the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 02 Apr 2020
| 02 Apr 2020
An instrument for in situ measurement of total ozone reactivity
Roberto Sommariva
Louisa J. Kramer
Leigh R. Crilley
Mohammed S. Alam
William J. Bloss
We present an instrument for the measurement of total ozone reactivity – the reciprocal of the chemical lifetime of ozone (O3) – in the troposphere. The Total Ozone Reactivity System (TORS) was developed with the objective to study the role of biogenic volatile organic compounds (BVOCs) as chemical sinks of tropospheric ozone. The instrument was extensively characterized and tested in the laboratory using individual BVOCs and small plants (lemon thyme, Thymus citriodorus) in a Teflon bag and proved able to measure reactivities corresponding to s−1 (at 5 min averaging time), with an estimated total uncertainty of ∼32%. Such reactivities correspond to >20 ppb of α-pinene or >150 ppb of isoprene in isolation – larger than typical ambient levels but observable in environmental chamber and enclosure experiments as well as in BVOC-rich environments. The functionality of TORS was demonstrated in quasi-ambient conditions with a deployment in a horticultural glasshouse containing a range of aromatic plants. The measurements of total ozone reactivity made in the glasshouse showed a clear diurnal pattern, following the emissions of BVOCs, and are consistent with mixing ratios of tens of parts per billion of monoterpenes and several parts per billion of sesquiterpenes.
- Article
(2292 KB) - Full-text XML
- BibTeX
- EndNote





Ozone (O3) is a key component of the troposphere: it is known to be damaging for human health and vegetation and to reduce crop yields, and it is an important greenhouse gas (Monks et al., 2015). Ozone is also a primary source of the OH radical, the main atmospheric oxidant, and acts as an oxidant itself (Johnson and Marston, 2008). Because of its importance to tropospheric chemistry, the ozone budget has long been a subject of considerable interest. Ozone is not directly emitted but is formed in the troposphere via photolysis of nitrogen dioxide (NO2), followed by reaction of atomic oxygen with molecular oxygen (Reactions R1 and R2).
Ozone is destroyed in the troposphere via a series of processes, both physical (e.g. dry and wet deposition) and chemical (Monks et al., 2015). The latter involve photolysis (Reactions R3 and R4) and reactions with a range of inorganic molecules and unsaturated volatile organic compounds (Reactions R5–R9).
Ozone photolysis forms atomic oxygen in ground (O(3P)) and excited (O(1D)) states, which primarily react with molecular oxygen in the atmosphere to reform ozone in a null cycle (Reactions R2, R10). However, a small fraction (∼10 % in the lower troposphere) of O(1D) reacts with water vapour to form OH radicals (Reaction R11), a key process for the oxidative capacity of the atmosphere.
The chemical losses of ozone are one of the least understood parts of the tropospheric ozone budget, in particular because of the many unknowns related to the abundance of volatile organic compounds (VOCs) and their reactivity with ozone (Johnson and Marston, 2008; Glasius and Goldstein, 2016). The organic compounds that react with ozone contain one or more double carbon bonds (e.g. alkenes and dialkenes), and many of these species are emitted by plants during their metabolic processes. These biogenic VOCs (BVOCs) constitute a large fraction of the carbon loading of the atmosphere: estimates suggest that the total biogenic sources of VOCs can be 8–10 times larger than the total anthropogenic sources (Williams, 2004; Glasius and Goldstein, 2016). Isoprene is by far the most important reactive BVOC, accounting for ∼50 % of the total natural emissions of non-methane hydrocarbons by mass, followed by monoterpenes (15 %), methanol (9 %), CO (7 %), and a range of other organic compounds which include acetone (4 %) and sesquiterpenes (3 %) (Guenther et al., 2012).
