gas-washing bottle. In addition, fires with high particulate loading that resulted in > 50 % reduction in flow rate are not considered for isotopic analysis because the low flow rate could induce incomplete collection with potential isotopic fractionation that might not represent BB emissions.
3.1 Temporal evolution of HONO and HNO3 from direct BB emissions
The time series of HONO and HNO3 concentrations measured by MC–IC at 5 min resolution for the majority of the stack burns are shown in Fig. 1, and original data can be found in the NOAA data archive (FIREX, 2016). HNO3 concentrations were nearly 2 orders of magnitude lower than typical HONO concentrations. The constant low concentration of HNO3 from fresh emissions across all fires is consistent with the findings in Keene et al. (2006), confirming that HNO3 is not a primary reactive nitrogen species in fresh smoke. Rather, it is largely produced secondarily in aged smoke and nighttime chemistry. Both HONO and HNO3 mixing ratios reach their peak in the first 5 min, except for fire no. 12 (Engelmann spruce–duff), for which the HONO concentration remains nearly constant over the course of the fire but is much lower than the HONO concentration of the rest of the fires. The largest HONO and HNO3 were emitted from burning subalpine fir in the Fish Lake canopy (fire no. 15), with an integrated concentration of up to 177 and 1.9 ppbv in the first 5 min sample, respectively. We note that fire no. 12 has the smallest MCE value of 0.868 (FIREX, 2016) and an abnormal flow rate (less than half of the typical flow rate during all other measurements) due to the inlet filter clogging from extraordinarily large particulate loadings. In general, the closer the MCE value is to 1, the more likely it is that N oxidation (e.g., NOx and HONO) dominates over N reduction (e.g., NH3 and HCN) as a result of flaming; when MCE approaches 0.8, more smoldering occurs such that N reduction becomes dominant (Ferek et al., 1998; Goode et al., 1999; McMeeking et al., 2009; Yokelson et al., 1996, 2008). Accordingly, the smoldering combustion condition of fire no. 12 leads to a lower concentration of oxidized nitrogen species than the rest of the fires in this study. Although fire no. 15 and no. 17 have relatively low MCE (∼0.89), the pulse of HONO in first 5–10 min suggests an active flaming phase followed by a longer smoldering phase. This indicates that both fires had combustion conditions that consisted of a mixture of flaming and smoldering, and thus significant HONO was still produced. In addition, the HONO∕NOx ratio ranged from 0.13 to 0.53 with a mean of 0.29±0.12 (1σ), comparable with previous results of laboratory experiments (0.11±0.04) and field experiments (0.23±0.09) (Akagi et al., 2013; Burling et al., 2010, 2011).
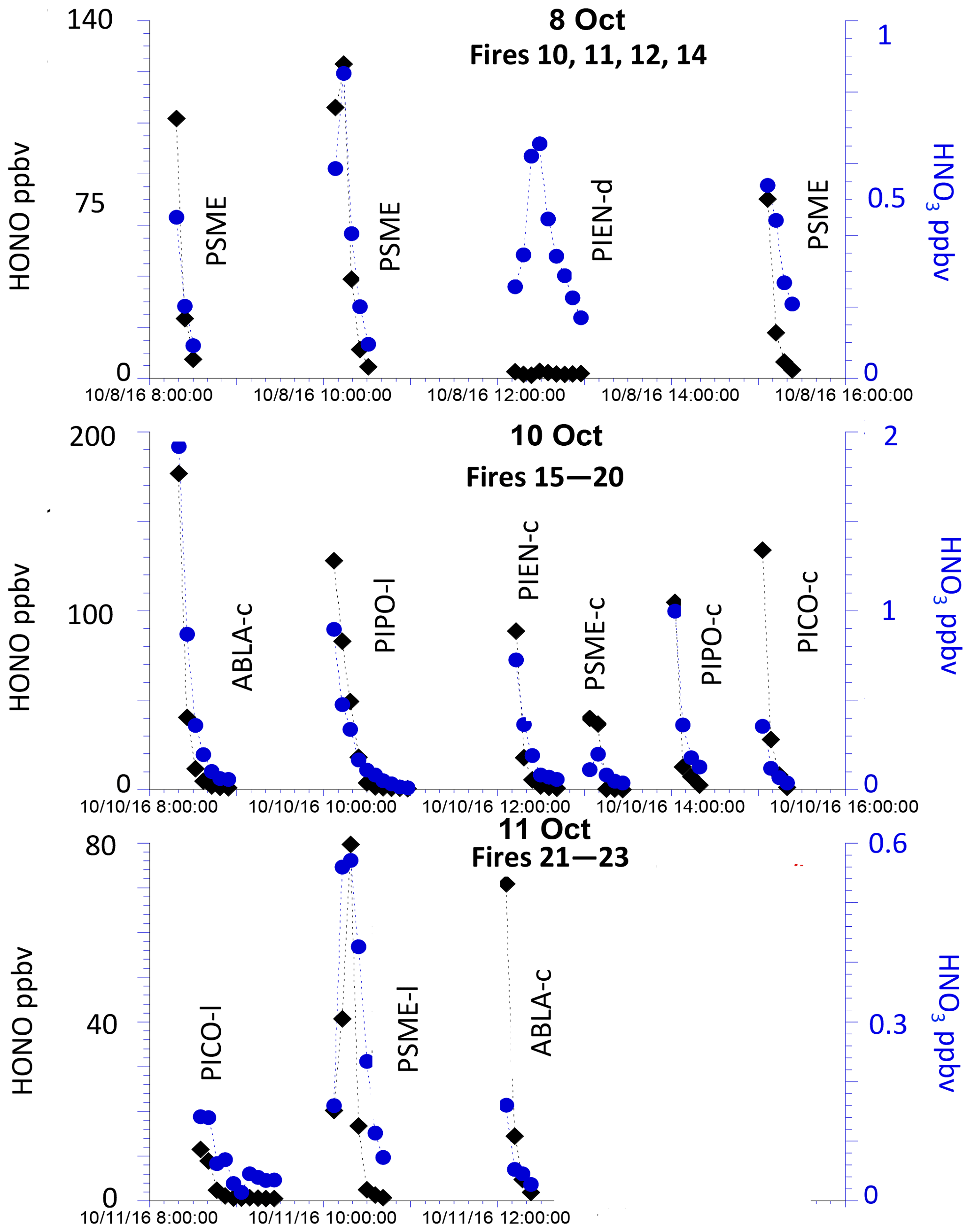
Figure 1Temporal profile of HONO (black diamond) and HNO3 (blue circle) concentration measured using the MC–IC method for various stack fires (fire numbers are given in Table 1).
3.2 Verification of ADS-collected HONO concentration
The HONO collected with the ADS represents a mean value over the course of each entire burn. We first compare the HONO concentration recovered from the ADS, denoted as [HONO]ADS, with that measured with the collocated MC–IC when both measurements were available (Fig. 2). The comparison demonstrates good consistency across all fires, with the [HONO]ADS of all available fires falling within the first and third quartile of MC–IC HONO data. Additionally, we made intercomparisons between [HONO]ADS with mean values of various high-resolution methods including MC–IC, OP-FTIR, ACES, and PTR-ToF that are also available from the NOAA data archive (Fig. 3; FIREX, 2016). The mean values used for the comparison are shown in Table S2. The linear regression results for all four comparisons are as follows:
We found significant linear correlations between each of the [HONO] techniques and [HONO]ADS with a slope of ∼1. Note that the y intercepts of Eqs. (1)–(4) are much smaller than the overall range of measured [HONO] (up to 121 ppbv). In addition, the p values of the intercepts for all four fittings are much greater than 0.05, suggesting the intercepts are not significantly different from zero. All data points except one fall within the 95 % prediction interval bounds of the overall fitting (Fig. 3). Therefore, we conclude that the ADS method has a high capture efficiency of HONO in the biomass combustion environment, which ensures the accuracy of the isotopic composition analysis and applicability of this method for field-based biomass combustion research.
