the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 19 Jul 2021
| 19 Jul 2021
On-line solid phase microextraction derivatization for the sensitive determination of multi-oxygenated volatile compounds in air
Esther Borrás
Luis A. Tortajada-Genaro
Milagro Ródenas
Teresa Vera
Thomas Speak
Paul Seakins
Marvin D. Shaw
Alastair C. Lewis
Multi-oxygenated volatile organic compounds are important markers of air pollution and precursors of ozone and secondary aerosols in both polluted and remote environments. Herein, their accurate determination was enhanced. The approach was based on an automated system for active sampling and on-fibre derivatization coupled with the gas chromatography–mass spectrometry (GC–MS) technique.
The method capability was determined for different compound families, such as aldehydes, ketones, α-dicarbonyls, hydroxy-aldehydes, hydroxy-ketones, and carboxylic acids. A good accuracy (<7 %) was demonstrated from the results compared to Fourier-transform infrared spectroscopy (FTIR). Limits of detection (LODs) of 6–100 pptV were achieved with a time resolution lower than 20 min. The developed method was successfully applied to the determination of multi-oxygenated compounds in air samples collected during an intercomparison campaign (EUROCHAMP-2020 project). Also, its capability and accuracy for atmospheric monitoring was demonstrated in an isoprene ozonolysis experiment. Both were carried out in the high-volume outdoor atmospheric simulation chambers (EUPHORE, 200 m3).
In summary, our developed technique offers near-real-time monitoring with direct sampling, which is an advantage in terms of handling and labour time for a proper quantification of trace levels of atmospheric multi-oxygenated compounds.
- Article
(1057 KB) - Full-text XML
-
Supplement
(581 KB) - BibTeX
- EndNote





The interest in the assessment of multi-oxygenated volatile organic compounds, so-called oxygenated volatile organic compounds (OVOCs), has increased significantly in recent years. These compounds exist naturally in the environment but mainly come from the photo-oxidation of hydrocarbons in the atmosphere (Mellouki et al., 2015). They play an important role in secondary organic aerosol (SOA) and ozone formation, can potentially influence climate change, and cause negative health effects on humans (Atkinson and Arey, 2003). Their tropospheric concentrations are highly variable, generally at trace levels, thus adding more complexity to their determination (Gomez-Alvarez et al., 2012). Moreover, OVOCs are polar and volatile, a fact that negatively affects their determination. Given their crucial role in tropospheric chemistry, there is an urgent need for sensitive and reliable techniques.
Several technologies have been proposed for the determination of OVOCs, including Fourier transform infrared spectroscopy (FTIR), proton transfer mass spectrometry (PTR-MS), selected-ion flow-tube mass spectrometry (SIFT-MS), broadband cavity-enhanced absorption spectroscopy (BBCEAS), and cavity-enhanced differential optical absorption spectroscopy (CE-DOAS) (Thalman et al., 2015). Atmospheric pressure interface time-of-flight chemical ionization mass spectrometers (APITOF-CIMS) have been recently proposed for the detection of OVOCs. Nevertheless, CIMS data present overlapping ions and large uncertainties in quantification of the wide variety of species (Riva et al., 2019). Moreover, some off-line methods, generally based on chromatographic techniques, have been applied (Legreid et al., 2007). They incorporate intensive sample pre-concentration steps by passing through high volumes of air. The sampling systems are denuders, bags, lab-on-a chip devices, impingers, and different types of solid-phase cartridges (Ras et al., 2009). In the recent years, solid-phase microextraction (SPME) has demonstrated an extraordinary potential for the sampling of volatile compounds, supporting a more accurate determination of multi-oxygenated compounds (Zhu et al., 2015). The advantages of SPME are low cost per sample, reusability, high selective sampling for target analytes, high sensitivity, no solvent extraction – meeting the requirement of green chemistry, high reproducibility, low time requirement, and the option of being automated (Chen and Pawliszyn, 2004; Gómez-Alvarez, 2007; Baimatova et al., 2016).
Because of high volatility and polarity of OVOCs, their latter quantification is quite complex, and usually it is necessary to derivatize prior to chromatographic analysis. Derivatization enhances chromatographic behaviour or detectability, and it enables a resolved separation of species which are not directly amenable to analysis due to inadequate volatility or stability (Edler et al., 2002). The main reagents for carbonyl determination include dinitrophenyl hydrazine (DNPH) – normally used in conjunction with high-performance liquid chromatography (HPLC) analysis – (Van Leeuwen et al., 2004) and O-(2,3,4,5,6-pentafluorobenzyl)hydroxylamine (PFBHA), which is usually applied in conjunction with gas chromatography (GC) (Yu et al., 1995). Silylant reagents such as N,O-bis(trimethylsilyl)trifluoroacetamide (BSTFA) or N-methyl-N-(trimethylsilyl)trifluoroacetamide (MSTFA) were employed for hydroxyl or carboxylic functional group derivatization (Borras and Tortajada-Genaro, 2012; Jaoui et al., 2012). These types of derivatization are performed after sampling and are followed by lengthy and aggressive sample treatment, which could potentially alter the sample composition. Moreover, some of them were questioned due to interferences with water vapour, ozone, or nitrogen dioxide (Mellouki et al., 2015).
The key to making major improvements in derivatization methodologies may lie in performing simple and automated sample preparation prior to analysis (Pang et al., 2013). In this way, PFBHA derivatization combined with SPME provides several exceptional advantages for carbonyl compounds (Bourdin and Desauziers, 2014). After the sampling, derivatization takes place immediately on-fibre, and the derivatives are thermally desorbed, avoiding dilution by solvents and sample alteration. However, previous studies were carried out at concentrations of analytes considerably higher (hundreds of parts per billion by volume) than those that would be present in atmospheric conditions. In addition, a decrease in fibre efficiency due to deterioration or depletion of the polymeric phase was also observed. Another problem is the competition among analytes and the consequent reduction of reaction yields (Larroque et al., 2006).
These facts have led us (1) to explore on-line variations in the SPME derivatization which could lead to improving its performance and (2) to conduct experiments aimed at evaluating the quantification approach for atmospheric studies. Therefore, a method based on active sampling with SPME fibres and quantitative GC–MS determination of OVOCs has been developed. The research aim was to investigate on-line derivatization for decreasing the sampling times, reaching an effective capture and a complete conversion and reducing competition between analytes. In order to evaluate the performances, the simultaneous determination of OVOCs in air samples was examined at the EUPHORE chamber facility, a highly instrumented, large-scale outdoor simulation chamber (Borras et al., 2015).
2.1 Reagents
The derivatization reagents O-(2,3,4,5,6-pentafluorobenzyl) hydroxylamine (PFBHA) (99 %), N,O-bis(trimethylsilyl)trifluoroacetamide (BSTFA), N-trimethylsilyl-N-methyl trifluoroacetamide (MSTFA), and trimethylchlorosilane (TMCs) as catalysts purchased from Sigma-Aldrich (Madrid, Spain) were used directly in this study without any further purification. Methylglyoxal at 30 % in water (MGLY), glyoxal (GLY), glutaraldehyde, methyl vinyl ketone (MVK), methacrolein, glycoladehyde, hydroxyacetone, succinic acid, and benzaldehyde were supplied by Fluka (Madrid, Spain). Other α-dicarbonyl compounds, such as 4-oxo-2-pentenal and E-butenedial, were synthetized (provided by Leeds University, Organic Chemistry Department).