However, due to the limitations of the analytical techniques used to measure VOCs, it is very likely that both the number and the mass of BVOCs in the troposphere are severely underestimated (Di Carlo et al., 2004; Sinha et al., 2010; Whalley et al., 2011). Estimates vary depending on the approach used, but it is thought that between 20 % and 60 % (and possibly more) of the total organic carbon pool in the troposphere is currently unaccounted for (Lewis et al., 2000; Goldstein and Galbally, 2007; Glasius and Goldstein, 2016). A significant fraction of these unmeasured organic compounds is constituted by biogenic VOCs: besides isoprene, only a few monoterpenes (e.g. α-pinene, β-pinene, limonene) and even fewer sesquiterpenes (e.g. β-caryophyllene) are routinely measured in the atmosphere (Bouvier-Brown et al., 2009; Hellén et al., 2018). Sesquiterpenes are particularly challenging to measure, due to their low vapour pressure, and therefore their ambient levels are most likely underestimated (Pollmann et al., 2005; Duhl et al., 2008; Kim et al., 2009).
Most BVOCs are reactive with OH and many of them, such as isoprene, monoterpenes, and sesquiterpenes, include double carbon bonds and therefore also react with O3: the existence of a significant pool of unknown and unmeasured BVOCs has important consequences for the quantification of the ozone budget, which are crucial to understanding the environmental and societal impacts of ozone pollution. The oxidation of BVOCs by ozone is especially important because it forms additional pollutants, such as secondary organic aerosol, and impacts key chemical processes, such as the conversion of NO to NO2 and therefore the formation of ozone via Reactions (R1) and (R2), as well as the radical budget (Lewis et al., 2000; Glasius and Goldstein, 2016). Missing a large fraction of BVOCs means that all these processes remain potentially underestimated.
One way to address this problem is to expand the number of VOCs that can be measured. This approach has achieved some success, thanks to comprehensive two-dimensional gas chromatographic techniques (Pankow et al., 2012; Edwards et al., 2013; Glasius and Goldstein, 2016). However, the chemical complexity of the composition of ambient air makes it difficult, if not impossible, to completely quantify the VOC loading of the atmosphere. An alternative approach is to measure an integrated chemical property of all VOCs, such as the chemical lifetime, which includes all the reactions that remove a given species, in this case O3 (Reactions R3–R9). Instruments to directly measure the total ozone reactivity have been demonstrated by Park et al. (2013) and Matsumoto (2011, 2014) and used for laboratory studies of gas-phase ozonolysis reactions (Matsumoto, 2016).
This paper presents an instrument designed to measure total ozone reactivity under ambient, environmental chamber, and branch enclosure conditions. The Total Ozone Reactivity System (TORS) was developed from the instrument described by Matsumoto (2014) with several modifications, as described below. Section 2 describes the theoretical and operating principles of TORS, while Sects. 3 and 4 describe the design and the laboratory characterization of the TORS instrument, respectively. In Sect. 5 the TORS instrument is evaluated with three types of experiments, increasingly approaching ambient conditions: laboratory measurements with known concentrations of selected BVOCs, laboratory measurements with small plants, and quasi-ambient measurements in a horticultural glasshouse.
2.1 Reactivity measurements
The atmospheric lifetime (τ) of a generic species A is defined as the inverse of its total chemical loss rate, i.e. of its chemical reactivity (R):
where [X]i is the concentration of a molecule reacting with A and ki is the corresponding bimolecular rate coefficient. The chemical lifetime of a species has long been considered a useful diagnostic parameter, especially to investigate the loss terms of key atmospheric oxidants, such as the OH radical, which are generally less well known than the respective production terms (Bell et al., 2003).
The comparison between the directly measured lifetime and the lifetime calculated from an independent determination of ki and [X]i (Eq. 1) allows us to understand whether all the loss terms for a given species have been accounted for. This approach has been used with success to investigate the budget of the OH radical (Kovacs and Brune, 2001; Ingham et al., 2009; Sinha et al., 2010; Whalley et al., 2016; Fuchs et al., 2017; Sanchez et al., 2018), of the HO2 radical (Miyazaki et al., 2013), and of the NO3 radical (Sobanski et al., 2016; Liebmann et al., 2018). Measurements of OH reactivity have also helped to discover previously unknown chemistry in terms of recycling and losses of the OH radical (Di Carlo et al., 2004; Lou et al., 2010; Whalley et al., 2011).