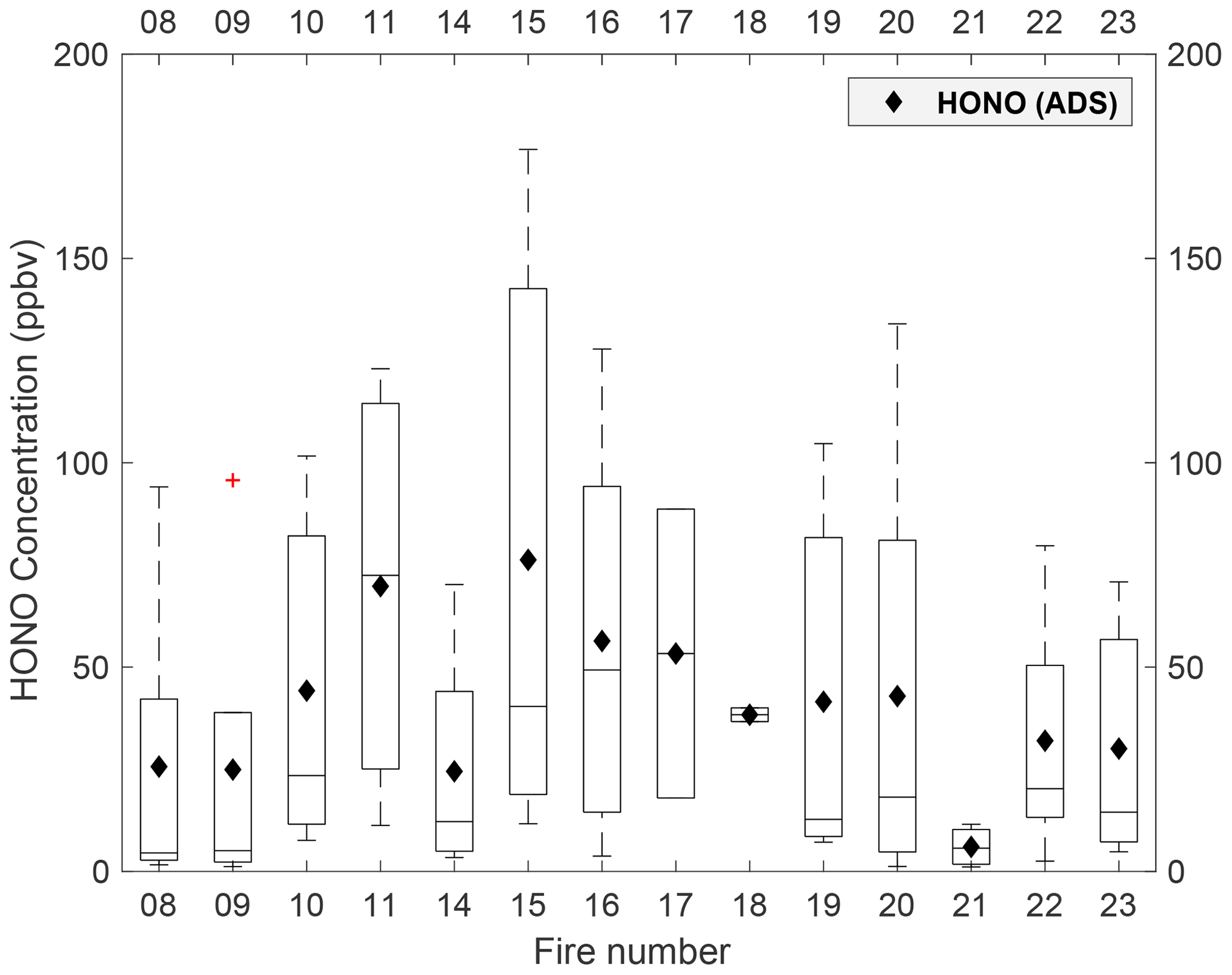
Figure 2Box plot of MC–IC HONO measurement with 5 min resolution over the course of each fire. Each box whisker represents the 5th, 25th, 50th, 75th, and 95th percentile of the HONO concentration during each collection period. The black diamond is the mean HONO concentration recovered from ADS collection. The red cross symbolizes outliers. Note that no isotopic analysis was performed for fire no. 12 (shown in Fig. 1) due to an insufficient amount of collected nitrite.
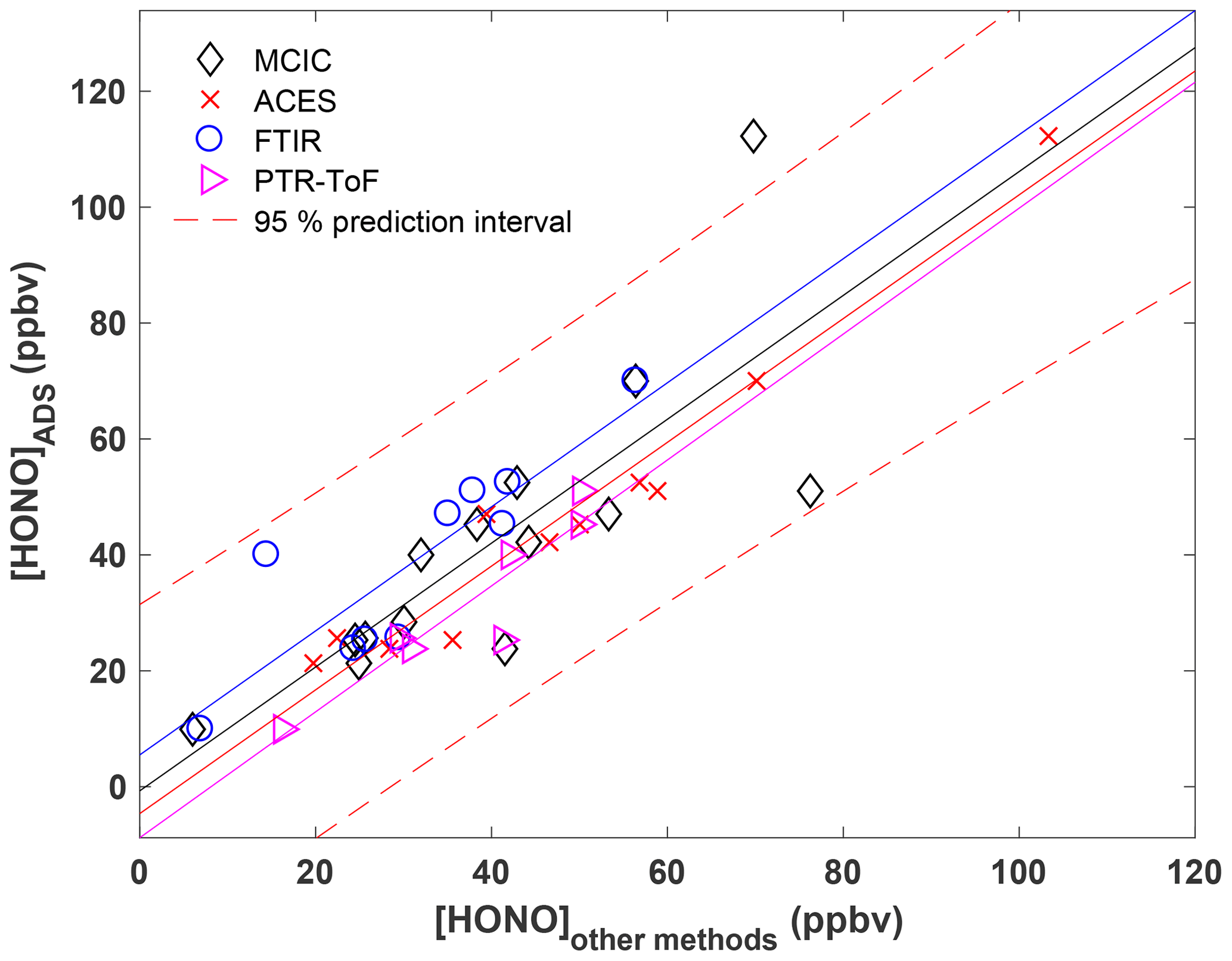
Figure 3Comparison of ADS-measured HONO concentration with the mean values of various high-resolution methods including MC–IC, FTIR, ACES, and PTR-ToF for available fires. Solid lines are linear regressions of each dataset with the same symbol color.
3.3 Isotopic composition of HONO and NOx from burning different biomass
δ15N of NOx and HONO emitted from burning various biomass types in this study ranged from −4.3 ‰ to +7.0 ‰ and −5.3 ‰ to +5.8 ‰, respectively (Table 1). There is no direct dependence of δ15N on the concentration of either HONO or NOx (Fig. S2). In Fig. 4, δ15N values of NOx and HONO are shown for each biomass type. Each value represents a concentration-weighted mean (if multiple samples were collected for a biomass type), with error bars representing the propagation of replicate variation and method precision. For biomass types burned in replicate (ponderosa pine, lodgepole pine, Engelmann spruce, and Douglas fir), the δ15N–NOx and δ15N–HONO variation within a given biomass type is smaller than the full range across all fuel types. Additionally, we note that the variations of δ15N–NOx and δ15N–HONO for ponderosa pine and δ15N–HONO for Engelmann spruce are larger than the method analytical precision of δ15N–NOx (1.5 ‰) and δ15N–HONO (0.5 ‰), respectively, which represents fire-by-fire variation, likely due to different combustion conditions and/or different fuel compositions. For example, fuel moisture content derived from the original biomass weight and dry biomass weight reveal that the ponderosa pine burned in fire no. 3 had more moisture content (48.1 %) than fire no. 2 (32.1 %), which could affect combustion temperature and thus product formation. Figure 4 also illustrates that burning different biomass parts from specific vegetation can result in fairly diverse δ15N–HONO and δ15N–NOx, e.g., among the ponderosa pine mixture, canopy, and litter, as well as between the Engelmann spruce mixture and duff.