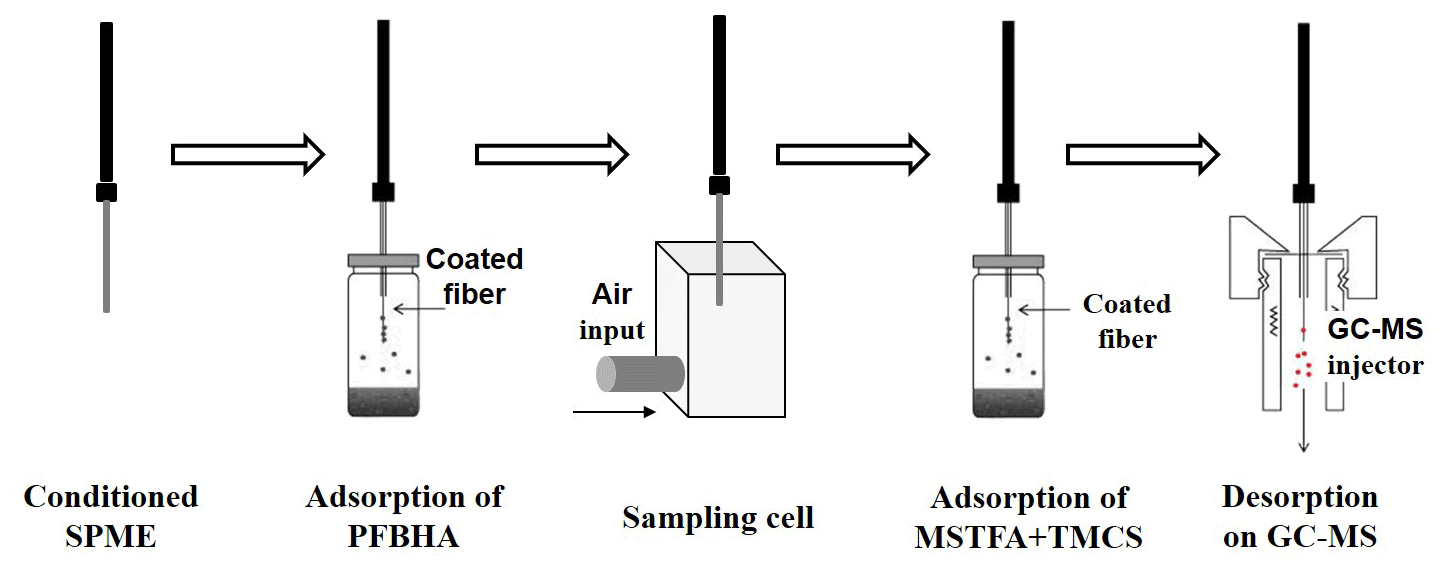
2.2 Automated active SPME sampling coupled to the GC–MS system
This methodology was carried out in four steps. The first one was the chemical modification of the fibre with PFBHA (carbonyl reagent). The second proposed step was the sampling on a cell of multi-oxygenated compounds for “on-fibre derivatization” of carbonyl compounds. The third step was the chemical modification of the fibre with MSTFA plus TMCs (hydroxyl or carboxylic compounds reagent). Finally, derivatized OVOCs on SPME were injected on GC for thermal desorption and their analysis by mass spectrometry. These steps were carried out by an automated procedure achieved with a Combi Pal autosampler (CTC Analytics, Zwingen, Switzerland) controlled by Cycle Composer software and equipped with SPME sampling adaptors, sample trays, and a temperature-controlled agitator tray. Figure 1 shows a scheme of multi-step derivatization protocol.
For the development of our proposed methodology, a sampling system with an inert sampling line and a cell was designed. The sampling line was of sulfinert® material, and it was heated at 80 ∘C to avoid losses of sticky OVOCs compounds. The sampling cell of was made of stainless steel. The air passed through, in a turbulent regime at 10 L min−1, which corresponds to 50 cm s−1, to guarantee air velocity higher than 10 cm s−1, defined by some authors as a certain critical value (Gómez-Alvarez et al., 2012; Augusto et al., 2001). Moreover, the sampling cell was installed just below the simulator chamber but at laboratory conditions, i.e. without the influence of solar radiation and under controlled temperature at 20 ∘C, critical parameters for SPME sampling (Gomez-Alvarez, 2007).
The SPME selected fibre was polydimethylsiloxane/divinylbenzene (PDMS/DVB) coating, a stable flex fibre, with a 23 gauge needle size and coating thickness of 65 µm (Sigma Aldrich, USA). This type of fibre was chosen because of its high affinity to PFBHA (Zhu et al., 2015). A robotic injection system (PAL Auto Sampler, Agilent Technologies, USA) was used. The protocol started with the conditioning of SPME fibre in the GC injector port (30 min at 200 ∘C). By doing so, every analysis also constitutes pre-conditioning for the following sampling step, avoiding fibre contamination. Secondly, a PFBHA derivatization solution (87 mg of PFBHA in 2 mL pure) was prepared and located in a reagent tray position 1. Thirdly, PFBHA headspace vapour was generated in an incubation cell under optimized conditions of temperature, agitation time and speed, and adsorption time. Fourthly, SPME doped with PFBHA was located in the sampling cell where air is passed through during a selected sampling time. Fifthly, pure MSTFA plus catalyst (40 µL plus 10 µL of TMCs) was prepared and located in a reagent tray position 2. Silylant reagent head space vapour was also generated in an incubation cell under temperature, agitation time and speed, and adsorption time optimized conditions. Finally, the sample was analysed by GC–MS. The chemical reactions involved in these derivatization steps are shown in Fig. S1 in the Supplement.
2.3 Chromatographic conditions
The sample was thermally desorbed in a GC port of an Agilent GC–MS (Santa Clara, USA) equipped with a HP-5MS column of 30 m×0.25 mm I.D ×0.25 mm film (Agilent, Santa Clara, USA). The chromatograph was programmed at 80 ∘C for 2 min, then ramped at a rate of 12 to 240 ∘C and 100 to 280 ∘C. The injection port was held at 250 ∘C, and the transfer line from GC to MS was held at 300 ∘C. Samples were injected in splitless mode, using helium as carrier at a flow of 1 mL min−1. The EI voltage was 70 eV, the ion source temperature was set at 200 ∘C, and the quadrupole temperature was set at 100 ∘C. Full scan mode was used ( 50–650) to identify the most abundant ions of multi-oxygenated compounds. The selected ion chromatograms of the most abundant ions were used to quantify them.
2.4 Smog chamber facilities and reference methods
The on-line SPME–GC–MS optimization was carried out in the high-volume outdoor smog chamber EUPHORE (EUropean PHOtoREactor) (Valencia, Spain). These chambers consist of two half-spherical fluoropolymeric bags, each one of 200 m3, with integrated measuring systems for monitoring pressure, humidity, temperature, precursor species, and reaction products. Pressure, relative humidity, and temperature were measured using a pressure sensor (Air-DB-VOC, Sirsa, Madrid, Spain) and a dew point hydrometer (TS-2, Walz, Effeltrich, Germany). A Serenius 50 ozone monitor (Ecotech, Melbourne, Australia) was also used (Borrás and Tortajada-Genaro, 2012).