The same principle can be used to investigate the chemical loss of ozone. From Reactions (R1)–(R11), the rate of production and loss of tropospheric ozone can be calculated as
where α is the fraction of atomic oxygen that does not reform O3 via reaction with O2 (Reaction R11). Halogens (X = Cl, Br, I) can also react with ozone to form halogen oxides (XO), although this process is likely minor in continental environments far from the main halogen sources (Monks et al., 2015; Simpson et al., 2015). Under the typical unpolluted conditions that can be encountered in a forested environment (NO=50 ppt, NO2=500 ppt, molecule cm−3, molecule cm−3; Griffith et al., 2013), the loss of ozone to NO2, OH, and HO2 is a factor of 50–100 times slower than the loss of ozone to NO (Table 1). Under more polluted conditions, NO2, OH and HO2 reactions are even less important as ozone sinks compared to NO.
Table 1Rate coefficients of the reactions with ozone (, ), chemical lifetimes for [O3]=120 ppb (τ, min), absolute ozone reactivities (, s−1), and relative ozone reactivities (with respect to α-pinene) of selected BVOCs and inorganic compounds. The rate coefficients are from Atkinson et al. (2004, 2006) and calculated for standard conditions (298 K, 1 atm); the ozone reactivities are calculated for mixing ratios of 1 ppb of each species.
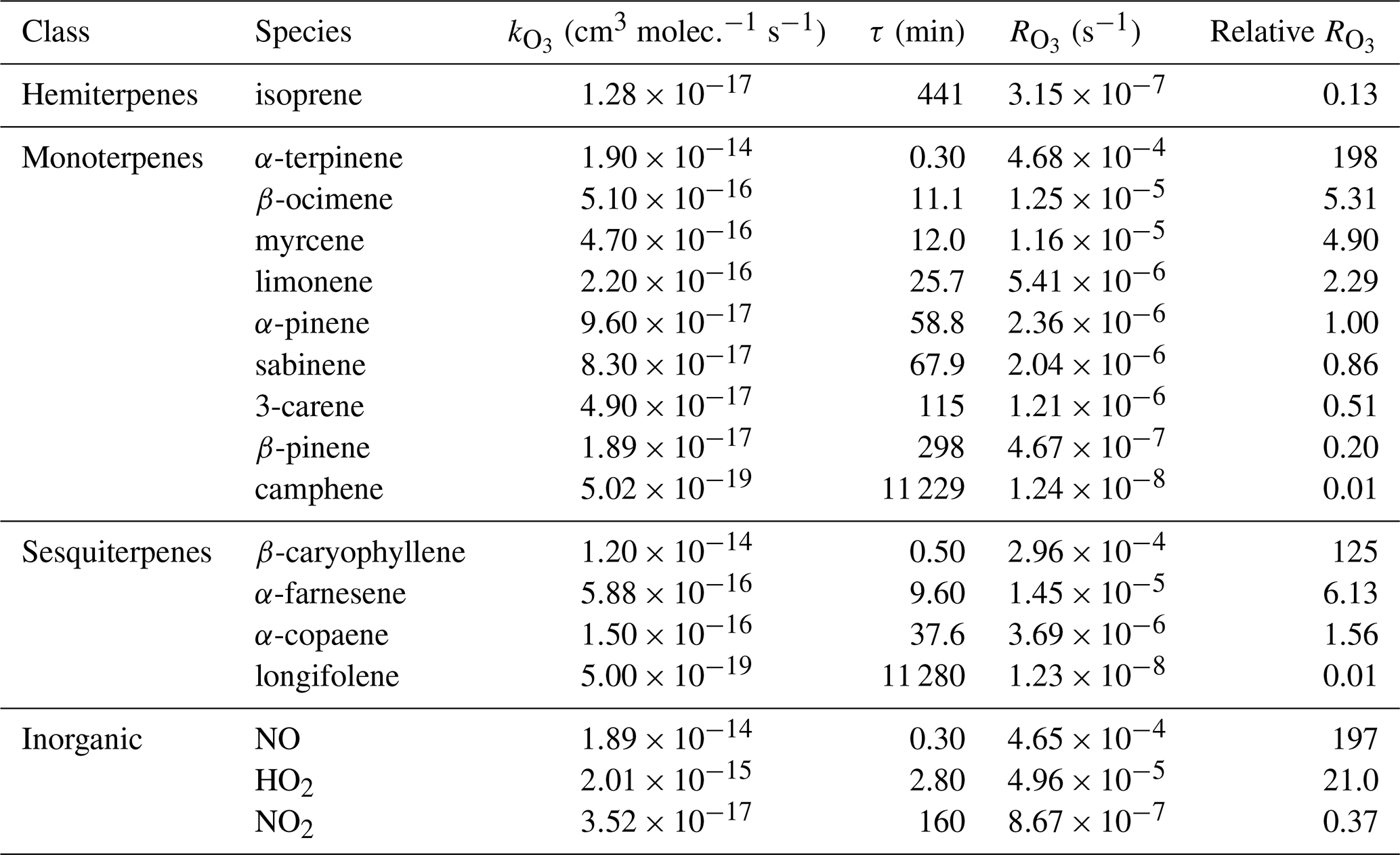
During the night – when photolysis rates are zero, and the concentrations of OH and HO2 are typically 2 orders of magnitude lower than during the day – NO is titrated to NO2 (Reaction R5) soon after sunset, leading to the formation of NO3 radicals (Reaction R6). The rate coefficient of O3+NO2 is small ( ; Table 1) compared to the rate coefficients of most monoterpenes and sesquiterpenes with O3 (since its emissions are light dependent, isoprene is normally not present at night; Guenther et al., 2012): for example, 1 ppb of NO2 has the same O3 reactivity as 0.37 ppb of α-pinene and 0.16 ppb of limonene (at 298 K). This means that NO2 can be a significant ozone loss during the night if its concentration is high – which is not usually the case in unpolluted forested environments. Equation (2) can thus be simplified to
