the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 18 Oct 2018
| 18 Oct 2018
Field measurements of methylglyoxal using proton transfer reaction time-of-flight mass spectrometry and comparison to the DNPH–HPLC–UV method
Vincent Michoud
Stéphane Sauvage
Thierry Léonardis
Isabelle Fronval
Alexandre Kukui
Nadine Locoge
Sébastien Dusanter
Methylglyoxal (MGLY) is an important atmospheric α-dicarbonyl species for which photolysis acts as a significant source of peroxy radicals, contributing to the oxidizing capacity of the atmosphere and, as such, the formation of secondary pollutants such as organic aerosols and ozone. However, despite its importance, only a few techniques exhibit time resolutions and detection limits that are suitable for atmospheric measurements.
This study presents the first field measurements of MGLY by proton transfer reaction time-of-flight mass spectrometry (PTR-ToF-MS) performed during the ChArMEx SOP2 field campaign. This campaign took place at a Mediterranean site characterized by intense biogenic emissions and low levels of anthropogenic trace gases. Concomitant measurements of MGLY were performed using the 2,4-dinitrophenylhydrazine (DNPH) derivatization technique and high performance liquid chromatography (HPLC) with UV detection. PTR-ToF-MS and DNPH–HPLC measurements were compared to determine whether these techniques can perform reliable measurements of MGLY.
Ambient time series revealed levels of MGLY ranging from 28 to 365 pptv, with a clear diurnal cycle due to elevated concentrations of primary biogenic species during the daytime, and its oxidation led to large production rates of MGLY. A scatter plot of the PTR-ToF-MS and DNPH–HPLC measurements indicates a reasonable correlation (R2=0.48) but a slope significantly lower than unity (0.58±0.05) and a significant intercept of 88.3±8.0 pptv. A careful investigation of the differences between the two techniques suggests that this disagreement is not due to spectrometric interferences from H3O+(H2O)3 or methyl ethyl ketone (or butanal) detected at m∕z 73.050 and m∕z 73.065, respectively, which are close to the MGLY m∕z of 73.029. The differences are more likely due to uncorrected sampling artifacts such as overestimated collection efficiency or loss of MGLY into the sampling line for the DNPH–HPLC technique or unknown isobaric interfering compounds such as acrylic acid and propanediol for the PTR-ToF-MS.
Calculations of MGLY loss rates with respect to OH oxidation and direct photolysis indicate similar contributions for these two loss pathways.
- Article
(2121 KB) - Full-text XML
-
Supplement
(385 KB) - BibTeX
- EndNote





Methylglyoxal (MGLY, CH3C(O)CHO) is an important α-dicarbonyl species in the atmosphere. It is mainly produced during the oxidation of volatile organic compounds (VOCs) of which isoprene and acetone are the main contributors. Fu et al. (2008) calculated production rates of 110 and 10 Tg year−1 from the oxidation of isoprene and acetone, respectively. Other precursors of MGLY are C3-C5 isoalkanes (Jacob et al., 2002), aromatic compounds (Volkamer et al., 2001; Pan and Wang, 2014; Wu et al., 2014), and monoterpenes (Fick et al., 2003; Nunes et al., 2005). Due to the anthropogenic and biogenic natures of MGLY precursors, this compound can therefore be found at significant levels (low tens to hundreds of pptv) in urban, rural or even remote and marine environments (Henry et al., 2012 and references therein).
The principal sink of MGLY is thought to be photolysis (Fu et al., 2008), which can significantly contribute to the formation of ROx radicals in the troposphere (Dusanter et al., 2009), which in turn can enhance the formation rates of secondary pollutants, including ozone and secondary organic aerosol (SOA). In addition, MGLY has been identified as a direct precursor of SOA (Altieri et al., 2008; Hallquist et al., 2009) due to aqueous reactions in clouds leading to the formation of oligomers and oxalic acids, which can then form SOA upon cloud droplet evaporation (Altieri et al., 2008).
Despite the important role of MGLY in the atmosphere, there are only a few measurement techniques, most of which are expensive, requiring highly skilled operators or suffering from low time resolution. A common method relies on chemical derivatization and chromatographic analysis. Several derivatization agents can be used to trap carbonyl compounds such as 2,4-dinitrophenylhydrazine (DNPH) (Lee et al., 1998; K. F. Ho et al., 2014; Lawson et al., 2015), o-(2,3,4,5,6-pentafluorobenzyl)hydroxylamine (PFBHA) (Spaulding et al., 1999; Ho and Yu, 2002; Ortiz et al., 2006, 2013; Temime et al., 2007) and pentafluorophenylhydrazine (PFPH) (Ho and Yu, 2004; Pang and Lewis, 2011; Pang et al., 2011; Dai et al., 2012). Methods relying on chemical derivatization imply active sampling through cartridges or liquid solutions containing the selected reagent and a subsequent offline analysis using gas chromatography – mass spectrometry (GC–MS) or high performance liquid chromatography with ultraviolet detection (HPLC-UV). Low detection limits are reached for these techniques with the advantages of monitoring several carbonyl compounds simultaneously. Indeed, Ho and Yu (2004) reported detection limits below 0.3 ppbv for a large range of carbonyl compounds, including formaldehyde, acetaldehyde, propanal, acrolein, glyoxal, MGLY and others. These authors used cartridges loaded with PFPH on a Tenax sorbent tube, a sampling time of 4 h and a sampling flow rate of 100 mL min−1. Ait-Helal et al. (2014) even reported lower detection limits ranging from 10 to 60 pptv for C1-C9 aldehydes and ketones, including MGLY, using a sampling duration of 3 h for DNPH cartridges and a sampling flow rate of 1.5 L min−1. The cartridges were analyzed by HPLC-UV. However, the main drawback of these methods is the low time resolution of typically 3–4 h, which is too long to investigate photochemical processes.
Alternative techniques based on mist chambers and derivatization solutions such as PFBHA (Spaulding et al., 2002) or DNPH (Munger et al., 1995) were used to measure MGLY with a faster time resolution of approximately 10 min and low limits of detection (LOD). Spaulding et al. (2002) reported a LOD of 7.7 pptv at a sampling flow rate of 25–70 L min−1 (Spaulding et al., 2002). More recently, a microfluidic derivatization approach using PFBHA and a planar glass micro-reactor was developed to measure glyoxal and MGLY at a time resolution of 30 min and sampling flow rate ranging from 100 to 600 mL min−1 (Pang et al., 2014). This setup exhibits LODs of 76 and 185 pptv (3σ) for glyoxal and MGLY, respectively. The authors also report the use of a solid-phase microextraction (SPME) method, previously described by Gomez Alvarez et al. (2012), capable of measuring MGLY with a LOD of 150 pptv (3σ) and a measurement time of 25 min. The SPME technique relies on a derivatization of aldehyde species into oximes on a fibre loaded with PFBHA and a subsequent analysis by gas chromatography flame ionization detection (GC–FID). Gomez Alvarez et al. (2007) also mentioned the possibility to measure MGLY using a SPME instrument as well as a gas chromatography – electron capture detector (GC–ECD), both calibrated against Fourier transform infrared spectroscopy (FTIR).
In addition to these chemical derivatization methods, optical/spectroscopic approaches have also been employed to measure MGLY. Henry et al. (2012) reported a laser-induced phosphorescence (LIP) instrument capable of simultaneous measurements of glyoxal and MGLY with a time resolution of 5 min and LODs of 4.4 and 243 pptv (3σ), respectively. Thalman and Volkamer (2010) developed a blue LED (light-emitting diodes) cavity-enhanced differential optical absorption spectroscopy (CE-DOAS) instrument for in situ measurements of MGLY among other compounds (nitrogen dioxide, glyoxal, iodine oxide and water vapor). This instrument exhibits a LOD of 170 pptv (2σ) at a time resolution of 1 min. Incoherent broadband cavity-enhanced absorption spectroscopy (IBBCEAS) has also been used to measure both glyoxal (Washenfelder et al., 2011) and MGLY (Pang et al., 2014), with a LOD of 1 ppbv (3σ) for a measurement time of 20 s for the latter. FTIR is another spectroscopic method capable of measuring MGLY (Talukdar et al., 2011). However, FTIR exhibits a LOD in the ppbv range, which is not low enough for ambient measurements, even with a long path length of hundreds of meters (Pang et al., 2014). Overall, while these spectroscopic techniques usually exhibit performances that are suitable for atmospheric measurements, they also require highly skilled operators and the use of fragile pieces of equipment (light sources, mirrors, etc.).
The use of proton transfer reaction time-of-flight mass spectrometry (PTR-ToF-MS) has been attempted for Glyoxal measurements by Stönner et al. (2017). However, these authors showed that the sensitivity of PTR-ToF-MS instruments was too low to monitor ambient concentrations. MGLY measurements by PTR-ToF-MS have been reported by Pang et al. (2014) and Thalman et al. (2015) during intercomparison experiments. Pang et al. (2014) observed a significant disagreement between PTR-ToF-MS measurements and results from other techniques (Microfluidic derivatization, IBBCEAS, FTIR, SPME) during photo-oxidation experiments of isoprene under low NOx conditions in the EUPHORE chamber. According to the authors, this disagreement was due to interferences from (H2O)3 H3O+ at m∕z 73 (no deconvolution of peaks within this mass unit). Thalman et al. (2015) also reported interferences from the (H2O)3 H3O+ cluster and the fragmentation of larger compounds upon protonation. However, blank measurements made at the same relative humidity than in ambient air should contain the contribution of (H2O)3 H3O+ and frequent blank measurements, as usually done during field campaigns, could easily be subtracted to reduce the impact of (H2O)3 H3O+ on the MGLY measurements. De Gouw and Warneke (2007) reported measurements of methyl ethyl ketone (MEK) at the same unit mass using a PTR-MS equipped with a quadrupole. However, time-of-flight mass spectrometers provide the opportunity to deconvolve signals of MGLY (m∕z 73.029) and MEK (m∕z 73.065), which are separated by 0.036 Daltons. Thus, if the mass resolution of the PTR-ToF-MS instrument is sufficient, an adequate peak-fitting procedure and frequent blank measurements should allow a selective detection of methylglyoxal. While PTR-ToF-MS instruments also require highly skilled operators and are more expensive than other techniques allowing MGLY measurements, a growing number of research groups are deploying this type of instrumentation during intensive field campaigns, making it of great interest for MGLY measurements. It is expected that PTR-MS should allow a lower LOD than any other technique reported in the literature so far.