Table 1Optimization experiments of on-fibre PFBHA derivatization.

The OVOCs were determined by two techniques. First, samples for the on-line SPME–GC–MS technique were taken over 6 h at a sampling frequency of 20 min (10 L min−1, sampling time: 5 min). Second, a White-type mirror system (path length of 553.5 m) coupled to a FTIR with a MCT detector (Nicolet 670, Thermo Scientific, USA) was used. Spectra were collected at 1 cm−1 resolution by averaging 300 scans (sampling time: 5 min). The quantification was based on aldehydic C–H stretching for methylglyoxal, within the spectral region of 2750–3000 cm−1 and C−C and CH2 bands for isoprene, methyl vinyl ketone, and methacrolein in 900–1047 cm−1 by using ANIR software (Ródenas, 2008).
For the optimization of the on-line SPME–GC–MS methodology, methylglyoxal, glyoxal, glutaraldehyde, methyl vinyl ketone, methacrolein, benzaldehyde, glycoldehyde, hydroxyacetone, succinic acid, 4-oxo-2-pentenal, and E-butenedial – selected carbonyl, α-dicarbonyl, hydroxyl-carbonyl compounds, and carboxylic acids – were injected in the smog chamber using an impinger. A stream of hot air (>150 ∘C) produced by a hot gun enabled their volatilization and the transference inside the chamber at a flow of 10 L min−1. Moreover, our technique was applied in an intercomparison campaign and in the monitoring of isoprene ozonolysis. Reactants and products are diluted during experiments, and to determine the correct concentration values, they must be corrected. The dilution rate in the chamber was calculated from the decay of SF6 by FTIR areas in the IR range of 762–956 cm−1. The specific dilution process was determined by FTIR adding 120 µg m−3 of SF6 as a non-reactive tracer (value of s−1) to the reaction mixtures at the start of the experiments.
Milli-Q water was added by a sprayer system (died, cleaned air at 2 bar) into the EUPHORE chamber. The dew point system was used to confirm relative humidity values.
A large intercomparison campaign of oxygenated organic compound measurements was held at EUPHORE from mid-May to 1 June 2018 (Muñoz et al., 2019; Ródenas et al., 2021). Taking advantage of it, our proposed SPME–GC–MS plus derivatization technique was compared to a number of instruments: four on-line instruments together with one off-line analytical method. The on-line techniques were on-line SPME–GC–MS plus derivatization, FTIR, two proton-transfer time-of-flight mass spectrometers (PTR-ToF-MS), and a selected-ion flow-tube mass spectrometer (SIFT-MS). The off-line techniques were DNPH cartridges analysed by liquid chromatography–mass spectrometry (LC–MS). These techniques and methodologies were operated by the University of York, University of Leeds, Forschungszentrum Jülich, and Fundación CEAM. A detailed summary of techniques and institutions is in Table S1 in the Supplement.
Measurement of OVOCs at unknown concentrations were done under different relative humidity conditions (0 %–50 % RH). After overnight cleaning, all instruments sampled background air for 1 h. After that, OVOCs were added into the simulation chamber. I.e, methacrolein, acetone, 2-butanone, hydroxyacetone, glycolaldehyde, formaldehyde, benzaldehyde, acetaldehyde, methyl vinyl ketone, glyoxal and methylglyoxal were added in a range of 40–60 ppbV. Samples were taken over 1 h followed by the addition of water to increase the relative humidity up to 50 %. Subsequently samples were taken during a 30 min period, and the gas mixture was diluted within 1 h down to 50 % of the initial concentration. This protocol was repeated twice.
Ozonolysis of isoprene. The ozonolysis experiment consisted in adding isoprene: 220 ppbV and 160 µL were fed to the photoreactor via heated air stream. O3 was added at 23 ppbV min−1 from an O2 purity of 5.0 using a UV lamp, until reaching 215 ppbV. CO was added as an OH radical scavenger (230 ppm of CO, 5000 ppmV at 10 L min−1). Later, all the reactants were mixed for 10 min, and all the instrumentation and techniques took samples over almost 6 h throughout the experiment.
3.1 Set-up experiments of on-fibre PFBHA derivatization
The first challenge was an effective on-fibre derivatization of the carbonyl compounds to oxime products based on a substitution reaction by PFBHA. For assay development, methylglyoxal was selected as the model compound, and a synthetic air mixture was generated at the EUPHORE smog chamber at 50 ppbV (confirmed by FTIR). Complete factorial designs were performed to study the variable effect (Table 1).
Table 2Description of chromatographic peaks detected after the on-fibre derivatization using PFBHA as reagent for the carbonyl and α-dicarbonyl compounds selected.
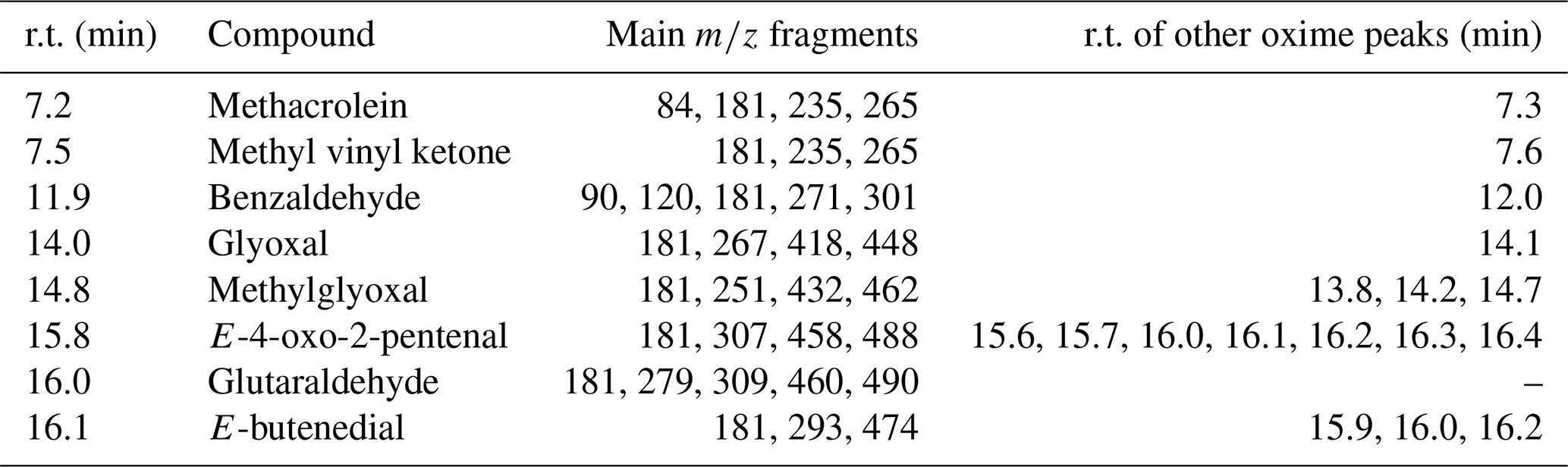
r.t.: retention time.