where the NO and photolysis terms are significant only during the day and the NO2 term is significant only during the night (and under relatively high-NOx conditions). The concentrations of O3, NO, and NO2, their rate coefficients with O3 and the photolysis rates of O3 and NO2 can be measured and/or are well known. Therefore, Eq. (3) can be used to evaluate the contribution of the volatile organic compounds (Σiki[VOC]i). As mentioned in Sect. 1, only a handful of the VOCs that react with ozone are routinely measured, which likely leads to underestimation of the VOC contribution to ozone loss. If the loss rate of ozone can be directly measured, it is possible to determine the total VOC loading and compare it with the measured VOCs, thus allowing an estimate of missing (i.e. unmeasured) VOCs.
2.2 TORS concept
The Total Ozone Reactivity System (TORS) is based on the technique developed by Matsumoto (2014). At its core, the system is a dark flow tube reactor in which a known amount of ozone is reacted with a sample (e.g. NO, unsaturated VOCs). If the change in the concentration of the co-reactants in the sample is small, the reaction follows pseudo-first-order kinetics and the change in ozone concentration is described by
where [O3]0 and [O3]t are, respectively, the initial and final ozone concentrations, k′ is the pseudo-first-order rate coefficient and t is the reaction time, which corresponds to the reactor residence time and can be determined experimentally (Sect. 4.1). Equation (4) can be solved analytically provided the reaction time and the initial and final O3 concentrations are known:
TORS provides a direct measurement of k′, which includes the chemical reactions inside the reactor (described by Eq. 3) plus other O3 removal processes, such as the loss of ozone on the reactor wall. Since the reactor is completely dark, the photolysis terms in Eq. (3) can be neglected and the only contributors to the O3 chemical loss are NO and VOCs. The focus of this study is on the reactivity of VOCs (Sect. 1), and therefore the loss of O3 due to NO and to the reactor wall needs to be subtracted. For the purpose of this work, we define ozone reactivity () as
where RNO is the removal of O3 by reaction with NO and Rwall is the loss of O3 on the reactor wall. This definition of is directly comparable to the definition by Matsumoto (2014, 2016), since those experiments were conducted in zero air (i.e. at RNO=0). The ozone loss on the reactor wall (Rwall) is a potentially significant parameter in the TORS technique and requires accurate and precise determination (Sect. 4.2). Another potentially important factor is the effect of secondary reactions, which can increase the loss of ozone (Reactions R5–R8) causing overestimation of or can decrease the concentration of VOCs in the sample via reactions other than ozonolysis (e.g. if there is significant formation of NO3 in the reactor via Reaction R6), causing underestimation of .