In this study, we present online measurements of MGLY using proton transfer reaction time-of-flight mass spectrometry (PTR-ToF-MS). This study describes a procedure to conduct measurements of MGLY using PTR-ToF-MS, reports a comparison of PTR-ToF-MS and DNPH–HPLC measurements performed during an intensive field campaign in the Mediterranean basin, and presents an investigation of the MGLY loss rate during this campaign.
2.1 The Chemistry-Aerosol Mediterranean Experiment (ChArMEx)
The ChArMEx SOP2 (special observation period 2) field campaign took place from 15 July to 5 August at Corsica Cape (France) on a hilltop (altitude of 533 m) within a wind farm (42.969∘ N, 9.380∘ E). It is a coastal site surrounded by the sea a few kilometers away in all directions (2.5–6 km) (Zannoni et al., 2015). The site was covered by typical Mediterranean vegetation (maquis shrubland) (Zannoni et al., 2015), leading to large emissions of biogenic VOCs and elevated concentrations of isoprene (up to 1.3 ppbv) and monoterpenes (up to 2.2 ppbv) (Michoud et al., 2017). Since MGLY is an oxidation product of isoprene (first, second and third generation), this site is of interest for performing and investigating its budget. On the contrary, a small anthropogenic influence was observed at the measurement site, since the closest city, Bastia, is located ∼30 km away (Michoud et al., 2017).
2.2 PTR-ToF-MS measurements
Measurements of MGLY, among other species (Michoud et al., 2017), were conducted using a PTR-ToF-MS instrument from KORE Inc™ (second generation). Ambient air was sampled through a 5 m long line made of PFA (perfluoroalkoxy). The line was held at 50 ∘C and the flow rate was set at 1.2 L min−1 to reduce the residence time below 4 s. The PTR-ToF-MS was sampled from this line at a constant flow rate of 150 mL min−1. Reactor pressure and temperature were set at 1.33 mbar and 40 ∘C, respectively, leading to an E∕N value of 135 Td. The PTR-ToF-MS spectra were integrated over 10 min, leading to six measurements per hour.
An automatized zero procedure was performed for 10 min every hour to subtract potential contaminations from the lines and to suppress interferences from water clusters and other ions formed inside the glow discharge. Zero air was generated by passing ambient air through a catalytic converter ( stainless steel tubing filled with 2 g of Pt wool held at 350 ∘C) allowing the instrument to be zeroed at the same relative humidity as in ambient air. In order to test the efficiency of the catalytic converter, mixtures of several tens of hydrocarbons at the ppb level were passed through the converter and the remaining VOCs were measured by GC analyzers. Levels lower than the detection limits of the GCs (5–10 pptv) were observed, indicating efficient removal of the VOCs.
VOC signals were extracted from the 10 min mass spectra by summing the number of counts detected within m∕z windows centered on the exact masses of the VOCs of interest (). These signals were normalized by the signals of H3O+ and the ionic water cluster H3O+(H2O) as proposed by de Gouw and Warneke (2007). VOC concentrations were then calculated using Eq. (1).
where iRH_net is the net VOC signal (difference of signals recorded when sampling ambient and zero air), the signal from H3O+ ions at m∕z 21, the signal from H3O+(H2O) at m∕z 39, Xr a factor to account for the effect of humidity on the PTR-ToF-MS sensitivity (de Gouw and Warneke, 2007), Rf the sensitivity determined by calibration (in ncps ppb−1) and 150 000 the corresponding number of primary H3O+ ions in the PTR-ToF-MS reactor (in counts per second). The instrument was calibrated every 3 days during the campaign using a gas calibration unit (IONICON®) and various standards (RESTEK, PRAXAIR) made of hydrocarbons (isoprene, benzene, toluene, o-xylene, ethylbenzene, α-Pinene) and monofunctional oxygenated VOCs (methanol, acetaldehyde, acetone, methyl ethyl ketone). Information about individual mixing ratios of VOCs in the calibration gases can be found in Michoud et al. (2017; Supplement Sect. S1). Mixing ratios were in the range 0.9–4.5 ppm for the abovementioned species and ranged from 3 to 15 ppb after dilution with zero air. Uncertainties associated with these mixing ratios range from 5 % to 10 % (1σ). These calibrations were performed at a relative humidity of 50 % at 20 ∘C without passing the entire 5 m long heated sampling line. XR was determined by conducting additional calibrations at various relative humidity values before and after the campaign. The calibration factor, Rf in Eq. (1), was normalized to 150 000 cps of reagent ions. Specific calibrations performed for methylglyoxal are described in Sect. 3.1.
As mentioned in the introduction, MGLY and MEK are detected at m∕z 73.029 and 73.065, respectively. A Gaussian peak-fitting operation was performed to deconvolve the two peaks observed in the m∕z window 72.95–73.15 during ambient sampling using the curvefit tools from Grams™ software (Thermo Scientific™) (see Supplement Fig. S1). The signals recorded in this window were accumulated over 1 h to reduce the time needed for this procedure, which was done manually. An automatic peak-fitting operation is planned in the future via software development. The MEK-to-methylglyoxal ratio of areas observed for the 1 h cumulated signals was then applied to each 10 min recorded signals (total number of counts recorded within the m∕z window 72.95–73.15) providing measurements of methylglyoxal and MEK at a 10 min time resolution. It is worth noting that the MGLY lifetime of at least 1 h and the longer lifetime of MEK ensure that the MGLY-to-MEK ratio do not change significantly over an hour. Once the signals were deconvolved for each compound, the procedure described in the previous paragraph was applied to calculate their ambient concentrations using sensitivity and humidity dependence factors determined during calibration (Xr=0.5 and 0.49 for MGLY and MEK, respectively).
The 3σ detection limits were calculated from the hourly blank measurements. The average detection limit for methylglyoxal during the whole campaign is 22 pptv (3σ) at the time resolution of 10 min. The total uncertainty was estimated following the “Aerosols, Clouds, and Trace gases Research InfraStructure network” guidelines (ACTRIS Measurement Guideline VOC, 2012), taking into account precision and systematic errors. The repeatability of MGLY measurements was calculated as the square root of the net signal (iRH_net) since the statistic for PTR-ToF-MS signals follows a Poisson distribution (de Gouw and Warneke, 2007) and was on average 9±3 %. The systematic errors concerned the calibration factor (Rf) and the peak-fitting procedure and are estimated to be 22 % for methylglyoxal (19 % and 10 % respectively for the individual errors associated with the calibration factor (Rf) and the peak-fitting procedure).
2.3 Active sampling on DNPH cartridges
Measurements of carbonyl compounds from C1 to C8, including MGLY and MEK, were performed using DNPH cartridges (Waters™) and an automatic sampler (ACROSS-TERA Environment™), based on the US EPA TO-11A method. The analysis of the cartridges was performed in the laboratory using HPLC-UV (Waters 2695 & 2487). This deployment has already been described by Ait-Helal et al. (2014) and Michoud et al. (2017). Ambient air was sampled through a 3 m long PFA line () at a height of 1.5 m above the roof of the trailer next to the PTR-ToF-MS sampling line. This air was collected for 3 h on each cartridge at a flow rate of 1.5 L min−1. A potassium iodide (KI) ozone scrubber and a stainless steel particle filter (porosity: 2 µm) were set up on the sampling line before the automatic sampler. The 3σ detection limit was determined to be 6 pptv for MGLY from blank cartridges (unused cartridges stored under similar conditions than exposed cartridges). The systematic error is estimated to be 25 % for these measurements.
The HPLC-UV instrument used to analyze the DNPH samples was calibrated using a standard solution of hydrazone compounds (TO11/IP-6A) commercialized by Supelco. However, MGLY-DNPH is not present in this solution and a hydrazone standard was made by mixing a known volume of an aqueous solution of MGLY (40 % in water, Acros Organics™) into an excess of acidified DNPH solution. It is worth noting that calibrating the HPLC-UV using a liquid standard of hydrazones is based on the assumption that the collection efficiency of carbonyl compounds through DNPH cartridges is 100 %.
2.4 Investigation of the methylglyoxal loss rate
Two sinks were considered in the steady-state loss calculations: reaction of MGLY with OH and MGLY photolysis. The loss from the reaction with OH was calculated using concentrations of both MGLY and OH, the latter being measured by chemical ionization mass spectrometry (Kukui et al., 2008), and the recommended rate constant of 1.50×1011 cm3 molecule−1 s−1 (Atkinson et al., 2006). J(NO2), and the photolysis frequencies for some other species were derived from the actinic flux measured with an actinic flux spectroradiometer METCON 6007 (Meteorologie Consult GmbH). However, photolysis frequencies for MGLY were not derived from these measurements. The approach described in Dusanter et al. (2009) was therefore employed to calculate J(MGLY) and J(NO2) as a function of the solar zenith angle for the measurement site (lat: 42.969∘ N, long: 9.380∘ E) using the master chemical mechanism (MCM) parameterization (Jenkin et al., 1997; Saunders et al., 2003). This parameterization was derived for an ozone column of 345 Dobson, an altitude of 500 m and clear-sky conditions. Calculated values of J(MGLY) were then corrected for differences in altitude, cloud covering, aerosol and O3 column densities using a scaling factor derived from the measured-to-calculated J(NO2) ratio. The photolytic loss of MGLY was calculated using these scaled photolysis frequencies and PTR-ToF-MS measurements of MGLY.