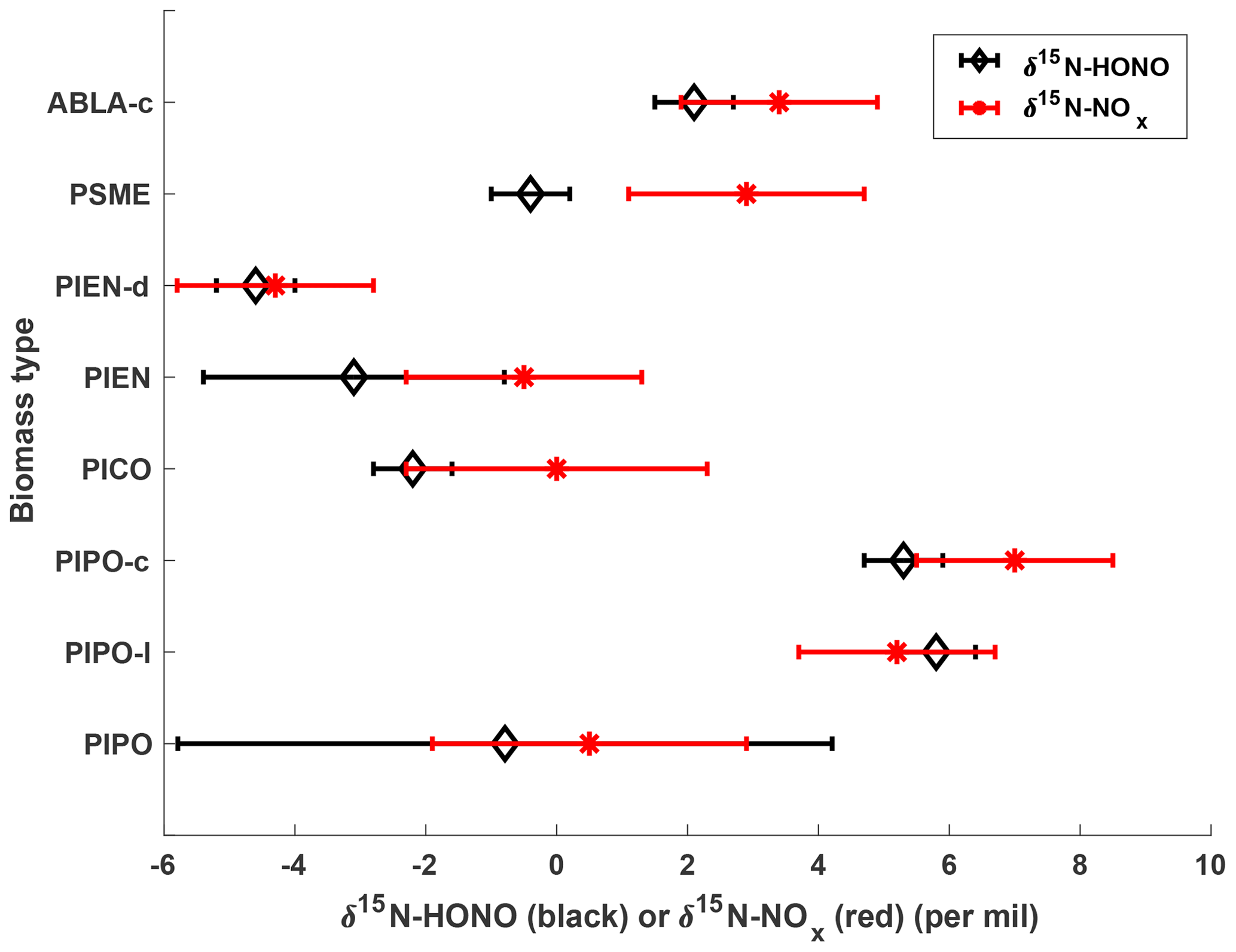
Figure 4Concentration-weighted mean δ15N of HONO and NOx versus biomass type. The error bars are the propagation of replicate ±1σ uncertainty (when n>1) and method uncertainty; otherwise, the error bars stand for method uncertainty. PIPO is ponderosa pine, PICO is lodgepole pine, PIEN is Engelmann spruce, PSME is Douglas fir, ABLA is subalpine (from Fish Lake, canopy); l indicates litter, c indicates canopy, d indicates duff.
Our δ15N–NOx range falls well within the range (−7 ‰ to +12 ‰) found in the FLAME-4 experiment (Fibiger and Hastings, 2016). The FLAME-4 study investigated NOx emissions from burning a relatively large range of vegetation biomass from all over the world and found a linear relationship (Eq. 5), indicating that 83 % of the variation of δ15N–NOx is explained by δ15N–biomass. The biomass types burned in this work focused on vegetation in the western US and differ greatly from that in FLAME-4, with ponderosa pine being the only common biomass between the two studies. Specifically, the δ15N–biomass range (−4.2 ‰ to +0.9 ‰) for this work is much narrower than that of the FLAME-4 experiment (−8 ‰ to +8 ‰).
To compare with the relationship found in Fibiger and Hastings (2016) we mass-weighted the contributions from different components of the same biomass type. For the same type of biomass, δ15N–biomass varies amongst different parts of the vegetation, with differences as great as 4.1 ‰, 2.4 ‰, 4.6 ‰, and 2.6 ‰ for ponderosa pine, lodgepole pine, Douglas fir, and Engelmann spruce, respectively (Table S1). In the FIREX experiments, many of the burns were conducted for mixtures of various vegetation parts. For instance, one ponderosa pine fire contains canopy (∼30 %), litter (∼28 %), and other parts (∼42 %) including duff and shrub, and the compositions vary slightly amongst each burn. Therefore, the δ15N of a particular biomass mixture is mass-weighted according to its composition contribution from each part (Table S1). Similarly, the δ15N–NOx and δ15N–HONO from fires of different biomass parts are weighted by concentrations for each biomass type, i.e., ponderosa pine (including mixture, canopy, and litter) and Engelmann spruce (including mixture and duff), to produce a signature associated with the combustion of that biomass type.
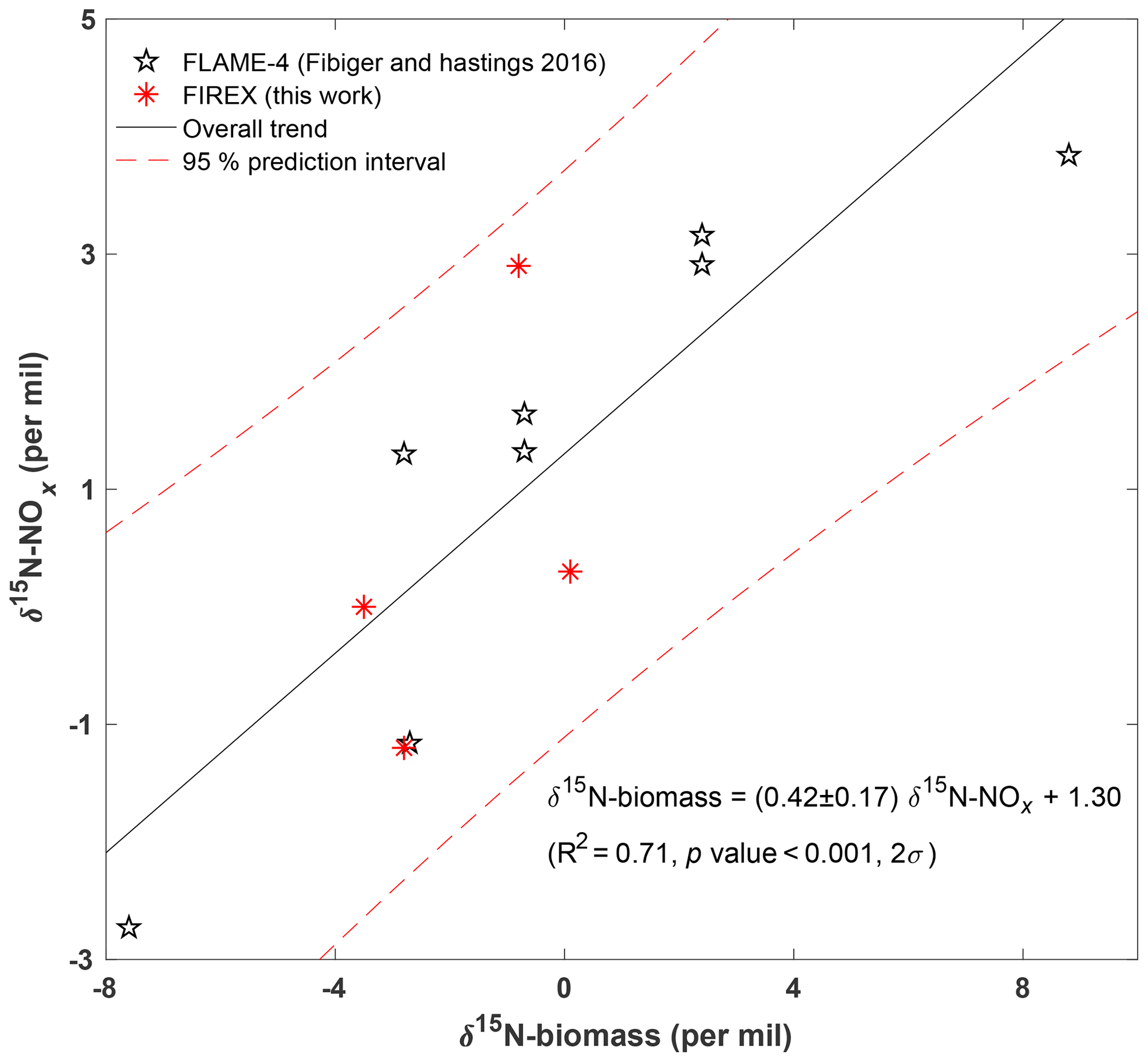
Figure 5Dependence of δ15N–NOx on δ15N–biomass. Star data points represent results from the FLAME-4 study (Fibiger and Hastings, 2016); asterisk data points represent results from this work; the solid line is a linear regression between δ15N–NOx and δ15N–biomass for the combined dataset; dashed lines are 95 % prediction intervals (2σ).