Particularly important can be the formation of OH radicals from VOC ozonolysis (Paulson et al., 1999; Rickard et al., 1999; Johnson and Marston, 2008): to remove this interference, an OH scrubber, such as cyclohexane, can be added to the system. Cyclohexane does not react with O3, but reacts quickly with OH ( ) and forms organic peroxy radicals which combine with HO2 to form products that do not react with O3 (Alam et al., 2011). Therefore, cyclohexane acts as an efficient OH scrubber when present at high mixing ratios (parts per million level). The effects of the ozone wall loss and of the secondary chemistry of ozonolysis products were investigated with a box-model simulation of the TORS chemistry (Sect. 2.3) and with laboratory characterization experiments (Sect. 4.2).
2.3 Simulation of TORS chemistry
A box model was used to simulate the chemical reactions occurring in the TORS reactor. The main objective of the model was to assess the effect of the OH radicals generated by the ozonolysis of VOCs and the role of the OH scrubber, as well as the impact of potential interfering chemistry such as removal of ozone by OH, HO2, and NO2 (Reactions R6–R8) and removal of VOCs by OH and NO3 radicals.
The model was assembled using the AtChem2 modelling toolkit (Sommariva et al., 2020) with the inorganic chemistry and the oxidation mechanisms of α-pinene and cyclohexane taken from the Master Chemical Mechanism (MCM v3.3.1; Saunders et al., 2003). The cyclohexane mechanism was updated to include a more accurate representation of the ring-opening path of the cyclohexoxy radical, following Alam et al. (2011), although the model results were not substantially different from those of a model using the MCM standard cyclohexoxy radical scheme (≤0.1 %). It must be noted that the model results depend on the VOC used, as the OH yield from ozone + alkene reactions can vary from 0.16 for ethene to 0.90 for 2,3-dimethyl-2-butene (Johnson and Marston, 2008). The choice of α-pinene for the model, as well as for the laboratory experiments (Sect. 5.1), is due to the fact that α-pinene is one of the most abundant BVOCs (Guenther et al., 2012). It also has a high OH yield (0.8; Johnson and Marston, 2008), allowing an estimate of the upper bounds of the potential interferences caused by OH chemistry.
The model was initialized with a range of α-pinene mixing ratios (0.1–50 ppb) and, for each, with a range of cyclohexane mixing ratios (0–20 ppm). The initial mixing ratios of NO and NO2 were 50 and 500 ppt, respectively, representative of an unpolluted forested environment (Griffith et al., 2013). The initial O3 mixing ratio was set to 120 ppb, as used during the experimental work (Sect. 4.3). The model runtime was 300 s, covering the range of potential instrument residence times (Sect. 4.1). A summary of the model results is shown in Fig. 1.

Figure 1(a, b) Modelled removal of α-pinene and O3 (relative to their initial concentrations) as a function of cyclohexane level. (c) Average modelled concentrations of OH as a function of cyclohexane level. (d) Sensitivity of modelled to cyclohexane level as a function of α-pinene mixing ratio.