3.1 PTR-ToF-MS calibrations for MGLY
These calibrations were not performed during the field campaign but a few months later using a liquid calibration unit (LCU, IONICON™) and an aqueous solution of MGLY (40 %, Acros Organics™) (see Fig. 1). The LCU allows a standard mixture containing the targeted compounds to be generated at known mixing ratios by evaporating an aqueous solution of these compounds into a large flow of zero air (1.0 L min−1 in our case). The standard solution flow was varied between 1 and 20 µL min−1 to generate MGLY concentrations ranging from 0.6 to 11 ppbv.
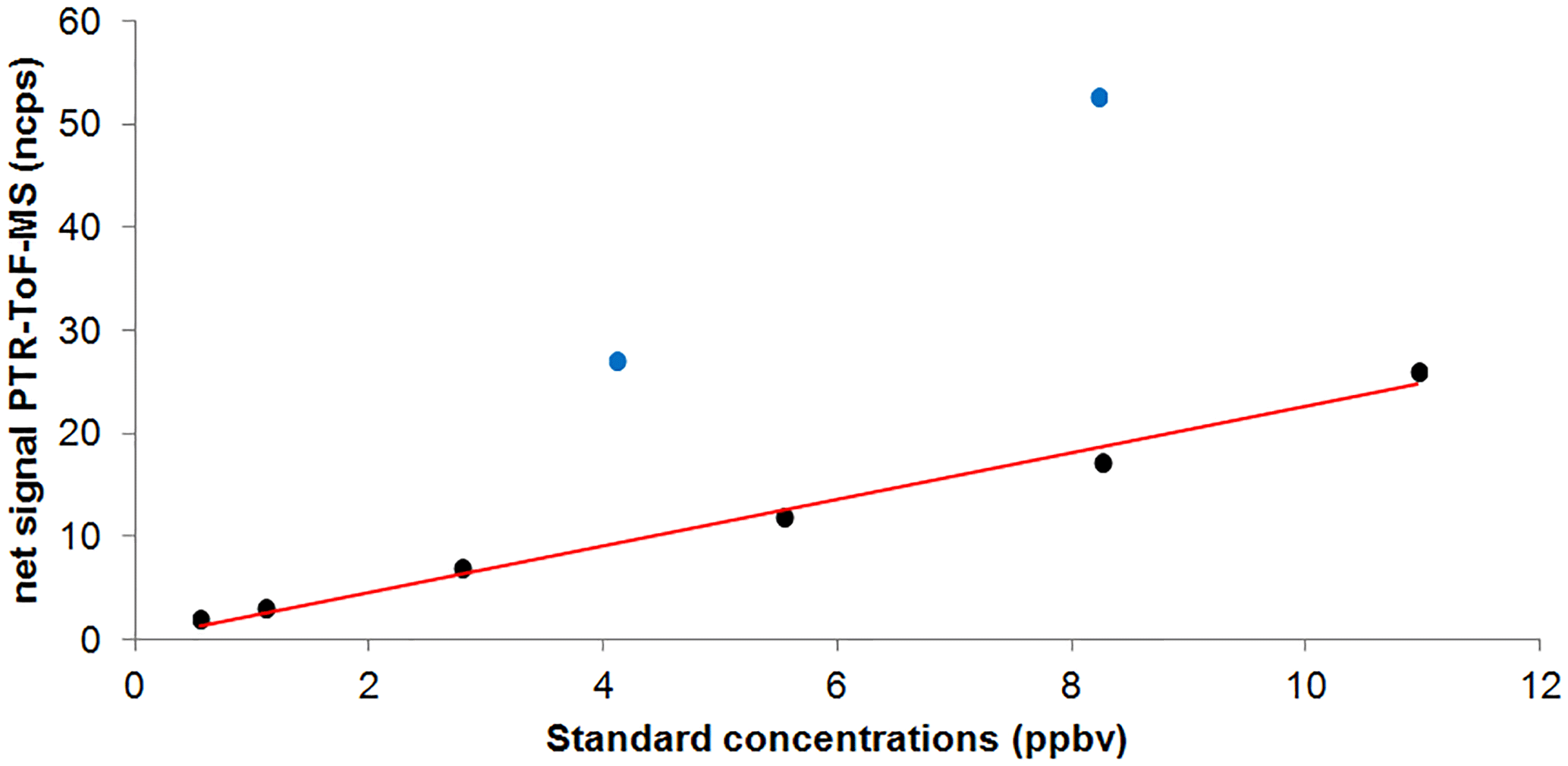
Figure 1MGLY (black circles) and MEK (blue circles) calibration plot for PTR-ToF-MS measurements: normalized net signals at m∕z 73.029 (MGLY, ncps) and 73.065 (MEK, ncps) vs. generated mixing ratio (ppbv).
Figure 1 shows that the PTR-ToF-MS response is linear with the MGLY concentration over the tested range, with no significant offset. While the lower limit of the tested range is larger than observed ambient concentrations (0.05–0.3 ppbv, Fig. 2), it has to be noted that the PTR-MS response has always been observed to be linear with the analyte concentration and a linear response is expected for MGLY for mixing ratios below 0.6 ppbv. These calibration experiments indicate an averaged calibration factor of 2.54±0.49 ncps ppb−1 when normalized to 150 000 cps of reagent ions and using a Xr factor set to 0.5. It is worth noting that changing the flow rate of the liquid standard solution to generate various MGLY concentrations leads to a change in humidity in the gas exiting the LCU, tracked by the m∕z 37–m∕z 19 ratio (varying from 0.1 to 0.5) during these calibration experiments. The good linearity observed in Fig. 1 gives confidence in the Xr factor value used to determine the calibration factor. Therefore, the same Xr value of 0.5 was used for ambient measurements of MGLY.
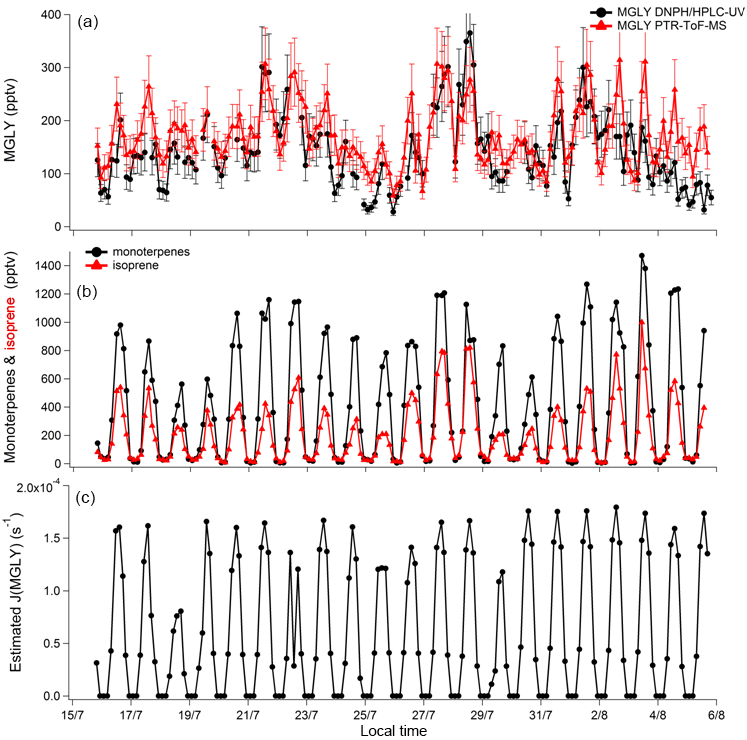
Figure 2Time series of MGLY measured by PTR-ToF-MS (red) and active sampling on DNPH cartridges (black) (a); sum of monoterpenes (black) and isoprene (red) measured by PTR-ToF-MS (b); and estimated J(MGLY) (black) (c). Error bars for MGLY measurements (a) correspond to systematic errors of 22 % and 25 % for PTR-ToF-MS and DNPH cartridge measurements, respectively.
To account for a potential drift in sensitivity between the field measurements and the calibration experiments performed later in the laboratory, calibrations of MEK were also performed during the laboratory experiments using a gas calibration unit (GCU, IONICON™) and a standard mixture provided by IONICON (Restek™). A comparison between MEK response factors observed during field measurements and laboratory experiments allows a drift of the PTRMS sensitivity to be accounted for as further discussed below. The Restek mixture contains 15 compounds including 0.99±0.05 (2σ) ppmv of MEK. Calibrations of MEK were performed every 3 days during the field experiments using the GCU and the same Restek mixture. Since MEK is detected at the same mass unit than MGLY, a change in sensitivity for MGLY between the field measurements and the laboratory calibrations due to a change in ion transmission inside the mass spectrometer would also be observed for MEK. A ratio of the calibration factors measured for MGLY and MEK during the laboratory experiments was used to calculate the MGLY calibration factor from the calibration factor measured for MEK during the field campaign. The laboratory calibrations led to an averaged sensitivity factor of 6.60±0.16 ncps ppb−1 for MEK when normalized to 150 000 cps of reagent ions, leading to a MGLY-to-MEK sensitivity ratio of 0.38. During the ChArMEx field campaign, an averaged calibration factor of 7.57±0.52 ncps ppb−1 was observed for MEK when normalized to 150 000 cps of reagent ions, indicating a decrease of approximately 13 % between the field and laboratory measurements. However, as mentioned above, using the MGLY-to-MEK sensitivity ratio determined in the laboratory allows this change to be corrected.