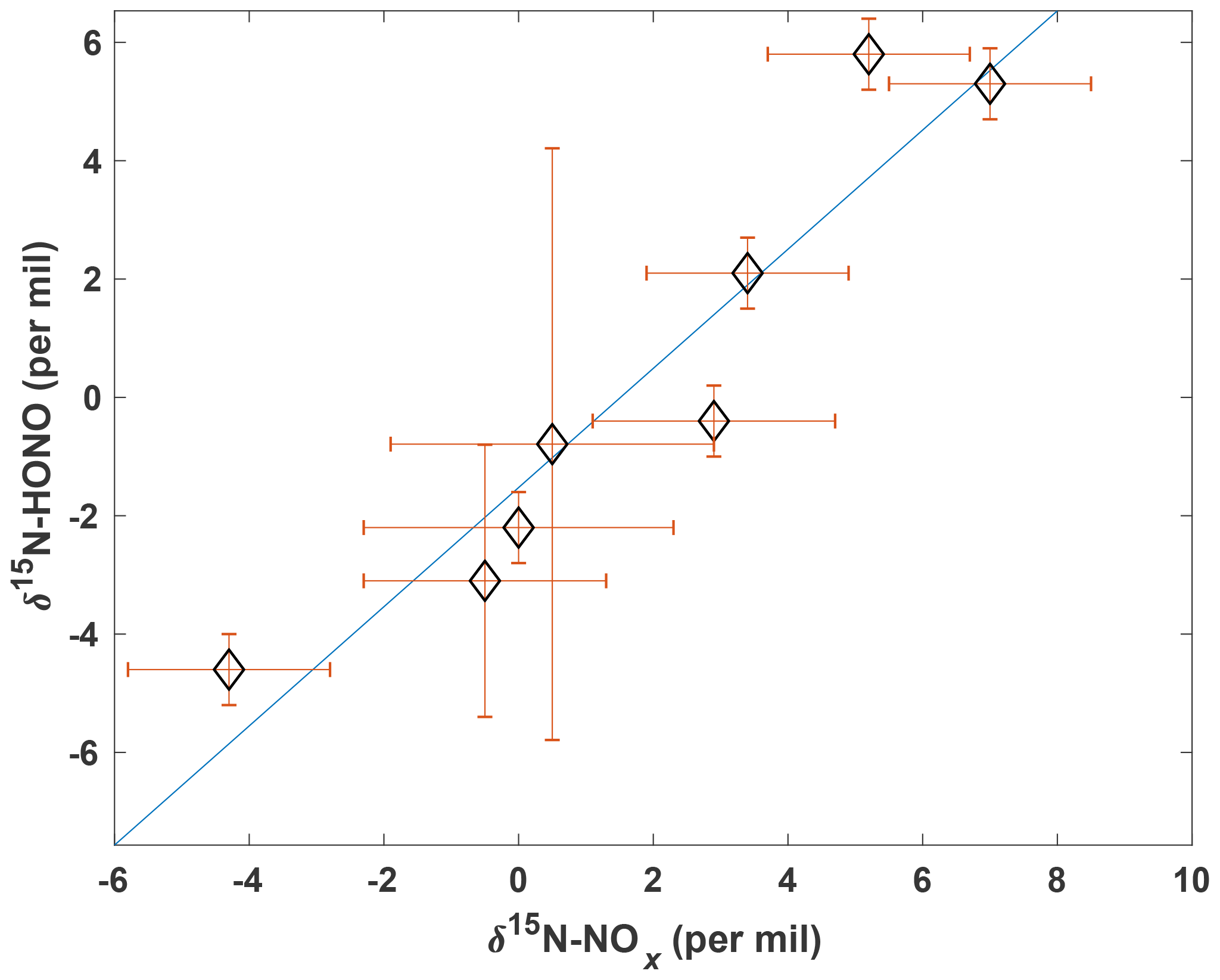
Figure 6Scatter plot between δ15N–HONO and δ15N–NOx. All error bars are the propagation of replicate uncertainty (±1σ) and method uncertainty. Linear regression follows , p<0.001).
For purposes of comparison among different biomass types, we average δ15N–NOx (δ15N–HONO) weighted by concentrations for each biomass type, i.e., ponderosa pine (including mixture, canopy, and litter) and Engelmann spruce (including mixture and duff) (all data are listed in Table S3). Linear regressions between δ15N–HONO and δ15N–biomass, as well as that between δ15N–NOx and δ15N–biomass, show that both δ15N–HONO and δ15N–NOx increase with δ15N–biomass in general (Fig. S3). However, the linear regressions performed here are limited by small datasets (four data points each) and unsurprisingly yield insignificant linear correlations for δ15N–HONO (or δ15N–NOx) versus δ15N–biomass (p values are 0.1 and 0.5, respectively). Still, combining our results of δ15N–NOx versus δ15N–biomass from this work with those from the FLAME-4 study (Fibiger and Hastings, 2016) results in a significant linear correlation (Eq. 6) and is shown in Fig. 5. Despite differences in burned biomass types between the two studies, our δ15N–NOx values reasonably overlap the FLAME-4 results within our δ15N–biomass range. The relationship between δ15N–NOx and δ15N–biomass (Eq. 6) for the combined data highly reproduces that obtained solely from the FLAME-4 study (Eq. 5) and confirms the dependence of δ15N–NOx on δ15N–biomass.
The mean values weighted by concentration plotted in Fig. 4 show that 15N of HONO is consistently slightly more depleted than that of NOx (δ15N–HONO < δ15N–NOx) across all the biomass types, except for ponderosa pine (litter), which results in an opposite relationship between δ15N–HONO and δ15N–NOx. Furthermore, δ15N–HONO is linearly correlated with δ15N–NOx following a relationship of Eq. (7) within the δ15N–NOx and δ15N–HONO range obtained in the current study (Fig. 6). This provides potential insights into HONO–NOx interactions and HONO formation pathways in fresh emissions from biomass burning. Although a number of studies on wildfire biomass burning have suggested that partitioning of N emissions between NOx and NH3 depends on combustion conditions represented by MCE (Ferek et al., 1998; Goode et al., 1999; McMeeking et al., 2009; Yokelson et al., 1996, 2008), HONO formation pathways remain unclear (Alvarado et al., 2009, 2015; Nie et al., 2015).
Previous mechanistic studies on the combustion of biomass–biofuel model compounds in a well-controlled closed system have investigated detailed nitrogen chemistry in the gas phase, suggesting that NOx and HONO are formed from chain reactions involving the oxidation of the precursors NH3 and HCN, which are produced via the devolatilization and pyrolysis of amines and proteins in biomass–biofuel (Houshfar et al., 2012; Lucassen et al., 2011). When the combustion conditions favor the oxidation of NH3 and HCN, NO is first formed and the chain reactions control the cycling of reactive nitrogen species (NO, NO2, and HONO). Detailed and mechanistic nitrogen chemistry for the chemical relationship between NOx and HONO in the combustion environment has been discussed in earlier works (Chai and Goldsmith, 2017; Shrestha et al., 2018; Skreiberg et al., 2004). In addition, Houshfar et al. (2012) performed biomass combustion kinetic modeling with a reduced mechanism via sensitivity analysis. From these works, we extract major pathway Reactions (R1)–(R11) that are likely responsible for fast gas-phase interconversion between NOx and HONO within the combustion system. It has been found that whether HONO is preferably converted from NO or NO2 in series during nitrogen transformation (referred to as nitrogen flow) critically depends on temperature. Specifically, within 1 s of residence time, at moderate temperatures (e.g., 700 ∘C), preferable nitrogen flow following NO formation in biomass combustion is , and major reactions involving NOx–HONO conversion are listed in Reactions (R1)–(R6); at high temperatures (e.g., 850 ∘C and above), the nitrogen flow cycle becomes preferable, and major reactions involving NOx–HONO are Reactions (R7)–(R11).
Although our studied fuels are more complicated in composition than a model system involving no more than a few starting species, results from the above studies provide fundamental underpinnings for biomass combustion. Also note that heterogeneous chemistry after these species were emitted was not considered here as the residence time of the fresh plume in our study was ∼5 s, which is of the same magnitude as that predicted in the nitrogen flow analysis (Houshfar et al., 2012). Kinetic isotope effects (KIEs) of these reactions have not been characterized, so only a semi-quantitative prediction is presented here. At low temperatures, Reactions (R1)–(R5) are all H-abstraction reactions involving loose transition states that have significant activation energy; a primary KIE is expected for such conditions and leads to 15N depletion in the product (HONO) (Chai et al., 2014; Matsson and Westaway, 1999, and references therein). Additionally, Reaction (R6) is a unimolecular dissociation reaction with no reaction barrier, and hence Reaction (R6) could be expected to have a small kinetic isotope effect enriching 15N in HONO, somewhat offsetting the depletion that arose from Reactions (R1)–(R5). Consequently, the overall isotope effect of Reactions (R1)–(R6) would lead to δ15N–HONO < δ15N–NOx by a small difference, consistent with our results (Fig. 4). On the other hand, the KIE for the Reactions (R7)–(R11) at higher temperatures (> 850 ∘C) is expected to enrich 15N in HONO relative to NOx (Chai and Dibble, 2014), leading to an opposite isotope effect to that predicted at lower temperatures.