The generation of PFBHA reagent vapour in a headspace mode was studied, varying reagent amount, temperature, agitation time, and speed. The selection criteria were (1) maximum oxime signal, (2) minimal artefacts for the reagent depletion, and (3) minimal standard deviation between replicates. Results showed that the signal saturation was achieved at PFBHA solution of 87 mg L−1. In fact, higher PFBHA concentrations reduced the derivatization capacity and increased the interfering peaks as was previously described by Yu et al. (2017). Regarding the incubation temperature, the oxime formation slightly improved when incubation temperature cell increased. The best values were reported at 50 ∘C. The modification of agitation time and speed factors was also tested, leading an improvement of reaction yield and reproducibility. Once an effective generation of the derivatizing reagent in the headspace was achieved, the fibre exposition was evaluated. The chosen adsorption time was 4 min, in accordance with previous studies (Gómez-Alvarez et al., 2012).
The next experiments were aimed at the on-fibre derivatization of carbonyl compounds. A sampling flow of 10 L min−1 over 5 min and no incubation time produced maximum capture of analyte at a fast sampling rate. Finally, derivatized compounds were desorbed and directly transferred to the GC–MS. Injector desorption conditions (10 min at 250 ∘C) assured total desorption, calculated from peak areas of methylglyoxal derivatives (13.9, 14.2, 14.4, and 14.5 min), with the presence of a underivatized chromatographic peak of methylglyoxal also being negligible (retention time 7.65 min). Thus, the on-line SPME PFBHA derivatization was completed for up to 95 % methylglyoxal in a reduced time (total time 18 min) and low reagent consumption (0.17 mg per assay). Also, memory effect on the sampling cell was evaluated, including a blank derivatized sample in the sampling sequence. No compounds were observed, confirming the absence of memory effect.
The reusability of PDMS/DVB fibres was tested, performing replicate experiments (>50 assays). Since reproducible responses were registered, on-fibre derivatization is a cost-effective approach for the detection of OVOCs in air samples.
3.2 Determination of carbonyl and α-dicarbonyl compounds after on-fibre PFBHA derivatization
The multiplexed automated on-fibre derivatization was approached, studying the methodology for a mixture of eight carbonyl compounds, including aldehydes, ketones, aromatic aldehydes, and α-dicarbonyls. Then, the air sample contained the selected compounds: methylglyoxal, glyoxal, glutaraldehyde, methyl vinyl ketone, methacrolein, benzaldehyde, 4-oxo-2-pentenal, and E-butenedial.
The chromatograms obtained from air mixtures confirmed the correct on-fibre derivatization and later chromatographic separation process. The chromatographic oxime peaks of each carbonyl compounds, and its corresponding mass spectra are reported in Figs. S2 and S3. The identification of derivatives was based on the molecular ion in the EI mass spectra. The fragments were 265 for both methacrolein and methyl vinyl ketone: 301, 448, 462, 490, 488, and 476, for benzaldehyde, glyoxal, methylglyoxal, glutaraldehyde, 4-oxo-2-pentenal, and E-butenedial, respectively. Also, several fragments were examined, such as ions with M-30, M-181, and M-211, resulting from loss of NO, C6F5CH2, and C6F5−CH2NO, respectively. Table 2 summarizes the main chromatographic features for the specific oximes formed.
Table 3Analytical performances for the determination of carbonyl and α-dicarbonyl compounds.

The number of underivatized compounds was negligible (<1.5 %), and the number of PFBHA interference residue peaks was reduced and perfectly resolved (resolution >1.5). The calculated recoveries ranged from 91 % (glutaraldehyde) to 99.7 % (methylglyoxal) as shown in Table S2. Therefore, the suitability of the developed method was demonstrated for a wide range of compounds, considering organic functionalities, molecular sizes, boiling temperatures, and reactivities. Therefore, our method avoided the main limitation of SPME-based methods for multiplexed purposes. Often, the simultaneous determination of several analyte families led to a wide range of recoveries, because these methods are highly sensitive to interferences and experimental conditions, such as sampling time, analyte concentrations, passive or active systems, reagent concentration, temperature, operator handling, etc. (Koziel and Novak, 2002).
The effect of humidity was examined because several techniques such as proton-transfer-reaction mass spectrometry – time of flight (PTRMS-TOF) or cavity-enhanced absorption spectroscopy (CEAS) showed an erroneous determination for air samples with high water content, depending on the applied data evaluation routine (Thalman et al., 2015). Air mixtures of the model organic compounds were prepared and mixed with water vapour. A statistical test demonstrated that the humidity effect was negligible (t test, p values <0.005).
3.3 Analytical performances of on-fibre PFBHA derivatization
OVOC air mixtures at different concentrations were analysed – ranging from 5 pptV to 100 ppbV. As a selective separation was achieved, appropriate calibration curves were obtained. The regression coefficients were 0.990–0.998.
The evaluation of sensitivity was performed analysing blank samples and serial dilutions of standard air mixtures (dilution factor 1:10). Detection limits (LOD) were calculated as 3 times the standard deviation of blank samples at the retention time corresponding to each compound (Table 3). The estimated detection limits ranged from 6 pptV (glyoxal) to 100 pptV (methacrolein), equivalent to 14 to 237 ng m−3. Limit of quantification (LOQs) were calculated as 10 times the standard deviation of blank samples at the retention time corresponding to each compound. The limits were from 20 pptV (glyoxal) to 300 pptV (methacrolein). The LOD values were compared to those reported in previously published papers. For glyoxal, the mist chamber (Cofer scrubber) obtained a LOD of 2.7 pptV (Spaulding et al., 2002), the DOAS system obtained 2 pptV, and CEAS systems obtained 19 pptV (Pang et al., 2013) or 75 pptV in Pang et al. (2014). For methylglyoxal, other studies obtained a LOD of 170 pptV using CEAS systems (Pang et al., 2013), 185 pptV in Pang et al. (2014), or 89 pptV using PTR-ToF-MS (Michoud et al., 2018). Our detection limit was 97 pptV. The detection limit of our technique is comparable to most spectrometric methods and improves the values for small molecules such as glyoxal and methylgyoxal. This result is particularly relevant because these α-dicarbonyl compounds play a key role in SOA formation in the atmosphere. Although this research was performed in a high-volume simulation chamber (200 m3), the detection limits were below to typical concentrations of these compounds observed in ambient air.
Method precision was estimated from replicate experiments at 1, 10, 25, 50, and 100 ppbV (n=5). The percentage of relative standard deviation (RSD) was 0.2 %–7 %. Other techniques reported higher errors. In Pang et al. (2013), the error was 22 % using microfluidic lab-on-a-chip derivatization methodology. On the other hand, in Pang et al. (2014) the reproducibility was 6.6 % for glyoxal and 7.5 % for methylglyoxal using the same methodology. Therefore, we can conclude that our method presented comparable, even better, results.
Table 4Analytical performances for the determination of hydroxyl carbonyl and carboxylic acid model compounds.

3.4 Extension to on-fibre PFBHA plus MSTFA derivatization
The on-line derivatization of hydroxyl or carboxylic groups, directly or after the described process for carbonyl groups, was studied based on a silylation reaction. For that, MSTFA was chosen as the silylant. In addition to previously selected ketones and aldehydes, hydroxyacetone, glycolaldehyde, and succinic acid were selected as hydroxyl carbonyl and carboxylic acid model compounds, due to their atmospheric relevance (Pospisilova et al., 2020; Mellouki et al., 2015). The derivatization sequence studied implied adsorption of PFBHA, sample loading, and MSTFA plus catalyst adsorption. Thus, the oximes ( groups) were generated before the formation of silanes (−OH and −COOH groups). Alternatively, BSTFA was tested as a silylant reagent, but the chromatograms showed more residual peaks and lower response for product peaks. These data suggested that short chain or branched compounds were hindered by steric impediment, as was observed in solution derivatization (Borrás and Tortajada-Genaro, 2012).