3.2 Time series of MGLY
Concurrent measurements of MGLY by PTR-ToF-MS and the DNPH–HPLC–UV method in an environment characterized by intense biogenic emissions represent a good opportunity to test how the two techniques compare for this compound. Figure 2 presents time series of MGLY measurements from PTR-ToF-MS (red) and active sampling on DNPH cartridges (black) from 15 July to 6 August. The PTR-ToF-MS measurements performed at a time resolution of 10 min were averaged over 3 h around the sampling middle time of each cartridge measurement to allow a direct comparison between the two techniques. This figure shows that significant levels of MGLY were observed during the campaign, with concentrations ranging from 30 to 370 pptv. In addition, these measurements indicate clear diurnal variations, which are consistent with similar variations of MGLY precursors of biogenic origin observed during the campaign, e.g., isoprene and monoterpenes (see Fig. 2, middle panel). Figure 3 displays campaign-averaged diurnal profiles for MGLY and indicates daily maxima observed around 13:45 LT (Central European Summer Time +02:00 UTC) when the photochemistry is the most intense (see Fig. 2, bottom panel).
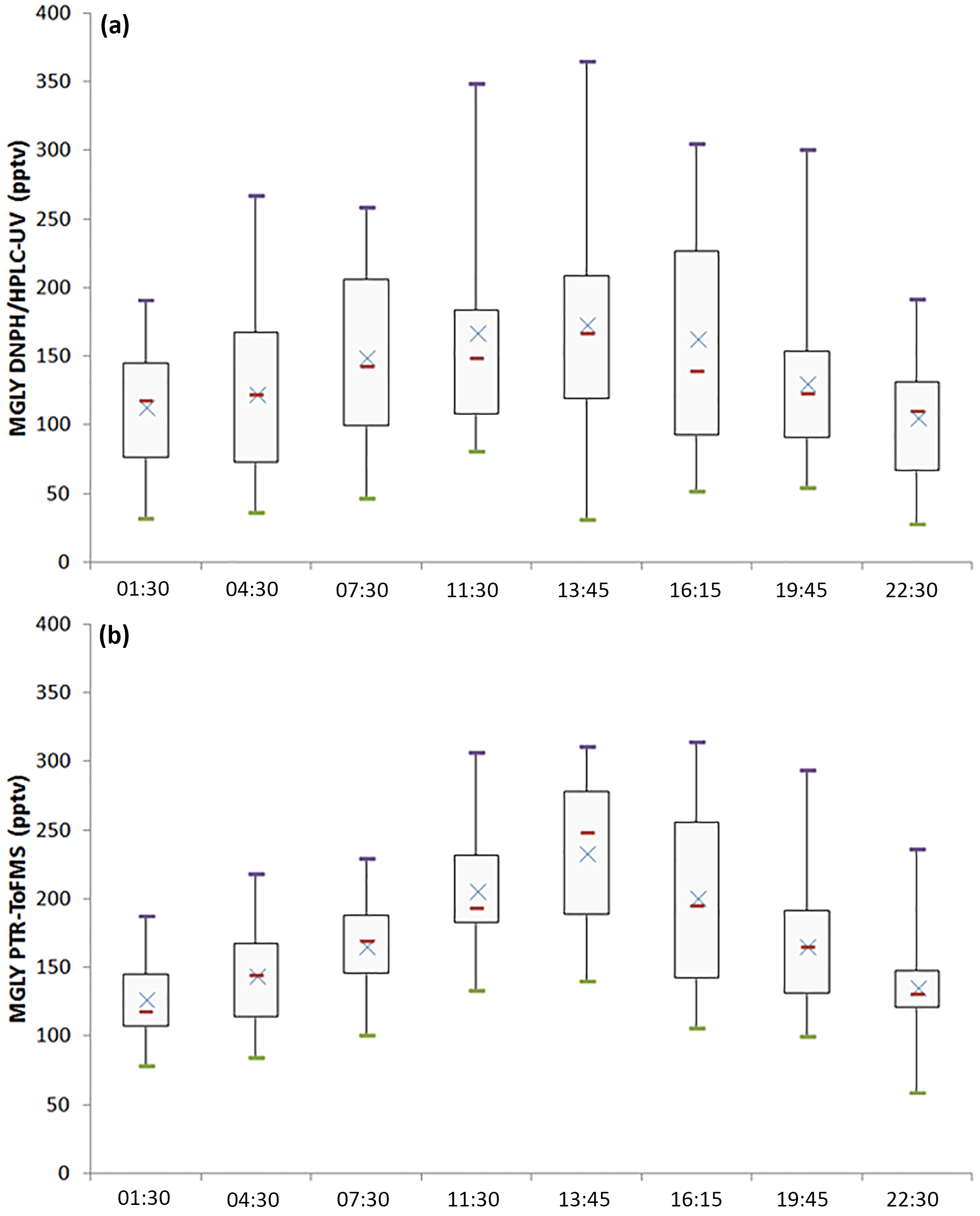
Figure 3Diurnal profiles (box plots) of MGLY measured by both PTR-ToF-MS (b) and active sampling on DNPH cartridges (a) for the campaign average. Purple bars represent the maxima, green bars the minima, red bars the medians, blue crosses the averages, and the sides of the boxes are the first (b) and the third (a) quartiles.
3.3 Comparison of MGLY measurements
Overall, reasonable agreement is observed between the two techniques (see Fig. 2), except for 17 July, 25 July and the last 4 days on which the measured PTR-ToF-MS concentrations were higher by 16 %–148 %. A close look at Fig. 2 also indicates that PTR-ToF-MS measurements are usually higher at night and the concentrations do not decrease as low as that observed for the cartridges. For example, 3 h averaged PTR-ToF-MS concentrations measured at 01:30, 04:30 and 22:30 LT for the overall campaign (Fig. 3) are 127, 144 and 136 pptv, respectively, which are approximately 14, 21 and 30 pptv higher (11 %–22 %) than cartridge measurements, respectively.
Figure 4 displays a scatter plot of PTR-ToF-MS vs. DNPH–HPLC–UV measurements, with a coefficient of determination of 0.48 (R2). A significant intercept of 88±16 pptv (1σ) confirms the higher concentrations observed by PTR-ToF-MS at night, suggesting a positive offset on the PTR-ToF-MS measurements, a negative offset on the cartridge measurements or both. In contrast, a slope significantly lower than unity (0.58±0.10, 1σ) seems to indicate a negative bias in the response of the PTR-ToF-MS measurements, a positive bias for the cartridge measurements or both.
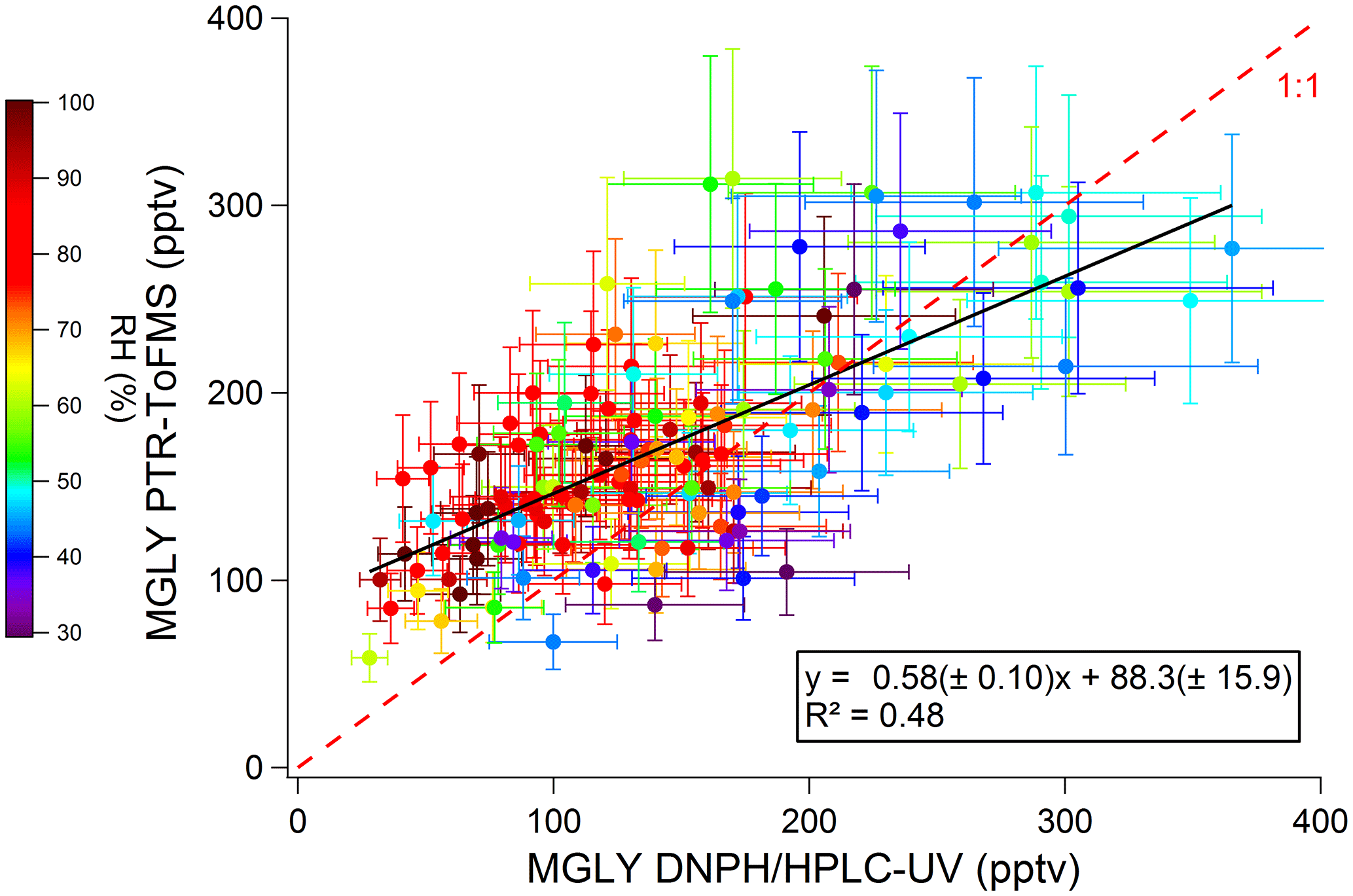
Figure 4Scatter plot of MGLY concentrations measured by PTR-ToF-MS vs. concentrations measured by active sampling on DNPH cartridges. Black line and insert represent the linear regression. Systematic errors associated with the PTR-ToF-MS (22 %) and DNPH cartridge (25 %) measurements are accounted for in the regression analysis. The scatter plot has been color coded according to the relative humidity.