The removal of α-pinene during the residence time in the reactor was ∼7 % and 4 %–5 % for initial α-pinene mixing ratios of 0.1 ppb and ≥0.5 ppb, respectively (Fig. 1a). In the absence of cyclohexane, the removal of α-pinene was slightly higher (1–2 percentage points) due to reaction with OH radicals. These numbers indicate that the consumption of α-pinene inside the reactor was over an order of magnitude smaller than its initial concentration, and, therefore, the chemical system approached the pseudo-first-order conditions required for the TORS method (Sect. 2.2). The error in the determination of ozone reactivity caused by the assumption of pseudo-first-order conditions can be estimated at <4 % for α-pinene mixing ratios >10 ppb. As expected (Alam et al., 2011), the reactions with oxidation products of cyclohexane were not significant sinks for ozone: apart from the wall loss, which is not included in the model, the loss of O3 inside the reactor was less than 1.5 % for α-pinene mixing ratios up to 50 ppb and was independent of the concentration of cyclohexane (Fig. 1b).
The average modelled concentrations of the OH radical at different levels of cyclohexane are shown in Fig. 1c. In the absence of cyclohexane, the model calculated OH concentrations between 1.3×106 and 4×106 molecule cm−3 for α-pinene mixing ratios of 0.1 and 50 ppb, respectively. With addition of the OH scrubber, the simulated concentration of OH in the reactor decreased by 2 orders of magnitude at mixing ratios of cyclohexane between 1 ppm (for α-pinene <5 ppb) and 10 ppm (for α-pinene =50 ppb). Increasing the level of cyclohexane above 10 ppm did not cause further significant decrease in the calculated concentration of OH or a reduction in the loss of ozone and α-pinene (Fig. 1a–c), at least within the range of α-pinene concentrations explored by the model. Figure 1d shows that the ozone reactivities determined with 1 and 20 ppm of cyclohexane were essentially the same for α-pinene initial mixing ratios up to 50 ppb. Moreover, the model results show that the differences between the ozone reactivity calculated with and without cyclohexane were between +1 % and −6 %, depending on the α-pinene level (Fig. 1d). This demonstrates that OH chemistry has a small overall impact on the determination of total ozone reactivity, a conclusion that is supported by the laboratory experiments (Sect. 5.1).
Model-calculated HO2 concentrations were less than 1×108 molecule cm−3, meaning that ozone reactivity with HO2 was 2 orders of magnitude lower than ozone reactivity with α-pinene (at 10 ppb of α-pinene). Only at very low concentrations of α-pinene (<0.1 ppb) was HO2 a significant sink for ozone. The presence of NO3 radicals in the reactor is a potentially important interference for the TORS technique, both because its formation consumes O3 (Reaction R6) and because NO3 reacts quickly with α-pinene ( ). Ambient NO3 is likely lost in the inlet before the reactor, since the transmission of NO3 through the inlet – a 6 mm diameter, 5 m long Teflon tube with a residence time of ∼4 s – is poor (Dubé et al., 2006). However, NO3 can be formed inside the reactor via Reaction (R6) and the model calculated NO3 formation of the order of 106 molecule cm−3 for α-pinene mixing ratios >5 ppb. Although the rate coefficient of α-pinene +NO3 is 5 orders of magnitude larger than the rate coefficient of α-pinene +O3, the ozone concentration in the reactor is 6–7 orders of magnitude higher than the concentration of NO3. Therefore, the reactivity of α-pinene with O3 was 1–2 orders of magnitude larger than its reactivity with NO3 (Table 1). It must also be noted that NO3 formation in the reactor is only an issue for ambient measurements under moderate- or high-NOx conditions, not for laboratory, enclosure, and environmental chamber experiments under low- or zero-NOx conditions.