The temperatures of the biomass combustion process span a large range involving different processes, including preheating, drying, distillation, pyrolysis, gasification (also called “glowing combustion”), and oxidation in turbulent diffusion flames at a range of temperatures associated with changing flame dynamics (Yokelson et al., 1996). Despite this complexity, our measured slight 15N enrichment in NOx compared to HONO (Table 1, Fig. 4) suggests that Reactions (R1)–(R6) played a more important role than Reactions (R7)–(R11) in HONO formation during the FIREX Fire Lab experiments.
3.4 Isotopic composition of nitrates collected on particle filters
All Nylasorb filter extract solutions showed no detectable and concentrations, indicating that no significant amount of HNO3 was collected on these filters, which is consistent with the very low concentrations measured by MC–IC (note that a low concentration and limited sample volume also preclude further isotopic analysis of HNO3 collected by MC–IC). By contrast, we found that 5 out of 20 particulate filter extract solutions had detectable concentrations that were sufficient (10 nmol N) for isotopic composition analysis (Table 1). δ15N and δ18O reported here are considered to represent p. δ15N–p of the five samples (burns) ranges from −10.6 ‰ to −7.4 ‰, all of which are more 15N-depleted than that of HONO and NOx. In addition, the smaller range of than that of δ15N–HONO and δ15N–NOx rules out the possible transformation of NOx and HONO to nitrate on the filters, which could distort the isotopic composition of NOx and HONO.
In the FLAME-4 experiments, only one particulate filter had captured p above the concentration detection limit, whereas the HNO3 collected on Nylasorb filters from seven experiments was above the concentration detection limit, and therefore only δ15N–HNO3 (−0.3 ‰ to 11.2 ‰) was reported (Fibiger and Hastings, 2016). The contrast with our filter results is likely attributed to different formation mechanisms under different conditions, in addition to variation of fuel types. Of the seven detectable HNO3 collections from FLAME-4, five represented room burns for which samples were collected from smoke aged for 1–2 h in the lab, and the sampled HNO3 was likely a secondary product. By contrast, all our observed p was in fresh emissions and may have been derived from plant nitrate (Cárdenas-Navarro et al., 1999) and/or combustion reactions. There have been no other studies on δ15N of p and HNO3 directly emitted from fresh plumes to the best of our knowledge, so more investigation using both laboratory work (isotope effect) and kinetic modeling will be needed in order to understand the formation mechanisms of HNO3 and p in the biomass combustion process and their respective isotope effects.
In addition to δ15N, we report δ18O of HONO and p directly emitted from biomass burning plumes with ranges of 5.2 ‰ to 15.2 ‰ and 11.5 ‰ to 14.8 ‰, respectively. These are the first observations reported for δ18O of reactive nitrogen species directly emitted from biomass burning, and low values are expected for the δ18O, which in this case is mainly extracted from that of molecular oxygen (δ18O = ∼23.5 ‰) (Kroopnick and Craig, 1972), biomass–cellulose (δ18O = 15 ‰–35 ‰), and/or biomass-contained water (δ18O = ∼0 ‰–16 ‰) (Keel et al., 2016). In field studies for which photochemistry and O3 are inevitably involved in the reactive nitrogen cycle in various stages of aged plumes, we expect to see much more elevated δ18O values of HONO and p due to the extremely high value of δ18O–O3 (∼110 ‰) (Vicars and Savarino, 2014). Therefore, the δ18O found in the lab is helpful in understanding the conditions under which photochemistry would not apply (e.g., nighttime fresh smoke) and should be distinguishable from the expected higher δ18O that would be found in aged smoke and/or daytime fresh smoke.
In this study we applied new methods for characterizing the isotopic composition of reactive nitrogen species, including NOx (δ15N), HONO (δ15N and δ18O), and p (δ15N and δ18O), emitted directly from biomass burning. We measured fresh (stack) emissions from 20 laboratory fires of different fuels during the 2016 FIREX Fire Lab experiments. NOx, HONO, and HNO3 emitted in fresh smoke reached their peak in most of our fires within 5 min of ignition of biomass (i.e., when flaming combustion peaked). The HONO mixing ratio was typically ∼2 orders of magnitude larger than HNO3, and the HONO∕NOx ratio ranged from 0.13 to 0.53.
Our HONO collection method (ADS) for isotopic analysis was applied to biomass burning (BB) for the first time. The good agreement for concentration comparisons between our method and four high-time-resolution HONO concentration methods suggests high collection efficiency of HONO from BB emissions, which ensures an accurate isotopic compositional analysis. Comparisons with concurrent observations and a previous study show that the combination of our HONO and NOx collection methods are compatible, allowing for simultaneous determination of the isotopic composition of both HONO and NOx. This provides important potential for investigating the photochemical and non-photochemical relationships between HONO and NOx in a variety of environments, especially in BB plumes.
δ15N–NOx emitted from burning various western US biomass types in this study ranged from −4.3 ‰ to +7.0 ‰, falling well within the range found by Fibiger and Hastings (2016), although the vegetation types were much broader in the earlier study. We report the first δ15N–HONO emitted directly from burning, ranging from −5.3 ‰ to +5.8 ‰. The δ15N–NOx and δ15N–HONO range derived from BB can be further compared with that from other sources using the same methods presented here and provide insights into source signatures for both NOx and HONO. This study also showed the important capability of determining δ18O–HONO and δ18O–p from BB plumes, and we expect that the δ18O of both HONO and p produced under photochemical conditions will be much higher than the lab results due to the important role of O3 in reactive nitrogen oxidation.
Interestingly, the linear correlation between δ15N–HONO and δ15N–NOx for the biomass we studied suggests that the systematic coproduction of NOx and HONO occurs during biomass combustion, and both of them are released as primary pollutants in fresh smoke. The relationship between δ15N–HONO and δ15N–NOx likely reflects the fact that HONO was produced to a larger extent at moderate combustion temperatures (< ∼800 ∘C) than higher temperatures on the basis of a simplified mechanism for the flow of reactive nitrogen species. However, we note that this relationship is derived from all measured δ15N–HONO and δ15N–NOx in fires ranging from smoldering to flaming, so is not necessarily representative of a particular combustion condition. Still, it is likely that a compilation over a range of conditions is more useful for potentially distinguishing HONO sources and formation pathways in the environment since it will always be a challenge to assess exact combustion temperatures. Determining these relationships in real wildfire smoke will be essential for better constraint on NOx and HONO budgets, and it may eventually improve ozone and secondary aerosol predictions for regional air quality.
The data from the laboratory tests are available on request from the corresponding authors. Data from the 2016 Missoula Fire Sciences Lab are available here: https://esrl.noaa.gov/csd/groups/csd7/measurements/2016firex/FireLab/DataDownload/ (National Oceanic and Atmospheric Administration, 2019).
The supplement related to this article is available online at: https://doi.org/10.5194/amt-12-6303-2019-supplement.
JC, MH, and JD designed this work. JC and DJM conducted the sample collections at the Fire Lab, with additional support from MH, JD, and ES. JC carried out the isotopic composition measurements; DJM supported the isotopic research and interpretation. ES helped analyze the MC–IC data. VS and RY provided the OP-FTIR data. KJZ and SSB provided the ACES data. ARK and CW provided the PTR-ToF data. JC wrote the paper, and all authors provided edits and feedback.
The authors declare that they have no conflict of interest.
We acknowledge financial support from the National Oceanic and Atmospheric Administration (AC4 Award NA16OAR4310098 to MH and JED) and the National Science Foundation (AGS-1351932 to MH). We are grateful to Ruby Ho for laboratory support and Marshall Otter for the biomass δ15N analysis. We also thank James Roberts and Matthew Coggon for helpful discussions. We are thankful to the two anonymous reviewers for their helpful comments.
This research has been supported by the National Oceanic and Atmospheric Administration (grant no. NA16OAR4310098) and the National Science Foundation, Division of Atmospheric and Geospace Sciences (grant no. 1351932).
This paper was edited by Maria Dolores Andrés Hernández and reviewed by two anonymous referees.
Akagi, S. K., Craven, J. S., Taylor, J. W., McMeeking, G. R., Yokelson, R. J., Burling, I. R., Urbanski, S. P., Wold, C. E., Seinfeld, J. H., Coe, H., Alvarado, M. J., and Weise, D. R.: Evolution of trace gases and particles emitted by a chaparral fire in California, Atmos. Chem. Phys., 12, 1397–1421, https://doi.org/10.5194/acp-12-1397-2012, 2012.
Akagi, S. K., Yokelson, R. J., Burling, I. R., Meinardi, S., Simpson, I., Blake, D. R., McMeeking, G. R., Sullivan, A., Lee, T., Kreidenweis, S., Urbanski, S., Reardon, J., Griffith, D. W. T., Johnson, T. J., and Weise, D. R.: Measurements of reactive trace gases and variable O3 formation rates in some South Carolina biomass burning plumes, Atmos. Chem. Phys., 13, 1141–1165, https://doi.org/10.5194/acp-13-1141-2013, 2013.