A calibration issue cannot explain an intercept in the scatter plot but could explain part of the disagreement observed for the slope. Three potential reasons may lead to a calibration issue: (i) the generation of unreliable calibration standards, a humidity dependence of (ii) the PTR-ToF-MS response or (iii) the DNPH derivatization. The procedures used to calibrate the PTR-ToF-MS and the HPLC-UV are described in the experimental section. Two different commercial methylglyoxal solutions were used to generate both the gas-phase standard for the PTR-ToF-MS and the liquid standard for the HPLC-UV. While we cannot rule out an issue with the MGLY solutions, it seems unlikely that the disagreement between the two techniques is only due to unreliable MGLY solutions since the good agreement observed on some days (21–22, 27, 31 July and 1 August) contrasts with the poor agreement observed on other days (17 July, 2–5 August) when MGLY peaks during the daytime (200–300 ppt).
As previously mentioned for the PTR-ToF-MS calibration, varying the concentration of MGLY in the range 0.6–11 ppbv with the LCU led to a change in RH. Calculating the calibration factor at each concentration, i.e., at different RHs, from the ratio of the measured normalized signal to the MGLY concentration and plotting it as a function of the m∕z 37–m∕z 19 ratio (Fig. S2) does not indicate a significant water dependence of the PTR-ToF-MS response. The humidity dependence of the DNPH–HPLC–UV method has been recently investigated for some ketone compounds, including acetone and MEK (S. S. H. Ho et al., 2014). It was shown that the collection efficiency is inversely related to relative humidity, with up to 35 %–80 % of the ketones being lost for RH values higher than 50 % at 22 ∘C. While MGLY exhibits a ketone function, it also exhibits an aldehyde function and it is not clear whether this compound will behave as simple ketones. The color coding shown in Fig. 4 indicates that, when higher RH values are observed (60 %–100 %), lower MGLY concentrations and larger relative differences between the two techniques are also observed. Figure 5 displays a scatter plot of the difference between the PTR-ToF-MS and DNPH–HPLC–UV measurements and relative humidity, showing a weak linear correlation with a negative slope. This trend with humidity seems to support that the collection efficiency of MGLY on DNPH cartridges decreases with RH. It is interesting to note that a collection efficiency lower than 100 %, even at low RH values, may explain lower concentrations measured by the DNPH–HPLC–UV method, on average, for the overall campaign.
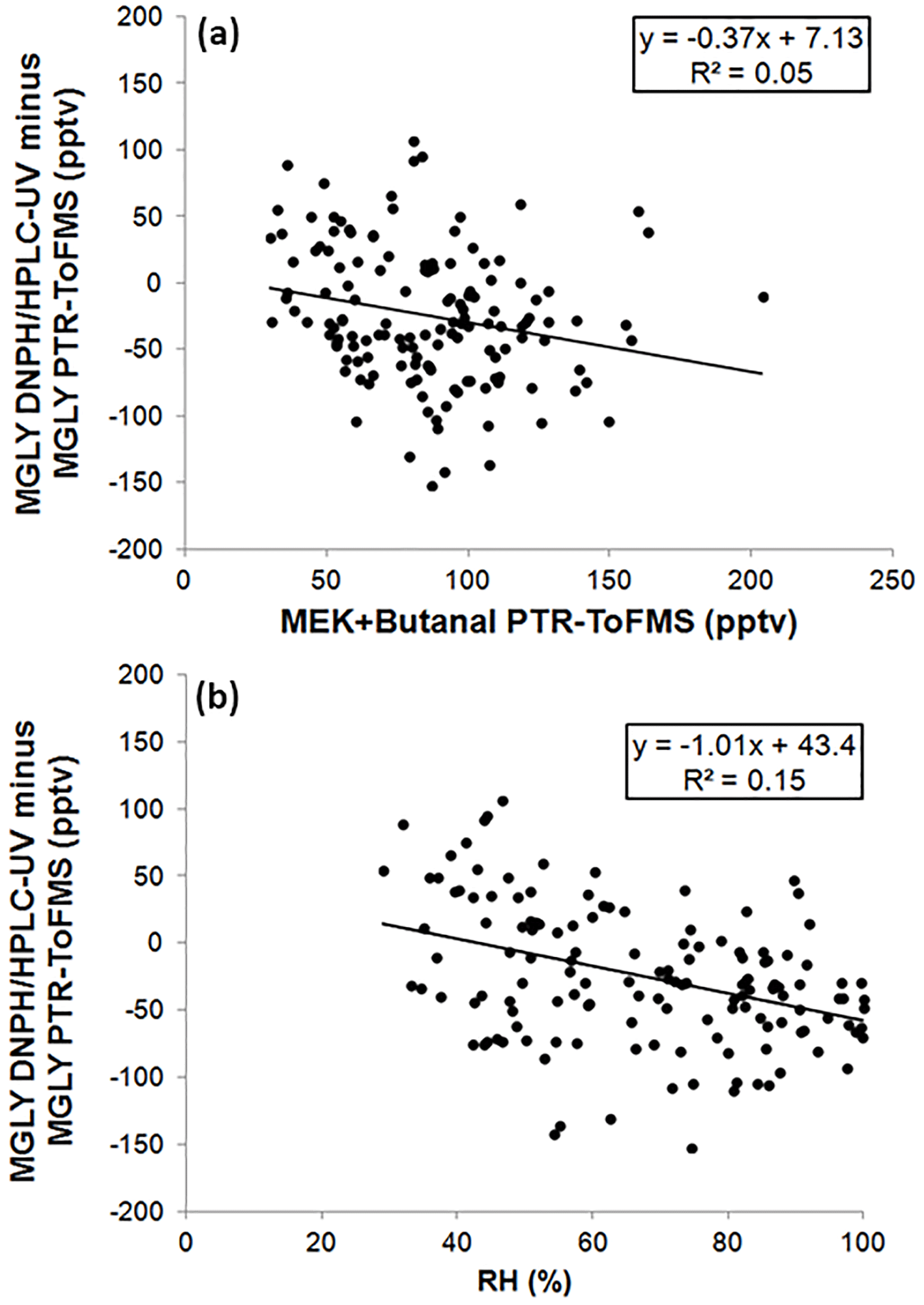
Figure 5Scatter plots of the difference between the two techniques and MEK + butanal (a) or relative humidity (RH, b).
A positive or negative bias in the PTR-ToF-MS measurements could be due to an inadequate peak-fitting procedure to separate the signals detected at m∕z 73.029 (MGLY) and m∕z 73.065 (MEK + butanal) (see Sect. 2.2). To check whether the peak-fitting procedure can lead to a bias in the measurements, Fig. 5 also presents a scatter plot of the difference between the PTR-ToF-MS and DNPH–HPLC–UV measurements and the MEK + butanal concentration measured by PTR-ToF-MS. This scatter plot indicates a very weak correlation (R2 of 0.05), suggesting that the fitting procedure was able to deconvolute the signals from MGLY and MEK + butanal. A weaker correlation is even found when the difference is plotted as a function of butanal, which was measured by DNPH–HPLC–UV (see Supplement Sect. S3).
An ionic water cluster, (H2O)3 H3O+, can also be detected at m∕z 73.050. However, as mentioned previously, the signal from this cluster is recorded during blank measurements and subtracted from ambient measurements. As a consequence, the detection of (H2O)3 H3O+ at m∕z 73 should not impact MGLY measurements reported in this study. Since the abundance of (H2O)3 H3O+ is highly dependent on the ambient water concentration, relative humidity was used as a proxy to investigate whether the cluster signal is efficiently recorded in the blank signal, which was performed hourly to ensure that RH does not change significantly between two blank measurements. A good correlation (, from daily analyses) observed between the blank signal at m∕z 73 and the m∕z 37–m∕z 19 ratio (proxy for humidity content), indicates that the (H2O)3 H3O+ water cluster signal is indeed recorded during blank measurements.
Scatter plots of the difference between PTR-ToF-MS and DNPH–HPLC–UV measurements with O3, acetaldehyde and nopinone were generated (see Supplement Sect. S3) to check whether the possible secondary formation of isobaric OVOCs (malondialdehyde, acrylic acid) in the atmosphere, from the oxidation of ambient VOCs or in the sampling line from reaction of O3 with unsaturated compounds adsorbed on surfaces, could lead to a positive bias in the PTR-ToF-MS measurements. The very weak correlations (R2 of 0.02, 0.01 and <0.01 for O3, acetaldehyde and nopinone, respectively) observed in Fig. S3 rule out this possibility.