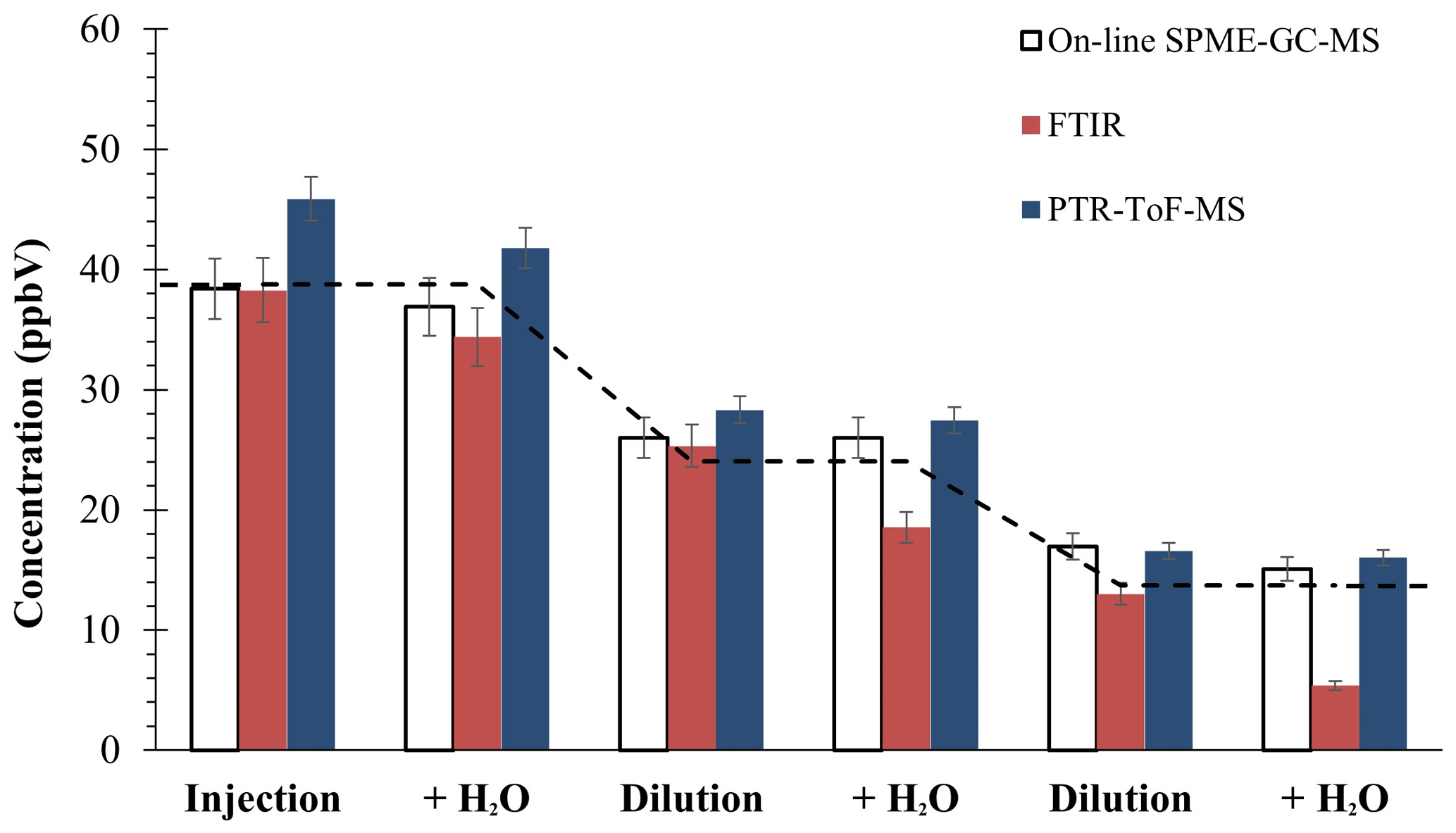
Figure 2Average concentration of methylglyoxal provided by on-line SPME–GC–MS, FTIR, and Ionicon PTR-ToF-MS (from Jülich). The dashed line indicates the theoretical value, according to the quantity injected into the chamber, and the steps show this adjusted value with the dilution of the chamber (according to SF6 values). Errors from SPME and FTIR include precision and accuracy; PTR-ToF-MS error shows 3σ of precision (did not include accuracy).
The main experimental variables of the MSTFA-based method were optimized from the corresponding chromatographic peaks (Table S3). Together with the molecular ions and PFBHA-associated fragments, the main ions were M-15 and M-73 resulting from loss of CH3 and Si(CH3)3. The results indicated that the direct loading of vaporized reagent and catalyst (TMCS) was an effective way to transform PFBHA products formed on the PDMS/DVB fibre. The absence of memory column effects and low number of artefacts were observed. Also, a successful thermal desorption of PFBHA MSTFA products was achieved given the quantitative recoveries (>95 %).
Regarding the analytical performances, the LOD was 0.08–0.15 ppbV, the linear range was 5–150 ppbV and reproducibility, expressed as standard deviation, was 2–4 % (see Table 4). In conclusion, double derivatization treatment allowed the proper determination of OVOCs, independently of the functionalized group (, −OH and/or −COOH), even carbonyl compounds with α-hydrogen.
3.5 Validation in an intercomparison study
An intercomparison campaign for the measurement of small multi-oxygenated compounds was carried out at the EUPHORE atmospheric simulator. The study included the variation in OVOC concentration and the impact that the presence of potential interferants, such as high humidity, and dilution steps can have on the methodologies evaluated in this work (see Table S1).
From the different OVOCs, we selected methylglyoxal since it was previously used as the OVOC model (see Sect. 3.1). Figure 2 shows the average concentration in each step. Some techniques (spectroscopic and off-line) presented large interferences. The methodologies provided consistent results of methylglyoxal quantification. As can be observed, the results from SPME–GC–MS plus the derivatization technique were in great agreement with the theoretical values – a known quantity of compound was introduced into EUPHORE chamber – and with the results obtained by other techniques, both optical and mass spectroscopic methodologies. In fact, we can affirm that it does not present interferences with relative humidity lower than 60 %. For that, a t test for paired samples was performed with a result of α<0.5 for our proposed methodology, confirming that high humidity did not significantly affect our measurements.