Alvarado, M. J. and Prinn, R. G.: Formation of ozone and growth of aerosols in young smoke plumes from biomass burning: 1. Lagrangian parcel studies, J. Geophys. Res.-Atmos., 114, D09306, https://doi.org/10.1029/2008JD011144, 2009.
Alvarado, M. J., Wang, C., and Prinn, R. G.: Formation of ozone and growth of aerosols in young smoke plumes from biomass burning: 2. Three-dimensional Eulerian studies, J. Geophys. Res.-Atmos., 114, D09307, https://doi.org/10.1029/2008JD011186, 2009.
Alvarado, M. J., Lonsdale, C. R., Yokelson, R. J., Akagi, S. K., Coe, H., Craven, J. S., Fischer, E. V., McMeeking, G. R., Seinfeld, J. H., Soni, T., Taylor, J. W., Weise, D. R., and Wold, C. E.: Investigating the links between ozone and organic aerosol chemistry in a biomass burning plume from a prescribed fire in California chaparral, Atmos. Chem. Phys., 15, 6667–6688, https://doi.org/10.5194/acp-15-6667-2015, 2015.
Böhlke, J. K., Mroczkowski, S. J., and Coplen, T. B.: Oxygen isotopes in nitrate: new reference materials for measurements and observations on nitrate-water equilibration, Rapid Commun. Mass Spectrom., 17, 1835–1846, https://doi.org/10.1002/rcm.1123, 2003.
Burling, I. R., Yokelson, R. J., Griffith, D. W. T., Johnson, T. J., Veres, P., Roberts, J. M., Warneke, C., Urbanski, S. P., Reardon, J., Weise, D. R., Hao, W. M., and de Gouw, J.: Laboratory measurements of trace gas emissions from biomass burning of fuel types from the southeastern and southwestern United States, Atmos. Chem. Phys., 10, 11115–11130, https://doi.org/10.5194/acp-10-11115-2010, 2010.
Burling, I. R., Yokelson, R. J., Akagi, S. K., Urbanski, S. P., Wold, C. E., Griffith, D. W. T., Johnson, T. J., Reardon, J., and Weise, D. R.: Airborne and ground-based measurements of the trace gases and particles emitted by prescribed fires in the United States, Atmos. Chem. Phys., 11, 12197–12216, https://doi.org/10.5194/acp-11-12197-2011, 2011.
Cárdenas-Navarro, R., Adamowicz, S., and Robin, P.: Nitrate accumulation in plants: a role for water, J. Exp. Bot., 50, 613–624, https://doi.org/10.1093/jxb/50.334.613, 1999.
Casciotti, K. L., Sigman, D. M., Hastings, M. G., Böhlke, J. K., and Hilkert, A.: Measurement of the Oxygen Isotopic Composition of Nitrate in Seawater and Freshwater Using the Denitrifier Method, Anal. Chem., 74, 4905–4912, https://doi.org/10.1021/ac020113w, 2002.
Casciotti, K. L., Böhlke, J. K., McIlvin, M. R., Mroczkowski, S. J. and Hannon, J. E.: Oxygen Isotopes in Nitrite:? Analysis, Calibration, and Equilibration, Anal. Chem., 79, 2427–2436, https://doi.org/10.1021/ac061598h, 2007.
Chai, J. and Dibble, T. S.: Pressure Dependence and Kinetic Isotope Effects in the Absolute Rate Constant for Methoxy Radical Reacting with NO2, Int. J. Chem. Kinet., 46, 501–511, https://doi.org/10.1002/kin.20865, 2014.
Chai, J. and Goldsmith, C. F.: Rate coefficients for fuel + NO2: Predictive kinetics for HONO and HNO2 formation, Proc. Combust. Inst., 36, 617–626, https://doi.org/10.1016/j.proci.2016.06.133, 2017.
Chai, J. and Hastings, M.: Collection Method for Isotopic Analysis of Gaseous Nitrous Acid, Anal. Chem., 90, 830–838, https://doi.org/10.1021/acs.analchem.7b03561, 2018.
Chai, J., Hu, H., Dibble, T. S., Tyndall, G. S., and Orlando, J. J.: Rate Constants and Kinetic Isotope Effects for Methoxy Radical Reacting with NO2 and O2, J. Phys. Chem. A, 118, 3552–3563, 2014.
Christian, T. J., Kleiss, B., Yokelson, R. J., Holzinger, R., Crutzen, P. J., Hao, W. M., Shirai, T., and Blake, D. R.: Comprehensive laboratory measurements of biomass-burning emissions: 2. First intercomparison of open-path FTIR, PTR-MS, and GC-MS/FID/ECD, J. Geophys. Res.-Atmos., 109, D02311, https://doi.org/10.1029/2003JD003874, 2004.
Cook, P. A., Savage, N. H., Turquety, S., Carver, G. D., O'Connor, F. M., Heckel, A., Stewart, D., Whalley, L. K., Parker, A. E., Schlager, H., Singh, H. B., Avery, M. A., Sachse, G. W., Brune, W., Richter, A., Burrows, J. P., Purvis, R., Lewis, A. C., Reeves, C. E., Monks, P. S., Levine, J. G., and Pyle, J. A.: Forest fire plumes over the North Atlantic: p-TOMCAT model simulations with aircraft and satellite measurements from the ITOP/ICARTT campaign, J. Geophys. Res.-Atmos., 112, D10S43, https://doi.org/10.1029/2006JD007563, 2007.
Crutzen, P. J. and Andreae, M. O.: Biomass burning in the tropics: impact on atmospheric chemistry and biogeochemical cycles, Science, 250, 1669–1678, https://doi.org/10.1126/science.250.4988.1669, 1990.
Dibb, J. E., Arsenault, M., Peterson, M. C., and Honrath, R. E.: Fast nitrogen oxide photochemistry in Summit, Greenland snow, Atmos. Environ., 36, 2501–2511, https://doi.org/10.1016/S1352-2310(02)00130-9, 2002.
Ferek, R., Reid, J., Hobbs, P., Blake, D., and Liousse, C.: Emission factors of hydrocarbons, halocarbons, trace gases and particles from biomass burning in Brazil, J. Geophys. Res.-Atmos., 103, 32107–32118, https://doi.org/10.1029/98JD00692, 1998.
Fibiger, D. L. and Hastings, M. G.: First Measurements of the Nitrogen Isotopic Composition of NOx from Biomass Burning, Environ. Sci. Technol., 50, 11569–11574, https://doi.org/10.1021/acs.est.6b03510, 2016.
Fibiger, D. L., Hastings, M. G., Lew, A. F., and Peltier, R. E.: Collection of NO and NO2 for Isotopic Analysis of NOx Emissions, Anal. Chem., 86, 12115–12121, https://doi.org/10.1021/ac502968e, 2014.
FIREX: FIREX 2016 Fire Lab Data Archive, available at: https://esrl.noaa.gov/csd/groups/csd7/measurements/2016firex/FireLab/DataDownload/index.php?page=/csd/groups/csd7/measurements/2016firex/FireLab/DataDownload/ (last access: 25 March 2019), 2016.
Goode, J. G., Yokelson, R. J., Susott, R. A., and Ward, D. E.: Trace gas emissions from laboratory biomass fires measured by open-path Fourier transform infrared spectroscopy: Fires in grass and surface fuels, J. Geophys. Res., 104, 21237–21245, https://doi.org/10.1029/1999JD900360, 1999.
Hastings, M. G., Casciotti, K. L., and Elliott, E. M.: Stable Isotopes as Tracers of Anthropogenic Nitrogen Sources, Deposition, and Impacts, Elements, 9, 339–344, https://doi.org/10.2113/gselements.9.5.339, 2013.
Houshfar, E., Skreiberg, Ø., Glarborg, P., and Løvås, T.: Reduced chemical kinetic mechanisms for NOx emission prediction in biomass combustion, Int. J. Chem. Kinet., 4, 219–231, https://doi.org/10.1002/kin.20716, 2012.
Jaeglé, L., Steinberger, L., Martin, R. V., and Chance, K.: Global partitioning of NOx sources using satellite observations: relative roles of fossil fuel combustion, biomass burning and soil emissions, Faraday Discuss., 130, 407–423, 2005.
Jaffe, D. and Briggs, N.: Ozone production from wildfires: A critical review, Atmos. Environ., 51, 1–10, https://doi.org/10.1016/j.atmosenv.2011.11.063, 2012.
Johnson, T. J., Profeta, L. T. M., Sams, R. L., Griffith, D. W. T., and Yokelson, R. L.: An infrared spectral database for detection of gases emitted by biomass burning, Vibrational Spectroscopy, 53, 97–102, https://doi.org/10.1016/j.vibspec.2010.02.010, 2010.
Johnson, T. J., Sams, R. L., Profeta, L. T. M., Akagi, S. K., Burling, I. R., Yokelson, R. J., and Williams, S. D.: Quantitative IR Spectrum and Vibrational Assignments for Glycolaldehyde Vapor: Glycolaldehyde Measurements in Biomass Burning Plumes, J. Phys. Chem. A, 117, 4096–4107, https://doi.org/10.1021/jp311945p, 2013.
Keel, S. G., Joos, F., Spahni, R., Saurer, M., Weigt, R. B., and Klesse, S.: Simulating oxygen isotope ratios in tree ring cellulose using a dynamic global vegetation model, Biogeosciences, 13, 3869–3886, https://doi.org/10.5194/bg-13-3869-2016, 2016.