Similar correlation plots were made for m∕z 137 (monoterpenes), 139 (nopinone), 151 (pinonaldehyde) and 155 (unidentified oxidation product of monoterpenes) measured by PTR-ToF-MS (see Supplement Sect. S4) to track whether differences observed between the techniques could be explained by interferences from the fragmentation of larger compounds observed at significant concentrations during the CharMEx field campaign. Poor correlations were found (R2<0.06) suggesting that MGLY measurements were free of interferences from the fragmentation of compounds measured at these four masses. Nevertheless, we cannot rule out interferences from the fragmentation of other higher m∕z compounds.
A closer look at the blank signals measured at m∕z 73 shows that this signal correlates with the total m∕z 73 signal on some days (21–22 July, 26–27 July, 1–3 August), with R2 factors ranging from 0.36 to 0.56. Lower correlations are observed on other days (R2<0.20). Interestingly, a scatter plot between the coefficients of determination for the above mentioned correlations and the daily averaged relative humidity exhibits an anticorrelation (negative slope, R2=0.58) (see Supplement Sect. S5). This type of correlation has also been observed by de Gouw et al. (2003), who explained this behavior by the sticky nature of MGLY, which could cause a memory effect in the sampling lines. Different sampling line lengths and characteristics (heated at 50 ∘C for PTR-ToF-MS and not heated for DNPH cartridges, presence of a stainless steel particle filter and a KI ozone scrubber for DNPH cartridges) could lead to different artifacts related to adsorption or heterogeneous reaction on line surfaces for the two techniques. It is worth noting that performing blank measurements every hour and the use of a high flow rate and heated sampling line likely reduces this artifact for the PTR-ToF-MS, while blank measurements for DNPH cartridges only take into account passive contamination of the cartridges, without considering any artifacts from lines. It is interesting to note that, while the difference between the two techniques is not correlated to the PTR-ToF-MS measurements (see Supplement Sect. S3), a fair correlation (R2=0.35) is observed with the cartridge measurements, which may suggest a bias on the cartridge measurements.
While reasonable agreement is observed between the two techniques, a close look at the correlation between the two measurement sets indicates that the DNPH–HPLC–UV methods measured lower concentrations than the PTR-ToF-MS technique by 18 % on average. The above discussion highlights several potential reasons for this disagreement: (i) calibration standards of MGLY are difficult to generate for both techniques and require further work to straighten out this aspect, (ii) the impact of artifacts from sampling lines needs to be further investigated to evaluate their significance, (iii) the collection efficiency of MGLY in DNPH cartridges needs to be investigated under ambient sampling conditions to assess whether MGLY is completely collected and whether there is a humidity dependence.
Finally, we cannot exclude that differences observed between PTR-ToF-MS and DNPH–HPLC–UV measurements of MGLY are partly due to differences in sampling sequences (3 h continuous sampling for DNPH–HPLC–UV, 3 h sampling minus 3 times 10 min of blank measurements for PTR-ToF-MS). However, the impact of differences in timescale for the two techniques should lead to random scatter when the measurements are compared and not to a systematic difference as observed in this study.
3.4 MGLY loss rate
Loss rates of MGLY are presented in Fig. 6. They were calculated as described in Sect. 2.4 using PTR-ToF-MS measurements since the DNPH–HPLC–UV measurements may suffer from inlet effects and an overestimated collection efficiency and since PTR-ToF-MS measurements have higher temporal resolution. The total loss rate peaks during the daytime around 14:00 LT at values ranging from 100 to 350 pptv h−1. The calculated loss rate is almost equally divided into photolysis and oxidation by OH, accounting for 53 % and 47 %, respectively, of the average diurnal loss from 10:00 to 19:00 LT.
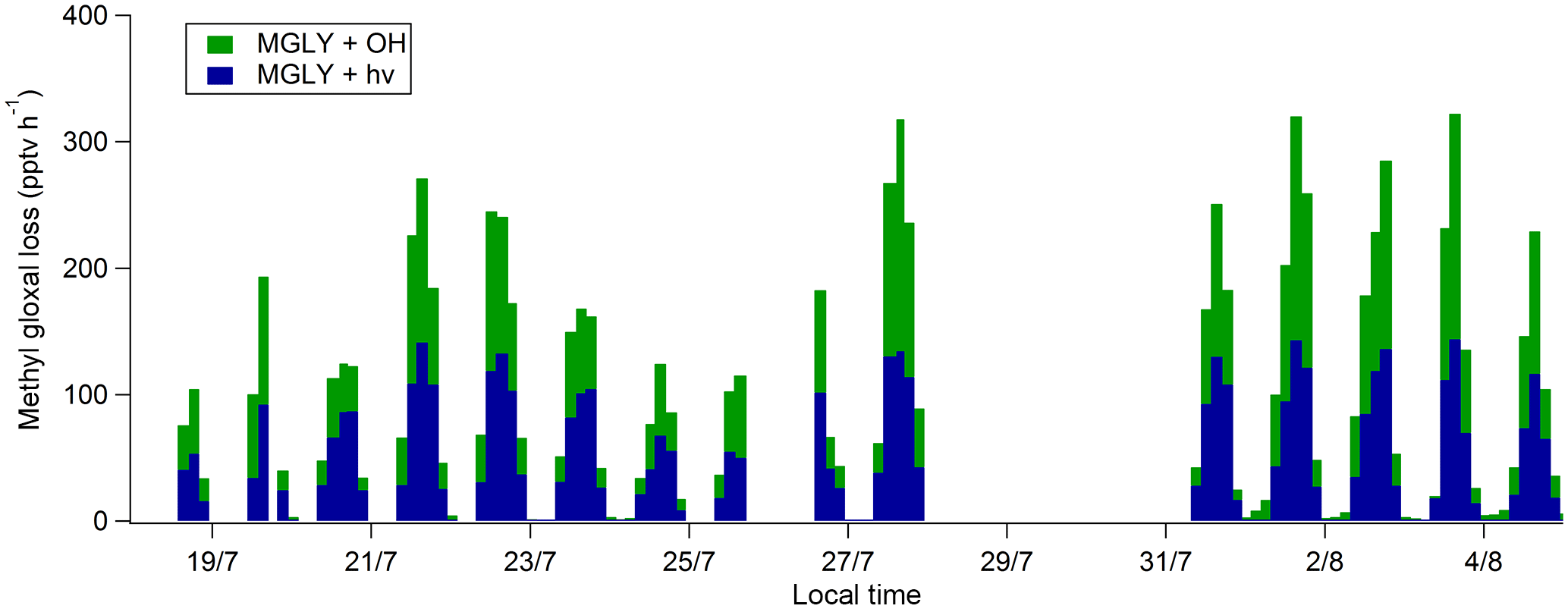
A thorough investigation of the MGLY budget would require calculating the total MGLY production rate from the oxidation of ambient VOCs for comparison to the total loss rate presented above. However, as mentioned in the introduction, MGLY is produced during the oxidation of many VOCs (isoprene, monoterpenes, acetone, aromatics, etc.) at average yields which are strongly dependent on ambient radical concentrations and NOx as recently reported for isoprene (Jenkin et al., 2015). It is also worth noting that calculating MGLY production rates based on ambient concentrations of precursors and average yield values would only be robust for first-generation oxidation products since no intermediate species is taken into account. Taking into account that MGLY is also a second- and higher-generation oxidation product in most degradation mechanisms would lead to a delayed formation. Indeed, MGLY production can take hours in NO-rich environments and even days in low-NOx environments such as in this study (Fu et al., 2008). As a consequence, it would be hazardous to try to calculate local MGLY production rates from the measured VOC precursors.
When a gaseous species exhibits a lifetime lower than a few seconds or minutes, such as radical species or highly photolabile compounds, this species should reach a photostationary state and chemical production and loss rates should balance each other out since transport processes such as advection and vertical dilution are too slow to significantly impact the local concentration of these short-lived species. MGLY exhibits a lifetime of approximately 1–2 h during the daytime, which may be short enough for the photostationary state to hold. In this case, production rates of MGLY should mimic the loss rate displayed in Fig. 6. However, Washenfelder et al. (2011) showed a breakdown of the photostationary state when applied to glyoxal, a dicarbonyl compound exhibiting a lifetime of the same order of magnitude as MGLY, and as a consequence the calculated loss rate reported in this study only provides a rough estimation of the local production rate.
To the best of our knowledge, this study presents the first ambient measurements of methylglyoxal by PTR-ToF-MS. This work aims to describe a simple and proper procedure with which to perform reliable measurements, relying on (i) the data processing proposed by de Gouw and Warneke (2007) to account for the impact of ambient humidity on the PTR-ToF-MS sensitivity, (ii) automatized blank measurements performed every hour to suppress potential memory effects and interferences from water clusters, and (iii) a Gaussian peak-fitting analysis to deconvolute the methylglyoxal signal from other compounds exhibiting a similar mass unit but different exact masses (i.e., butanone and butanal).
The ChArMEx SOP2 field campaign was conducted in an environment characterized by high biogenic emissions, including isoprene, at the extreme north of Corsica. This campaign therefore provides a good opportunity to study methylglyoxal measurements, since this compound is mainly formed via isoprene oxidation. Furthermore, concomitant measurements of methylglyoxal by PTR-ToF-MS and DNPH–HPLC–UV allowed an intercomparison of these two techniques to test their reliability.