To summarize, the model of the TORS reactor suggests that, under the typical operating conditions described in Sect. 4.3, the concentrations of HO2 and NO3 are too small to compete with BVOCs for reaction with O3. Additionally, the model provides no indication that the products of the oxidation of cyclohexane, when used as OH scrubber, can significantly affect the determination of total ozone reactivity. While parts-per-million levels of cyclohexane effectively eliminate the OH radicals formed by BVOC ozonolysis reactions, the model suggests that ozone reactivities determined with and without an OH scrubber differ by <6 %. The model results are in agreement with the discussion in Sect. 2.1, where it was concluded that the decay of O3 in the TORS reactor is predominantly due to the reactions with NO and VOCs, alongside loss on the reactor wall (Eq. 6). It is important to note, however, that the conclusions drawn from the model simulations may vary depending on the chemical conditions in the reactor, as several factors affect the chemistry inside the TORS reactors: the type and mixture of VOCs in the sample, their OH yields, the ambient concentrations of NO and NO2, and, to a lesser extent, ambient temperature and pressure (which influence the rate coefficients of chemical reactions). To date, the effect of temperature and pressure is negligible, as the system has been operated under near-ambient conditions.
3.1 Description of TORS
The operating principles of TORS are described in Sect. 2.1 and 2.2, and a diagram of the TORS instrument is shown in Fig. 2. The reactor is a 1 m long polytetrafluoroethylene (PTFE) tube with an external diameter of 90 mm and an internal diameter of 87.33 mm. Several different materials and geometries for the reactor were tested during the instrument development phase. This design was found to allow a residence time inside the reactor sufficient for the ozonolysis reactions to take place to a suitably measurable extent, while minimizing the consumption of VOCs – in order to maintain pseudo-first-order conditions – and the loss of ozone on the reactor wall (Sect. 4.1).
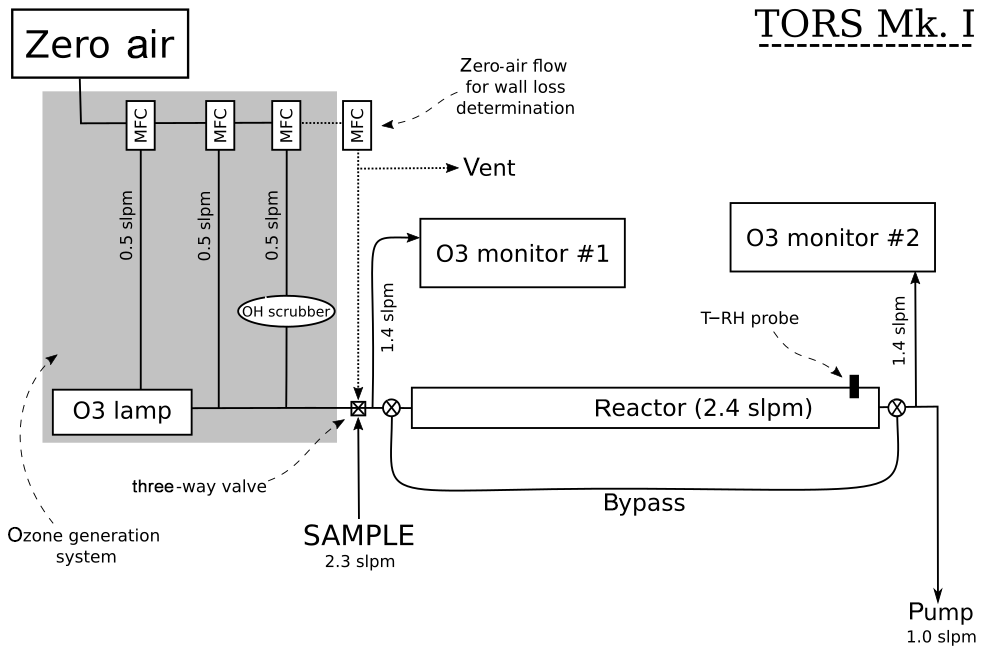
An ozone flow is generated by irradiating a flow of zero air with a UV mercury lamp (UVP Ltd., UK); a zero-air flow is added downstream of the mercury lamp to control the concentration of ozone (Fig. 2). The ozone flow is mixed with the sample flow just before the reactor, and the initial ozone concentration ([O3]0) is measured at this point, while the final ozone concentration ([O3]t) is measured at the exit point of the reactor (Fig. 2). Depending on the instrument settings, this setup produces an O3 mixing ratio in the reactor of 100–140 ppb. Ozone concentrations are measured using two identical UV photometric O3 monitors (model 49i, Thermo Fisher Scientific, USA). The model 49i O3 monitor has a stated detection limit of 1 ppb and a precision of 0.25 ppb at 1 min averaging time. The reactor can be bypassed using two three-way Teflon valves, so that the two ozone monitors can simultaneously measure the O3 concentration before it enters the reactor, thus allowing the ozone measurements to be corrected for any difference between the two monitors (Sect. 4.3). In addition, a temperature (T) and relative humidity (RH) probe (HMP110, Vaisala Oyj, Finland) is inserted in the reactor to monitor temperature and relative humidity. All the flows in the TORS instrument are controlled with mass flow controllers (Brooks Instrument LLC, USA) using a custom-built control box (IGI Systems Ltd, UK). The signals from the ozone monitors, the T–RH probe, and the mass flow controllers are logged on a laptop and processed with bespoke software in the R programming language.