Keene, W. C., Lobert, J. M., Crutzen, P. J., Maben, J. R., Scharffe, D. H., Landmann, T., Hély, C., and Brain, C.: Emissions of major gaseous and particulate species during experimental burns of southern African biomass, J. Geophys. Res.-Atmos., 111, D04301, https://doi.org/10.1029/2005JD006319, 2006.
Koss, A. R., Sekimoto, K., Gilman, J. B., Selimovic, V., Coggon, M. M., Zarzana, K. J., Yuan, B., Lerner, B. M., Brown, S. S., Jimenez, J. L., Krechmer, J., Roberts, J. M., Warneke, C., Yokelson, R. J., and de Gouw, J.: Non-methane organic gas emissions from biomass burning: identification, quantification, and emission factors from PTR-ToF during the FIREX 2016 laboratory experiment, Atmos. Chem. Phys., 18, 3299–3319, https://doi.org/10.5194/acp-18-3299-2018, 2018.
Kraus, S.: DOASIS: a framework design for DOAS, PhD thesis, University of Mannheim, Germany, 162 pp., 2006.
Kroopnick, P. and Craig, H.: Atmospheric oxygen: isotopic composition and solubility fractionation, Science, 175, 54–55, https://doi.org/10.1126/science.175.4017.54, 1972.
Lapina, K., Honrath R. E., Owen R. C., Val Martín M., Hyer E. J., and Fialho P.: Late summer changes in burning conditions in the boreal regions and their implications for NOx and CO emissions from boreal fires, J. Geophys. Res.-Atmos., 113, D11304, https://doi.org/10.1029/2007JD009421, 2008.
Liu, X., Zhang, Y., Huey, L. G., Yokelson, R. J., Wang, Y., Jimenez, J. L., Campuzano-Jost, P., Beyersdorf, A. J., Blake, D. R., Choi, Y., Clair, J. M. S., Crounse, J. D., Day, D. A., Diskin, G. S., Fried, A., Hall, S. R., Hanisco, T. F., King, L. E., Meinardi, S., Mikoviny, T., Palm, B. B., Peischl, J., Perring, A. E., Pollack, I. B., Ryerson, T. B., Sachse, G., Schwarz, J. P., Simpson, I. J., Tanner, D. J., Thornhill, K. L., Ullmann, K., Weber, R. J., Wennberg, P. O., Wisthaler, A., Wolfe, G. M., and Ziemba, L. D.: Agricultural fires in the southeastern U.S. during SEAC4RS: Emissions of trace gases and particles and evolution of ozone, reactive nitrogen, and organic aerosol, J. Geophys. Res.-Atmos., 121, 7383–7414, https://doi.org/10.1002/2016JD025040, 2016.
Lucassen, A., Labbe, N., Westmoreland, P. R., and Kohse-Höinghaus, K.: Combustion chemistry and fuel-nitrogen conversion in a laminar premixed flame of morpholine as a model biofuel, Combust. Flame, 158, 1647–1666, https://doi.org/10.1016/j.combustflame.2011.02.010, 2011.
Matsson, O. and Westaway, K. C.: Secondary Deuterium Kinetic Isotope Effects and Transition State Structure, in: Advances in Physical Organic Chemistry, vol. 31, edited by: Bethell, D., 143–248, Academic Press, Cambridge, Massachusetts, USA, 1999.
McMeeking, G. R., Kreidenweis, S. M., Baker, S., Carrico, C. M., Chow, J. C., Collett, J. L., Hao, W. M., Holden, A. S., Kirchstetter, T. W., Malm, W. C., Moosmüller, H., Sullivan, A. P., and Wold, C. E.: Emissions of trace gases and aerosols during the open combustion of biomass in the laboratory, J. Geophys. Res.-Atmos., 114, D19210, https://doi.org/10.1029/2009JD011836, 2009.
Miller, D. J., Wojtal, P. K., Clark, S. C., and Hastings, M. G.: Vehicle NOx emission plume isotopic signatures: Spatial variability across the eastern United States, J. Geophys. Res.-Atmos., 122, 4698–4717, https://doi.org/10.1002/2016JD025877, 2017.
Miller, D. J., Chai, J., Guo, F., Dell, C. J., Karsten, H., and Hastings, M. G.: Isotopic Composition of In Situ Soil NOx Emissions in Manure-Fertilized Cropland, Geophys. Res. Lett., 45, 12058–12066, https://doi.org/10.1029/2018GL079619, 2018.
Min, K.-E., Washenfelder, R. A., Dubé, W. P., Langford, A. O., Edwards, P. M., Zarzana, K. J., Stutz, J., Lu, K., Rohrer, F., Zhang, Y., and Brown, S. S.: A broadband cavity enhanced absorption spectrometer for aircraft measurements of glyoxal, methylglyoxal, nitrous acid, nitrogen dioxide, and water vapor, Atmos. Meas. Tech., 9, 423–440, https://doi.org/10.5194/amt-9-423-2016, 2016.
National Oceanic and Atmospheric Administration: Fire Lab Study, Earth System Laboratory, Chemical Sciences Division, available at: https://esrl.noaa.gov/csd/groups/csd7/measurements/2016firex/FireLab/DataDownload/index.php?page=/csd/groups/csd7/measurements/2016firex/FireLab/DataDownload/, last access: 28 November 2019.
Neuman, J. A., Trainer, M., Brown, S. S., Min, K.-E., Nowak, J. B., Parrish, D. D., Peischl, J., Pollack, I. B., Roberts, J. M., Ryerson, T. B., and Veres, P. R.: HONO emission and production determined from airborne measurements over the Southeast U.S., J. Geophys. Res.-Atmos., 121, 9237–9250, https://doi.org/10.1002/2016JD025197, 2016.
Nie, W., Ding, A. J., Xie, Y. N., Xu, Z., Mao, H., Kerminen, V.-M., Zheng, L. F., Qi, X. M., Huang, X., Yang, X.-Q., Sun, J. N., Herrmann, E., Petäjä, T., Kulmala, M., and Fu, C. B.: Influence of biomass burning plumes on HONO chemistry in eastern China, Atmos. Chem. Phys., 15, 1147–1159, https://doi.org/10.5194/acp-15-1147-2015, 2015.
Perrino, C., De Santis, F., and Febo, A.: Criteria for the choice of a denuder sampling technique devoted to the measurement of atmospheric nirous and nitric acids, Atmos. Environ., 24, 617–626, https://doi.org/10.1016/0960-1686(90)90017-H, 1990.
Pinto, J. P., Dibb, J., Lee, B. H., Rappenglück, B., Wood, E. C., Levy, M., Zhang, R.-Y., Lefer, B., Ren, X.-R., Stutz, J., Tsai, C., Ackermann, L., Golovko, J., Herndon, S. C., Oakes, M., Meng, Q.-Y., Munger, J. W., Zahniser, M., and Zheng, J.: Intercomparison of field measurements of nitrous acid (HONO) during the SHARP campaign, J. Geophys. Res.-Atmos., 119, 5583–5601, https://doi.org/10.1002/2013JD020287, 2014.
Reinhardt, E. D., Keane, R. E., and Brown, J. K.: First Order Fire Effects Model: FOFEM 4.0, user's guide, Gen Tech Rep INT-GTR-344 Ogden UT US Dep. Agric. For. Serv. Intermt. Res. Stn. 65 P, 344, https://doi.org/10.2737/INT-GTR-344, 1997.
Roberts, J. M., Veres, P., Warneke, C., Neuman, J. A., Washenfelder, R. A., Brown, S. S., Baasandorj, M., Burkholder, J. B., Burling, I. R., Johnson, T. J., Yokelson, R. J., and de Gouw, J.: Measurement of HONO, HNCO, and other inorganic acids by negative-ion proton-transfer chemical-ionization mass spectrometry (NI-PT-CIMS): application to biomass burning emissions, Atmos. Meas. Tech., 3, 981–990, https://doi.org/10.5194/amt-3-981-2010, 2010.
Rothman, L. S., Gordon, I. E., Barbe, A., Benner, D. C., Bernath, P. F., Birk, M., Boudon, V., Brown, L. R., Campargue, A., Champion, J.-P., Chance, K., Coudert, L. H., Dana, V., Devi, V. M., Fally, S., Flaud, J.-M., Gamache, R. R., Goldman, A., Jacquemart, D., Kleiner, I., Lacome, N., Lafferty, W. J., Mandin, J.-Y., Massie, S. T., Mikhailenko, S. N., Miller, C. E., Moazzen-Ahmadi, N., Naumenko, O. V., Nikitin, A. V., Orphal, J., Perevalov, V. I., Perrin, A., Predoi-Cross, A., Rinsland, C. P., Rotger, M., Šimečková, M., Smith, M. A. H., Sung, K., Tashkun, S. A., Tennyson, J., Toth, R. A., Vandaele, A. C., and Vander Auwera, J.: The HITRAN 2008 molecular spectroscopic database, J. Quant. Spectrosc. Ra., 110, 533–572, https://doi.org/10.1016/j.jqsrt.2009.02.013, 2009.