Time series of methylglyoxal measured by both PTR-ToF-MS and DNPH–HPLC–UV revealed concentration levels ranging from 28 to 365 pptv with a clear diurnal cycle due to the secondary nature of this compound. The visual comparison of the measured time series shows a reasonable agreement, with the DNPH–HPLC–UV methods measuring concentrations lower by 18 % on average compared to the PTR-ToF-MS technique. A linear regression analysis performed between the two measurement sets indicates a fair correlation with a determination coefficient (R2) of 0.48, a slope significantly different than unity (0.58±0.10, 1σ) and a nonzero intercept (88.3±15.9 pptv, 1σ). Interferences from (H2O)3 H3O+, butanone and butanal can be excluded for the PTR-ToF-MS measurements, validating the procedure used for data acquisition and analysis. Methylglyoxal formation into sampling lines due to heterogeneous reactions of O3 with adsorbed organic compounds is also not likely. Potential remaining uncorrected artifacts from lines on some days for both techniques could be partly responsible for measurements disagreements and this aspect needs to be further investigated to evaluate its significance. In addition, this work questions the collection efficiency of MGLY in DNPH cartridges and recommends an investigation under ambient sampling conditions to assess whether all the MGLY is collected and whether a humidity dependence exists. Comparisons of PTR-ToF-MS with other existing techniques in the field and/or in atmospheric simulation chambers would be of interest to identify potential artifacts causing the disagreement observed in this study. Nevertheless, PTR-ToF-MS seems promising for methylglyoxal measurements.
The methylglyoxal loss rate was studied at Corsica Cape, revealing that the contributions of direct photolysis and OH oxidation were almost similar.
Access to the data used in this article is restricted to registered users of the ChArMEx project. The data are available on the project website (http://mistrals.sedoo.fr/ChArMEx/, last access: 12 October 2018) and it should be used following the data and publication policies of the ChArMEx project; http://mistrals.sedoo.fr/ChArMEx/Data-Policy/ChArMEx_DataPolicy.pdf (last access: 12 October 2018).
The supplement related to this article is available online at: https://doi.org/10.5194/amt-11-5729-2018-supplement.
The first and last author contributed equally to this work. VM and DS prepared the paper with inputs from all co-authors. SS, TL and NL participated in the field campaign and were in charge of VOC measurements. AK was involved in the field measurements and provided OH data. IF performed the analysis of DNPH cartridges for carbonyls measurements.
The authors declare that they have no conflict of interest.
This article is part of the special issue “CHemistry and AeRosols Mediterranean EXperiments (ChArMEx) (ACP/AMT inter-journal SI)”. It is not associated with a conference.
This study received financial support from the Mistrals/ChArMEx program, ADEME, the French environmental ministry, and the CaPPA projects. The CaPPA project (Chemical and Physical Properties of the Atmosphere) is funded by the French National Research Agency (ANR) through the PIA (Programme d'Investissement d'Avenir) under contract ANR-11-LABX-0005-01 and by the Regional Council Nord-Pas de Calais and the “European Funds for Regional Economic Development” (FEDER). This research was also funded by the European Union Seventh Framework Programme under grant agreement number 293897, DEFIVOC project and CARBOSOR/Primequal. This study also received funding from the Région Hauts-de-France, the Ministère de l'Enseignement Supérieur et de la Recherche and the European Fund for Regional Economic Development through the CLIMIBIO project.
The authors also want to thank Eric Hamonou and François Dulac for
logistics management during the campaign as well as all the participants of
the ChArMEx SOP2 field campaign.
Edited by:
Nikolaos Mihalopoulos
Reviewed by: two anonymous referees
ACTRIS Measurement Guidelines VOC, WP4-NA4: Trace gases networking: Volatile organic carbon and nitrogen oxides Deliverable D4.1: Draft for standardized operating procedures (SOPs) for VOC measurements, available at: http://ebas-submit.nilu.no/Portals/117/media/SOPs/MG_VOC_draft_20120718.pdf (12 October 2018), 24–32, 2012.
Ait-Helal, W., Borbon, A., Sauvage, S., de Gouw, J. A., Colomb, A., Gros, V., Freutel, F., Crippa, M., Afif, C., Baltensperger, U., Beekmann, M., Doussin, J.-F., Durand-Jolibois, R., Fronval, I., Grand, N., Leonardis, T., Lopez, M., Michoud, V., Miet, K., Perrier, S., Prévôt, A. S. H., Schneider, J., Siour, G., Zapf, P., and Locoge, N.: Volatile and intermediate volatility organic compounds in suburban Paris: variability, origin and importance for SOA formation, Atmos. Chem. Phys., 14, 10439–10464, https://doi.org/10.5194/acp-14-10439-2014, 2014.
Altieri, K. E., Seitzinger, S. P., Carlton, A. G., Turpin, B. J., Klein, G. C., and Marshall, A. G.: Oligomers formed through in-cloud methylglyoxal reactions: Chemical composition, properties, and mechanisms investigated by ultra-high resolution FT-ICR mass spectrometry, Atmos. Environ., 42, 1476–1490, https://doi.org/10.1016/j.atmosenv.2007.11.015, 2008.
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., Troe, J., and IUPAC Subcommittee: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume II – gas phase reactions of organic species, Atmos. Chem. Phys., 6, 3625–4055, https://doi.org/10.5194/acp-6-3625-2006, 2006.
Dai, W. T., Ho, S. S. H., Ho, K. F., Liu, W. D., Cao, J. J., and Lee, S. C.: Seasonal and diurnal variations of mono- and di-carbonyls in Xi'an, China, Atmos. Res., 113, 102–112, 2012.
de Gouw, J. and Warneke, C.: Measurements of volatile organic compounds in the earth's atmosphere using proton-transferreaction mass spectrometry, Mass. Spectrom. Rev., 26, 223–257, https://doi.org/10.1002/mas.20119, 2007.
de Gouw, J. A., Goldan, P. D., Warneke, C., Kuster, W. C., Roberts, J. M., Marchewka, M., Bertman, S. B., Pszenny, A. A. P., and Keene, W. C.: Validation of proton transfer reaction-mass spectrometry (PTR-MS) measurements of gas-phase organic compounds in the atmosphere during the New England Air Quality Study (NEAQS) in 2002, J. Geophys. Res., 108, 4682, https://doi.org/10.1029/2003JD003863, 2003.
Dusanter, S., Vimal, D., Stevens, P. S., Volkamer, R., Molina, L. T., Baker, A., Meinardi, S., Blake, D., Sheehy, P., Merten, A., Zhang, R., Zheng, J., Fortner, E. C., Junkermann, W., Dubey, M., Rahn, T., Eichinger, B., Lewandowski, P., Prueger, J., and Holder, H.: Measurements of OH and HO2 concentrations during the MCMA-2006 field campaign – Part 2: Model comparison and radical budget, Atmos. Chem. Phys., 9, 6655–6675, https://doi.org/10.5194/acp-9-6655-2009, 2009.
Fick, J., Pommer, L., Nilsson, C., and Andersson, B.: Effect of OH radicals, relative humidity, and time on the composition of the products formed in the ozonolysis of alpha-pinene, Atmos. Environ., 37, 4087–4096, 2003.
Fu, T.-M., Jacob, D. J., Wittrock, F., Burrows, J. P., Vrekoussis, M., and Henze, D. K.: Global budgets of atmospheric glyoxal and methylglyoxal, and implications for formation of secondary organic aerosols, J. Geophys. Res., 113, D15303, https://doi.org/10.1029/2007JD009505, 2008.
Gómez Alvarez, E., Viidanoja, J., Muñoz, A., Wirtz, A., and Hjorth, J.: Experimental confirmation of the dicarbonyl route in the photo-oxidation of toluene and benzene, Environ. Sci. Technol., 41, 8362–8369, https://doi.org/10.1021/es0713274, 2007.
Gomez Alvarez, E., Moreno, M. V., Gligorovski, S., Wortham, H., and Cases, M. V. R.: Characterisation and calibration of active sampling Solid Phase Microextraction applied to sensitive determination of gaseous carbonyls, Talanta, 88, 252–258, 2012.
Hallquist, M., Wenger, J. C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N. M., George, C., Goldstein, A. H., Hamilton, J. F., Herrmann, H., Hoffmann, T., Iinuma, Y., Jang, M., Jenkin, M. E., Jimenez, J. L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, Th. F., Monod, A., Prévôt, A. S. H., Seinfeld, J. H., Surratt, J. D., Szmigielski, R., and Wildt, J.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009.
Henry, S. B., Kammrath, A., and Keutsch, F. N.: Quantification of gas-phase glyoxal and methylglyoxal via the Laser-Induced Phosphorescence of (methyl)GLyOxal Spectrometry (LIPGLOS) Method, Atmos. Meas. Tech., 5, 181–192, https://doi.org/10.5194/amt-5-181-2012, 2012.
Ho, S. S. H. and Yu, J. Z.: Feasibility of Collection and Analysis of Airborne Carbonyls by On-Sorbent Derivatisation and Thermal Desorption, Anal. Chem., 74, 1232–1240, 2002.
Ho, S. S. H. and Yu, J. Z.: Determination of Airborne Carbonyls: Comparison of a Thermal Desorption/GC Method with the Standard DNPH/HPLC Method, Environ. Sci. Technol., 38, 862–870, 2004.
Ho, K. F., Ho, S., Dai, W. T., Cao, J. J., Huang, R.-J., Tian, L., and Deng, W. J.: Seasonal Variations of Monocarbonyl and Dicarbonyl in Urban and Sub-Urban Sites of Xi'an, China, Environ. Monit. Assess., 186, 2835–2849, 2014.