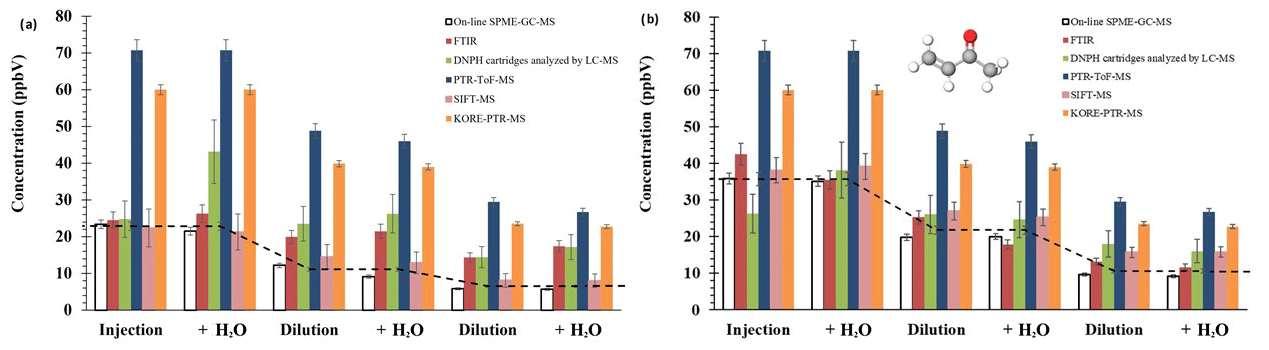
Figure 3Average concentration of methacrolein (a) and methyl vinyl ketone (b) for all compared techniques: on-line SPME–GC–MS, FTIR, DNPH-LC–MS, Ionicon PTR-ToF-MS, SIFT-MS, and KORE-PTR-ToF-MS. Errors from SPME, FTIR, and DNPH-LC–MS include precision and accuracy; Ionicon PTR-ToF-MS, SIFT-MS, and KORE-PTR-ToF-MS errors consider 3σ of precision (did not include accuracy).
The intercomparison included OVOCs identified as main degradation products of biogenic pollutants (Aschmann and Atkinson, 1994; Iannone et al., 2010). Figure 3 shows the determined concentration levels of methacrolein and methyl vinyl ketone in a mixture containing both compounds. As previously observed, the results from SPME–GC–MS plus the derivatization technique agreed with the theoretical values from chamber dilutions. Regarding other techniques, the concentrations were comparable to SIFT-MS, FTIR, and DNPH cartridges analysed by LC–MS. However, PTR-ToF-MS cannot discriminate between the structural isomers of both carbonyl compounds. In this case, the sum of MVK and methacrolein (MACR) is measured due to PTR-MS methods not being selective. Both compounds have a different sensitivity factor, imposing an additional inaccuracy on the data; for more details see Ródenas et al. (2021). On the contrary, the on-line SPME–GC–MS approach can be used for a reliable monitoring of both MVK and MACR in atmospheric reactions.
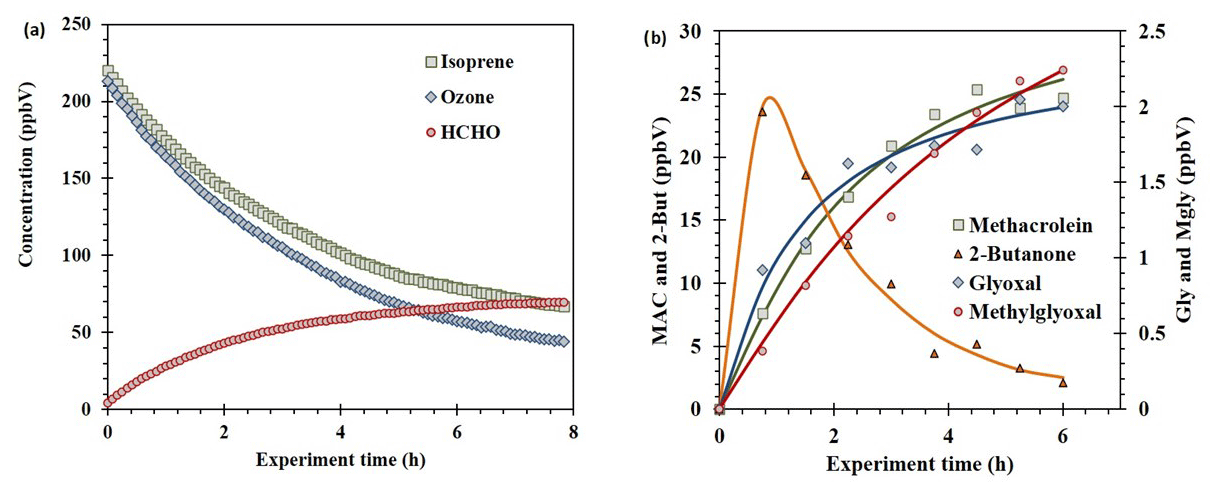
Figure 4Concentration profiles obtained from ozonolysis of isoprene at the high-volume atmospheric simulation chamber. Reagents and the main product determined by FTIR (a). Main multi-oxygenated organic compounds determined by on-line SPME–GC–MS (b). [Isoprene]0=220 ppV; [Ozone]0=215 ppbV; [CO scavenger]=230 ppmV; reaction volume =200 m3.
3.6 Application to monitoring of an ozonolysis reaction
The ozonolysis of isoprene, one of the main biogenic compounds emitted to the atmosphere, was studied at the EUPHORE atmospheric simulator. On-line SPME–GC–MS plus PFBHA derivatization and MSTFA derivatization was applied for tracing the formation of the multi-oxygenated compounds. Figure 4a shows the decay of isoprene and ozone and the major degradation product (formaldehyde) formation. The degradation rate of isoprene and ozone was fitted to first-order decay as previously described in Karl et al. (2004). Regarding minor products, the OVOCs determined were 2-butanone, methacrolein, methyl vinyl ketone, glycoladehyde, hydroxyacetone, glyoxal, and methylglyoxal. Some of them are plotted in Fig. 4b. The results were fitted to a standard growth for degradation products. In the case of 2-butanone, the formation was fast, and, after 1 h, a decay by chemical degradation was registered. The maximum concentrations were (23.6±1.2), (25.4±1.3), (4.2±0.2), (4.1±0.2), (3.8±0.2), (2.1±0.1), and (2.2±0.1) ppbV, for 2-butanone, methacrolein, methyl vinyl ketone, glycoladehyde, hydroxyacetone, glyoxal, and methylglyoxal, respectively. Other OVOCs such as methyl vinyl ketone were also detected. A total of seven OVOCs were identified and quantified in the isoprene ozonolysis, in good accordance with previous studies (Karl et al., 2004; Wennberg et al., 2018).
Therefore, our proposed on-line SPME–GC–MS plus derivatization enabled the accurate sensitive atmospheric monitoring of secondary pollutants. This technique will support their relevance since the ozonolysis of biogenic VOCs such as isoprene gives multifunctional oxygenated organic compounds that participate in the formation of aerosols. SOA formation during the atmospheric oxidation of biogenic organic compounds is estimated at 20–380 Tg r−1 globally, influencing human health and climate (Mellouki et al., 2015).
The oxygenated volatile organic compounds play an important role in the atmosphere, even at low concentrations, so their reliable determination is challenging. On-line SPME–GC–MS double derivatization has demonstrated an efficient approach for alcohols, aldehydes, ketones, carboxylic acids, and their combinations, independently of molecular size or structure. Compared to other techniques, such as FTIR and PTR-ToF-MS among others, this approach provides discontinuous data (20 min). However, the on-line SPME–GC–MS method shows excellent analytical performances for LOD (6–100 pptV), reproducibility (0.2 %–7 %), selectivity (high resolution), cost-effectiveness, and high throughput. It is important to highlight the advantages compared to other chromatographic techniques, derived from an automated format, such as null solvent consumption, low reagent amounts, hands-off preparation, reusability of SPME fibre, robustness, and high precision.