Scheuer, E., Talbot, R. W., Dibb, J. E., Seid, G. K., DeBell, L., and Lefer, B.: Seasonal distributions of fine aerosol sulfate in the North American Arctic basin during TOPSE, J. Geophys. Res.-Atmos., 108, TOP 18, https://doi.org/10.1029/2001JD001364, 2003.
Selimovic, V., Yokelson, R. J., Warneke, C., Roberts, J. M., de Gouw, J., Reardon, J., and Griffith, D. W. T.: Aerosol optical properties and trace gas emissions by PAX and OP-FTIR for laboratory-simulated western US wildfires during FIREX, Atmos. Chem. Phys., 18, 2929–2948, https://doi.org/10.5194/acp-18-2929-2018, 2018.
Sharpe, S. W., Johnson, T. J., Sams, R. L., Chu, P. M., Rhoderick, G. C., and Johnson, P. A.: Gas-phase databases for quantitative infrared spectroscopy, Appl Spectrosc., 58, 1452–1461, https://doi.org/10.1366/0003702042641281, 2004.
Shrestha, K. P., Seidel, L., Zeuch, T., and Mauss, F.: Detailed Kinetic Mechanism for the Oxidation of Ammonia Including the Formation and Reduction of Nitrogen Oxides, Energy Fuels, 32, 10202–10217, https://doi.org/10.1021/acs.energyfuels.8b01056, 2018.
Sigman, D. M., Casciotti, K. L., Andreani, M., Barford, C., Galanter, M., and Böhlke, J. K.: A Bacterial Method for the Nitrogen Isotopic Analysis of Nitrate in Seawater and Freshwater, Anal. Chem., 73, 4145–4153, https://doi.org/10.1021/ac010088e, 2001.
Skreiberg, Ø., Kilpinen, P., and Glarborg, P.: Ammonia chemistry below 1400 K under fuel-rich conditions in a flow reactor, Combust. Flame, 136, 501–518, https://doi.org/10.1016/j.combustflame.2003.12.008, 2004.
Stockwell, C. E., Yokelson, R. J., Kreidenweis, S. M., Robinson, A. L., DeMott, P. J., Sullivan, R. C., Reardon, J., Ryan, K. C., Griffith, D. W. T., and Stevens, L.: Trace gas emissions from combustion of peat, crop residue, domestic biofuels, grasses, and other fuels: configuration and Fourier transform infrared (FTIR) component of the fourth Fire Lab at Missoula Experiment (FLAME-4), Atmos. Chem. Phys., 14, 9727–9754, https://doi.org/10.5194/acp-14-9727-2014, 2014.
Stutz, J., Oh, H.-J., Whitlow, S. I., Anderson, C., Dibb, J. E., Flynn, J. H., Rappenglück, B., and Lefer, B.: Simultaneous DOAS and mist-chamber IC measurements of HONO in Houston, TX, Atmos. Environ., 44, 4090–4098, https://doi.org/10.1016/j.atmosenv.2009.02.003, 2010.
Tkacik, D. S., Robinson, E. S., Ahern, A., Saleh, R., Stockwell, C., Veres, P., Simpson, I. J., Meinardi, S., Blake, D. R., Yokelson, R. J., Presto, A. A., Sullivan, R. C., Donahue, N. M., and Robinson, A. L.: A dual-chamber method for quantifying the effects of atmospheric perturbations on secondary organic aerosol formation from biomass burning emissions, J. Geophys. Res.-Atmos., 122, 6043–6058, https://doi.org/10.1002/2016JD025784, 2017.
Travis, K. R., Jacob, D. J., Fisher, J. A., Kim, P. S., Marais, E. A., Zhu, L., Yu, K., Miller, C. C., Yantosca, R. M., Sulprizio, M. P., Thompson, A. M., Wennberg, P. O., Crounse, J. D., St. Clair, J. M., Cohen, R. C., Laughner, J. L., Dibb, J. E., Hall, S. R., Ullmann, K., Wolfe, G. M., Pollack, I. B., Peischl, J., Neuman, J. A., and Zhou, X.: Why do models overestimate surface ozone in the Southeast United States?, Atmos. Chem. Phys., 16, 13561–13577, https://doi.org/10.5194/acp-16-13561-2016, 2016.
Trentmann, J., Yokelson, R. J., Hobbs, P. V., Winterrath, T., Christian, T. J., Andreae, M. O., and Mason, S. A.: An analysis of the chemical processes in the smoke plume from a savanna fire, J. Geophys. Res.-Atmos., 110, D12301, https://doi.org/10.1029/2004JD005628, 2005.
Val Martín, M., Honrath, R. E., Owen, R. C., Pfister, G., Fialho, P., and Barata, F.: Significant enhancements of nitrogen oxides, black carbon, and ozone in the North Atlantic lower free troposphere resulting from North American boreal wildfires, J. Geophys. Res.-Atmos., 111, D23S60, https://doi.org/10.1029/2006JD007530, 2006.
Veres, P., Roberts, J. M., Burling, I. R., Warneke, C., de Gouw, J., and Yokelson, R. J.: Measurements of gas-phase inorganic and organic acids from biomass fires by negative-ion proton-transfer chemical-ionization mass spectrometry, J. Geophys. Res.-Atmos., 115, D23302, https://doi.org/10.1029/2010JD014033, 2010.
Vicars, W. C. and Savarino, J.: Quantitative constraints on the 17O-excess (Ä17O) signature of surface ozone: Ambient measurements from 50∘ N to 50∘ S using the nitrite-coated filter technique, Geochim. Cosmochim. Acta, 135, 270–287, https://doi.org/10.1016/j.gca.2014.03.023, 2014.
Wojtal, P. K., Miller, D. J., O'Conner, M., Clark, S. C., and Hastings, M. G.: Automated, High-resolution Mobile Collection System for the Nitrogen Isotopic Analysis of NOx, J. Vis. Exp. JoVE, 118, e54962, https://doi.org/10.3791/54962, 2016.
Ye, C., Zhou, X., Pu, D., Stutz, J., Festa, J., Spolaor, M., Tsai, C., Cantrell, C., Mauldin, R. L., Campos, T., Weinheimer, A., Hornbrook, R. S., Apel, E. C., Guenther, A., Kaser, L., Yuan, B., Karl, T., Haggerty, J., Hall, S., Ullmann, K., Smith, J. N., Ortega, J., and Knote, C.: Rapid cycling of reactive nitrogen in the marine boundary layer, Nature, 532, 489–491, https://doi.org/10.1038/nature17195, 2016.
Yokelson, R., Griffith, W. T. D., and Ward, D.: Open-path Fourier transform infrared studies of large-scale laboratory biomass fires, J. Geophys. Res., 101, 21067, https://doi.org/10.1029/96JD01800, 1996.
Yokelson, R. J., Karl, T., Artaxo, P., Blake, D. R., Christian, T. J., Griffith, D. W. T., Guenther, A., and Hao, W. M.: The Tropical Forest and Fire Emissions Experiment: overview and airborne fire emission factor measurements, Atmos. Chem. Phys., 7, 5175–5196, https://doi.org/10.5194/acp-7-5175-2007, 2007.
Yokelson, R. J., Christian, T. J., Karl, T. G., and Guenther, A.: The tropical forest and fire emissions experiment: laboratory fire measurements and synthesis of campaign data, Atmos. Chem. Phys., 8, 3509–3527, https://doi.org/10.5194/acp-8-3509-2008, 2008.
Yokelson, R. J., Crounse, J. D., DeCarlo, P. F., Karl, T., Urbanski, S., Atlas, E., Campos, T., Shinozuka, Y., Kapustin, V., Clarke, A. D., Weinheimer, A., Knapp, D. J., Montzka, D. D., Holloway, J., Weibring, P., Flocke, F., Zheng, W., Toohey, D., Wennberg, P. O., Wiedinmyer, C., Mauldin, L., Fried, A., Richter, D., Walega, J., Jimenez, J. L., Adachi, K., Buseck, P. R., Hall, S. R., and Shetter, R.: Emissions from biomass burning in the Yucatan, Atmos. Chem. Phys., 9, 5785–5812, https://doi.org/10.5194/acp-9-5785-2009, 2009.
Yuan, B., Koss, A., Warneke, C., Gilman, J. B., Lerner, B. M., Stark, H., and de Gouw, J. A.: A high-resolution time-of-flight chemical ionization mass spectrometer utilizing hydronium ions (H3O+ ToF-CIMS) for measurements of volatile organic compounds in the atmosphere, Atmos. Meas. Tech., 9, 2735–2752, https://doi.org/10.5194/amt-9-2735-2016, 2016.
Zarzana, K. J., Selimovic, V., Koss, A. R., Sekimoto, K., Coggon, M. M., Yuan, B., Dubé, W. P., Yokelson, R. J., Warneke, C., de Gouw, J. A., Roberts, J. M., and Brown, S. S.: Primary emissions of glyoxal and methylglyoxal from laboratory measurements of open biomass burning, Atmos. Chem. Phys., 18, 15451–15470, https://doi.org/10.5194/acp-18-15451-2018, 2018.
Zhou, S., Young, C. J., VandenBoer, T. C., Kowal, S. F., and Kahan, T. F.: Time-Resolved Measurements of Nitric Oxide, Nitrogen Dioxide, and Nitrous Acid in an Occupied New York Home, Environ. Sci. Technol., 52, 8355–8364, https://doi.org/10.1021/acs.est.8b01792, 2018.