Ho, S. S. H., Chow, J. C., Watson, J. G., Ip, H. S. S., Ho, K. F., Dai, W. T., and Cao, J.: Biases in ketone measurements using DNPHcoated solid sorbent cartridges, Anal. Methods-UK, 6, 967–974, https://doi.org/10.1039/C3AY41636D, 2014.
Jacob, D. J., Field, B. D., Jin, E., Bey, I., Li, Q., Logan, J., Yantosca, R. M., and Singh, H. B.: Atmospheric budget of acetone, J. Geophys. Res.-Atmos., 107, https://doi.org/10.1029/2001JD000694, 2002.
Jenkin, M. E., Saunders, S. M., and Pilling, M. J.: The tropospheric degradation of volatile organic compounds: A protocol for mechanism development, Atmos. Environ., 31, 81–104, 1997.
Jenkin, M. E., Young, J. C., and Rickard, A. R.: The MCM v3.3.1 degradation scheme for isoprene, Atmos. Chem. Phys., 15, 11433–11459, https://doi.org/10.5194/acp-15-11433-2015, 2015.
Kukui, A., Ancellet, G., and Le Bras, G.: Chemical ionisation mass spectrometer for measurements of OH and Peroxy radical concentrations in moderately polluted atmospheres, J. Atmos. Chem., 61, 133–154, 2008.
Lawson, S. J., Selleck, P. W., Galbally, I. E., Keywood, M. D., Harvey, M. J., Lerot, C., Helmig, D., and Ristovski, Z.: Seasonal in situ observations of glyoxal and methylglyoxal over the temperate oceans of the Southern Hemisphere, Atmos. Chem. Phys., 15, 223–240, https://doi.org/10.5194/acp-15-223-2015, 2015.
Lee, Y. N., Zhou, X., Kleinman, L. I., Nunnermacker, L. J., Springston, S. R., Daum, P. H., Newman, L., Keigley, W. G., Holdren, M. W., Spicer, C. W., Young, V., Fu, B., Parrish, D. D., Holloway, J., Williams, J., Roberts, J. M., Ryerson, T. B., and Fehsenfeld, F. C.: Atmospheric chemistry and distribution of formaldehyde and several multioxygenated carbonyl compounds during the 1995 Nashville Middle Tennessee Ozone Study, J. Geophys. Res.-Atmos., 103, 22449–22462, https://doi.org/10.1029/98jd01251, 1998.
Michoud, V., Sciare, J., Sauvage, S., Dusanter, S., Léonardis, T., Gros, V., Kalogridis, C., Zannoni, N., Féron, A., Petit, J.-E., Crenn, V., Baisnée, D., Sarda-Estève, R., Bonnaire, N., Marchand, N., DeWitt, H. L., Pey, J., Colomb, A., Gheusi, F., Szidat, S., Stavroulas, I., Borbon, A., and Locoge, N.: Organic carbon at a remote site of the western Mediterranean Basin: sources and chemistry during the ChArMEx SOP2 field experiment, Atmos. Chem. Phys., 17, 8837–8865, https://doi.org/10.5194/acp-17-8837-2017, 2017.
Munger, J. W., Jacob, D. J., Daube, B. C., Horowitz, L. W., Keene, W. C., and Heikes, B. G.: Formaldehyde, glyoxal, and methylglyoxal in air and cloudwater at a rural mountain site in central Virginia, J. Geophys. Res.-Atmos., 100, 9325–9333, 1995.
Nunes, F. M. N., Veloso, M. C. C., Pereira, P. A. D. P., and de Andrade, J. B.: Gas-phase ozonolysis of the monoterpenoids (S)- (+)-carvone, (R)-(−)-carvone, (−)-carveol, geraniol and citral, Atmos. Environ., 39, 7715–7730, 2005.
Ortiz, R., Hagino, H., Sekiguchi, K., Wang, Q. Y., and Sakamoto, K.: Ambient air measurements of six bifunctional carbonyls in a suburban area, Atmos. Res., 82, 709–718, https://doi.org/10.1016/j.atmosres.2006.02.025, 2006.
Ortiz, R., Shimada, S., Sekiguchi, K., Wang, Q., and Sakamoto, K.: Measurements of changes in the atmospheric partitioning of bifunctional carbonyls near a road in a suburban area, Atmos. Environ., 81, 554–560, 2013.
Pan, S. S. and Wang, L. M.: Atmospheric Oxidation Mechanism of m-Xylene Initiated by OH Radical, J. Phys. Chem. A, 118, 10778–10787, https://doi.org/10.1021/jp506815v, 2014.
Pang, X. and Lewis, A. C.: Carbonyl compounds in gas and particle phases of mainstream cigarette smoke, Sci. Total Environ., 409, 5000–5009, 2011.
Pang, X., Lewis, A. C., and Hamilton, J. F.: Determination of airborne carbonyls via pentafluorophenylhydrazine derivatisation by GC-MS and its comparison with HPLC method, Talanta, 85, 406–414, 2011.
Pang, X., Lewis, A. C., Rickard, A. R., Baeza-Romero, M. T., Adams, T. J., Ball, S. M., Daniels, M. J. S., Goodall, I. C. A., Monks, P. S., Peppe, S., Ródenas García, M., Sánchez, P., and Muñoz, A.: A smog chamber comparison of a microfluidic derivatisation measurement of gas-phase glyoxal and methylglyoxal with other analytical techniques, Atmos. Meas. Tech., 7, 373–389, https://doi.org/10.5194/amt-7-373-2014, 2014.
Saunders, S. M., Jenkin, M. E., Derwent, R. G., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): tropospheric degradation of non-aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 161–180, https://doi.org/10.5194/acp-3-161-2003, 2003.
Spaulding, R. S., Frazey, P. A., Rao, X., and Charles, M. J.: Measurement of Hydroxy Carbonyls and Other Carbonyls in Ambient Air Using Pentafluorobenzyl Alcohol as a Chemical Ionization Reagent, Anal. Chem., 71, 3420–3427, 1999.
Spaulding, R. S., Talbot, R. W., and Charles, M. J.: Optimisation of a Mist Chamber (Cofer Scrubber) for Sampling Water-Soluble Organics in Air, Environ. Sci. Technol., 36, 1798–1808, 2002.
Stönner, C., Derstroff, B., Klüpfel, T., Crowley, J. N., and Williams, J.: Glyoxal measurement with a proton transfer reaction time of flight mass spectrometer (PTR-TOF-MS): characterization and calibration, J. Mass Spectrom., 52, 30–35, 2017.
Talukdar, R. K., Zhu, L., Feierabend, K. J., and Burkholder, J. B.: Rate coefficients for the reaction of methylglyoxal (CH3COCHO) with OH and NO3 and glyoxal (HCO)2 with NO3, Atmos. Chem. Phys., 11, 10837–10851, https://doi.org/10.5194/acp-11-10837-2011, 2011.
Temime, B., Healy, R. M., and Wenger, J. C.: A Denuder-Filter Sampling Technique for the Detection of Gas and Particle Phase Carbonyl Compounds, Environ. Sci. Technol., 41, 6514–6520, 2007.
Thalman, R. and Volkamer, R.: Inherent calibration of a blue LED-CE-DOAS instrument to measure iodine oxide, glyoxal, methyl glyoxal, nitrogen dioxide, water vapour and aerosol extinction in open cavity mode, Atmos. Meas. Tech., 3, 1797–1814, https://doi.org/10.5194/amt-3-1797-2010, 2010.
Thalman, R., Baeza-Romero, M. T., Ball, S. M., Borrás, E., Daniels, M. J. S., Goodall, I. C. A., Henry, S. B., Karl, T., Keutsch, F. N., Kim, S., Mak, J., Monks, P. S., Muñoz, A., Orlando, J., Peppe, S., Rickard, A. R., Ródenas, M., Sánchez, P., Seco, R., Su, L., Tyndall, G., Vázquez, M., Vera, T., Waxman, E., and Volkamer, R.: Instrument intercomparison of glyoxal, methyl glyoxal and NO2 under simulated atmospheric conditions, Atmos. Meas. Tech., 8, 1835–1862, https://doi.org/10.5194/amt-8-1835-2015, 2015.
Volkamer, R., Platt, U., and Wirtz, K.: Primary and secondary glyoxal formation from aromatics: Experimental evidence for the bicycloalkyl-radical pathway from benzene, toluene, and p-xylene, J. Phys. Chem. A, 105, 7865–7874, https://doi.org/10.1021/Jp010152w, 2001.
Washenfelder, R. A., Young, C. J., Brown, S. S., Angevine, W. M., Atlas, E. L., Blake, D. R., Bon, D. M., Cubison, M. J., de Gouw, J. A., Dusanter, S., Flynn, J., Gilman, J. B., Graus, M., Griffith, S., Grossberg, N., Hayes, P. L., Jimenez, J. L., Kuster, W. C., Lefer, B. L., Pollack, I. B., Ryerson, T. B., Stark, H., Stevens, P. S., and Trainer, M. K.: The glyoxal budget and its contribution to organic aerosol for Los Angeles, California, during CalNex 2010, J. Geophys. Res., 116, D00V02, https://doi.org/10.1029/2011JD016314, 2011.
Wu, R. R., Pan, S. S., Li, Y., and Wang, L. M.: Atmospheric Oxidation Mechanism of Toluene, J. Phys. Chem. A, 118, 4533–4547, https://doi.org/10.1021/jp500077f, 2014.
Zannoni, N., Dusanter, S., Gros, V., Sarda Esteve, R., Michoud, V., Sinha, V., Locoge, N., and Bonsang, B.: Intercomparison of two comparative reactivity method instruments inf the Mediterranean basin during summer 2013, Atmos. Meas. Tech., 8, 3851–3865, https://doi.org/10.5194/amt-8-3851-2015, 2015.