Demonstrated for the ozonolysis of isoprene, the number of potential applications is extraordinarily wide. On-line SPME–GC–MS double derivatization methodology could support a better understanding of atmospheric chemistry processes and an accurate monitoring of their atmospheric levels. This technology is particularly useful in atmospheric simulation chambers for air chemistry studies. A relevant example is the research of OVOCs as secondary organic aerosols (SOA) and ozone precursors. This information should help a better assessment of their impact on human health and climate change. Moreover, after an adequate adaptation for mobile laboratories, the monitoring of atmospheric OVOC levels is feasible.
The experimental data is available via the EUROCHAMP-2020 data center (https://data.eurochamp.org/data-access/chamber-experiments/, last access: 2 July 2021). Specific links of the data used in this work are: Muñoz (2021a, https://doi.org/10.25326/BQK8-RZ90) and Muñoz (2021b, https://doi.org/10.25326/WP8B-VR27).
The supplement related to this article is available online at: https://doi.org/10.5194/amt-14-4989-2021-supplement.
EB conceived and designed the analysis, performed the measurements, processed the data, and wrote the paper. LATG wrote the paper, MR performed the measurements and wrote the paper, and TV performed the measurements and wrote the paper. TS performed the measurements; PS wrote the paper; MDS performed the measurements; ACL wrote the paper; and AM supervised the project, conceived and designed the analysis, and helped with paper preparation.
The authors declare that they have no conflict of interest.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the special issue “Simulation chambers as tools in atmospheric research (AMT/ACP/GMD inter-journal SI)”. It is not associated with a conference.
This project/work has received funding from the European Union's Horizon 2020 research and innovation programme through the EUROCHAMP-2020 Infrastructure Activity under grant agreement no. 730997 and from the IMAGINA-Prometeo project (PROMETEO/2019/110 from Generalitat Valenciana) Fundación CEAM is partly funded by the GVA. We especially want to thank Ralf Tillmann, Sergej Wedel, and David Reimer for the measurements made with PTRMS by the Forschungszentrum Jülich (FZJ) institution.
This research has been supported by the European Commission Horizon 2020 research and innovation programme (EUROCHAMP-2020 (grant no. 730997) and Generalitat Valenciana (grant no. PROMETEO/2019/110).
This paper was edited by Anna Novelli and reviewed by two anonymous referees.
Aschmann, S. M. and Atkinson, R.: Formation Yields of Methyl Vinyl Ketone and Methacrolein from the Gas-Phase Reaction of O3 with Isoprene, Environ. Sci. Technol., 28, 1539–1542, https://doi.org/10.1021/es00057a025, 1994.
Atkinson, R. and Arey, J.: Gas-phase tropospheric chemistry of biogenic volatile organic compounds: A review, Atmos. Environ., 37, 197–219, https://doi.org/10.1016/S1352-2310(03)00391-1, 2003.
Augusto, F., Koziel, J., and Pawliszyn, J.: Design and Validation of Portable SPME Devices for Rapid Field Air Sampling and Diffusion-Based Calibration, Anal. Chem., 73, 481–486, https://doi.org/10.1021/ac000629k, 2001.
Baimatova, N., Kenessov, B., Koziel, J. A., Carlsen, L., Bektassov, M., and Demyanenko, O. P.: Simple and accurate quantification of BTEX in ambient air by SPME and GC–MS, Talanta, 154, 46–52, https://doi.org/10.1016/j.talanta.2016.03.050, 2016.
Borrás, E. and Tortajada-Genaro, L. A.: Determination of oxygenated compounds in secondary organic aerosol from isoprene and toluene smog chamber experiments, Int. J. Environ. An. Ch., 92, 110–124, https://doi.org/10.1080/03067319.2011.572164, 2012.
Borrás, E., Tortajada-Genaro, L., Ródenas, M., Vera, T., Coscollá, C., Yusá, V., and Muñoz, A.: Gas-Phase and Particulate Products from the Atmospheric Degradation of the Organothiophosphorus Insecticide Chlorpyrifos-methyl, Chemosfere, 38, 888–894, https://doi.org/10.1016/j.chemosphere.2014.11.067, 2015.
Bourdin, D. and Desauziers, V.: Development of SPME on-fiber derivatization for the sampling of formaldehyde and other carbonyl compounds in indoor air, Anal. Bioanal. Chem., 406, 317–28, https://doi.org/10.1007/s00216-013-7460-6, 2014.
Chen, Y. and Pawliszyn, J.: Solid-Phase Microextraction Field Sampler, Anal. Chem., 76, 6823–6828, https://doi.org/10.1021/ac0490806, 2004.
Edler, M., Metze, D., Jakubowski, N., and Linscheid, M.: Quantification of silylated organic compounds using gas chromatography coupled to ICP-MS, J. Anal. Atom. Spectrom., 17, 1209–1212, https://doi.org/10.1039/B207227K, 2002.
Gómez-Alvarez, E.: Characterisation of absorptive sampling with SPME fibres in the EUPHORE photoreactor, Talanta, 72, 1757–1766, https://doi.org/10.1016/j.talanta.2007.02.013, 2007.
Gómez-Alvarez, E., Vázquez, M., Gligorovski, S., Wortham, H., and Valcárcel, M.: Characterisation and calibration of active sampling Solid Phase Microextraction applied to sensitive determination of gaseous carbonyls, Talanta, 88, 252–258, https://doi.org/10.1016/j.talanta.2011.10.039, 2012.
Iannone, R., Koppmann, R., and Rudolph, J.: Stable carbon kinetic isotope effects for the production of methacrolein and methyl vinyl ketone from the gas-phase reactions of isoprene with ozone and hydroxyl radicals, Atmos. Environ., 44, 4135–4141, https://doi.org/10.1016/j.atmosenv.2010.07.046, 2010.
Jaoui, M., Kleindienst, T. E., Offenberg, J. H., Lewandowski, M., and Lonneman, W. A.: SOA formation from the atmospheric oxidation of 2-methyl-3-buten-2-ol and its implications for PM2.5, Atmos. Chem. Phys., 12, 2173–2188, https://doi.org/10.5194/acp-12-2173-2012, 2012.
Karl, M., Brauers, T., Dorn, H. P., Holland, F., Komenda, M., Poppe, D., Rohrer, F., Rupp, L., Schaub, A., and Wahner, A.: Kinetic Study of the OH-isoprene and O3-isoprene reaction in the atmosphere simulation chamber, SAPHIR, Geophys. Res. Lett., 31, L05117, https://doi.org/10.1029/2003GL019189, 2004.
Koziel, J. A. and Novak, I.: Sampling and sample-preparation strategies based on solid-phase microextraction for analysis of indoor air, TRAC-Trend. Anal. Chem., 21, 840–850, https://doi.org/10.1016/S0165-9936(02)01204-9, 2002.
Larroque, V., Desauziers, V., and Mocho, P.: Development of a solid phase microextraction (SPME) method for the sampling of VOC traces in indoor air, J. Environ. Monitor., 1, 106–11, https://doi.org/10.1039/B511201J, 2006.
Legreid, G., BalzaniLööv, J., Staehelin, J., Hueglin, C., Hill, M., Buchmann, B., Prevot, A. S. H., and Reimann, S.: Oxygenated volatile organic compounds (OVOCs) at an urban background site in Zürich (Europe): Seasonal variation and source allocation, Atmos. Environ., 41, 8409–8423, https://doi.org/10.1016/j.atmosenv.2007.07.026, 2007.
Mellouki, A., Wallington, T. J., and Chen J.: Atmospheric Chemistry of Oxygenated Volatile Organic Compounds: Impacts on Air Quality and Climate, Chem. Rev., 115, 3984–4014, https://doi.org/10.1021/cr500549n, 2015.
Michoud, V., Sauvage, S., Léonardis, T., Fronval, I., Kukui, A., Locoge, N., and Dusanter, S.: Field measurements of methylglyoxal using proton transfer reaction time-of-flight mass spectrometry and comparison to the DNPH–HPLC–UV method, Atmos. Meas. Tech., 11, 5729–5740, https://doi.org/10.5194/amt-11-5729-2018, 2018.
Muñoz, A.: Atmospheric simulation chamber study: formaldehyde + acetaldehyde + acetone + glyoxal + methylglyoxal + glycoladehyde + 2-butanone + hydroxyacetone + benzaldehyde + None − Instruments intercomparison, AERIS [data set], https://doi.org/10.25326/BQK8-RZ90, 2021a (available at: https://data.eurochamp.org/data-access/chamber-experiments/, last access: 2 July 2021).
Muñoz, A.: Atmospheric simulation chamber study: isoprene + ozone + carbon monoxide + O3 − Gas-phase oxidation − product study, AERIS [data set], https://doi.org/10.25326/WP8B-VR27, 2021b (available at: https://data.eurochamp.org/data-access/chamber-experiments/, last access: 2 July 2021).
Muñoz, A., Ródenas, M., Borrás, E., Brenan, A., Dellen, J., Escalante, J. M., Gretien, A., Gómez, T., Herrmann, H., Kari, E., Michoud, V., Mutzel, A., Olariu, R., Seakins, P., Tillmann, R., Vera, T., Viertanen, A., and Wedel, S.: Intercomparison of instruments to measure OVOCs: assessment of performance under different relevant controlled conditions (EUPHORE chambers), EGU General Assembly, Vienna, Austria, 7–12 April 2019, EGU2019-17788, 2019.
Pang, X., Lewis, A. C., and Ródenas, M.: Microfluidic lab-on-a-chip derivatization for gaseous carbonyl analysis, J. Chromatogr. A, 1296, 93–103, https://doi.org/10.1016/j.chroma.2013.04.066, 2013.
Pang, X., Lewis, A. C., Rickard, A. R., Baeza-Romero, M. T., Adams, T. J., Ball, S. M., Daniels, M. J. S., Goodall, I. C. A., Monks, P. S., Peppe, S., Ródenas García, M., Sánchez, P., and Muñoz, A.: A smog chamber comparison of a microfluidic derivatisation measurement of gas-phase glyoxal and methylglyoxal with other analytical techniques, Atmos. Meas. Tech., 7, 373–389, https://doi.org/10.5194/amt-7-373-2014, 2014.
Pospisilova, V., Lopez-Hilfiker, F. D., Bell, D. M., El Haddad, I., Mohr, C., Huang, W., Heikkinen, L., Xiao, M., Dommen, J., Prevot, A. S. H., Baltensperger, U., and Slowik, J. G.: On the fate of oxygenated organic molecules in atmospheric aerosol particles, Science Advances, 6, eaax8922, https://doi.org/10.1126/sciadv.aax8922,2020.
Ras, M. R., Borrull, F., and Marcé, R. M.: Sampling and preconcentration techniques for determination of volatile organic compounds in air samples, TRAC-Trend. Anal. Chem., 28, 347–361, https://doi.org/10.1016/j.trac.2008.10.009, 2009.
Riva, M., Rantala, P., Krechmer, J. E., Peräkylä, O., Zhang, Y., Heikkinen, L., Garmash, O., Yan, C., Kulmala, M., Worsnop, D., and Ehn, M.: Evaluating the performance of five different chemical ionization techniques for detecting gaseous oxygenated organic species, Atmos. Meas. Tech., 12, 2403–2421, https://doi.org/10.5194/amt-12-2403-2019, 2019.
Ródenas, M.: Improvements in Spectroscopy Data Processing: Faster Production and Better Reliability of Lab Data, INTROP Report, available at: http://www.ceam.es/GVAceam/archivos/MRodenasINTROPReport.pdf (last access: 2 July 2021), 2008.
Ródenas, M.: Intercomparison of on-line and off-line techniques for the measurement of oxygenated organic compounds, in preparation, 2021.
Spaulding, R. S., Talbot, R. W., and Charles, M. J.: Optimization of a Mist Chamber (Cofer Scrubber) for Sampling Water-Soluble Organics in Air, Environ. Sci. Technol., 36, 1798–1808, https://doi.org/10.1021/es011189x, 2002.
Thalman, R., Baeza-Romero, M. T., Ball, S. M., Borrás, E., Daniels, M. J. S., Goodall, I. C. A., Henry, S. B., Karl, T., Keutsch, F. N., Kim, S., Mak, J., Monks, P. S., Muñoz, A., Orlando, J., Peppe, S., Rickard, A. R., Ródenas, M., Sánchez, P., Seco, R., Su, L., Tyndall, G., Vázquez, M., Vera, T., Waxman, E., and Volkamer, R.: Instrument intercomparison of glyoxal, methyl glyoxal and NO2 under simulated atmospheric conditions, Atmos. Meas. Tech., 8, 1835–1862, https://doi.org/10.5194/amt-8-1835-2015, 2015.
van Leeuwen, S., Hendriksen, L., and Karst, U.: Determination of aldehydes and ketones using derivatization with 2,4-dinitrophenylhydrazine and liquid chromatography-atmospheric pressure photoionization-mass spectrometry, J. Chromatogr. A, 1058, 107–12, https://doi.org/10.1016/j.chroma.2004.08.149, 2004.
Wennberg, P. O., Bates, K. H., Crounse, J. D., Dodson, L. G., McVay, R. C., Mertens, L. A., Nguyen, T. B., Praske, E., Schwantes, R. H., Smarte, M. D., St Clair, J. M., Teng, A. P., Zhang, X., and Seinfeld, J. H.: Gas-phase reactions of isoprene and its major oxidation products, Chem. Rev., 118, 3337–3390, https://doi.org/10.1021/acs.chemrev.7b00439, 2018.
Yu, J., Jeffries, H. E., and Le Lacheur, R. M.: Identifying Airborne Carbonyl Compounds in Isoprene Atmospheric Photooxidation Products by Their PFBHA Oximes Using Gas Chromatography/Ion Trap Mass Spectrometry, Environ. Sci. Technol., 29, 8, 1923–1932, https://doi.org/10.1021/es00008a009, 1995.
Yu, R., Duan, L., Jiang, J., and Hao, J.: An optimized two-step derivatization method for analyzing diethylene glycol ozonation products using gas chromatography and mass spectrometry, J. Environ. Sci., 53, 313–321, https://doi.org/10.1016/j.jes.2016.02.021, 2017.
Zhu, W., Koziel, J. A., Cai, L., Özsoy, H. D., and van Leeuwen, J.: Quantification of Carbonyl Compounds Generated from Ozone-Based Food Colorants Decomposition Using On-Fiber Derivatization-SPME-GC-MS, Chromatography, 2, 1–18, https://doi.org/10.3390/chromatography2010001 2015.