A potentially important factor for TORS is the stability of the ozone source: highly variable levels of O3 in the ozone flow (Fig. 2) can affect the determination of the ozone reactivity and increase the signal-to-noise ratio of the instrument. The ozone mixing ratio generated by the mercury lamp was found to vary, on average, by 0.4–0.6 ppb (5 min, 2σ), i.e. less than 1 %. The ozone reactivity measurements reported in Sect. 5 were all averaged to 5 min.
3.2 Supporting VOC measurements
A proton-transfer-reaction quadrupole-interface time-of-flight mass spectrometer (PTR-QiTOF-MS, Ionicon Analytik GmbH, Austria) was used to measure VOC concentrations during the laboratory experiments (Sect. 5.1 and 5.2). The instrument (Sulzer et al., 2014) was operated according to the standard operating conditions recommended by the manufacturer (drift pressure =3.8 mbar, drift tube temperature =80 ∘C and Td), using H3O+ as the reagent ion. Calibration was performed using a TO-14A aromatics standard mixture (Airgas Inc., USA). This mixture does not contain biogenic compounds, so a mass transmission curve calculated using the calibration gas was used for quantification. Recent work by Holzinger et al. (2019) showed that a PTR-MS operated under standard conditions is able to accurately measure concentration of uncalibrated compounds (to within 30 %) using a mass transmission curve, if these compounds have high proton affinity and do not undergo unknown fragmentation. This assumption is likely valid for BVOCs, such as isoprene and monoterpenes (Holzinger et al., 2019), which were the primary focus of this work. The limits of detection determined using zero air for calibrated compounds were on the order of 20–80 ppt.
Measurements of BVOCs can be problematic for monoterpenes and sesquiterpenes, as the PTR technique cannot distinguish between isomers (de Gouw and Warneke, 2007). Typically, when measured by PTR-MS the main fragment ions for monoterpenes and sesquiterpenes are at m∕z 81.07 and 149.1, respectively, and this fragmentation is independent of the structure of the isomers (Tani et al., 2003; Kim et al., 2009). Therefore total monoterpene and sesquiterpene concentrations were estimated using the abundances of the protonated parent ions (m∕z 137 and 205, respectively) and of the main fragment ions (m∕z 81 and 149, respectively). Other compounds associated with biogenic emissions (e.g. substituted monoterpene alcohols, after loss of a neutral H2O) can also be detected at these fragment ions (Kim et al., 2010) and thus the estimated concentrations shown here may be considered upper limits.
The PTR-MS instrument was not available for the experiments outside the laboratory (Sect. 5.3). Instead, samples were taken with adsorption tubes, which were desorbed using a TD UNITY 2 thermal desorption unit (Markes Int., UK) and subsequently analysed with a gas chromatograph (GC 7890B, Agilent Technologies, USA) interfaced with a BenchTOF-Select time-of-flight mass spectrometer with tandem ionization (Markes Int., UK). Further information on the analytical technique and the GC protocol can be found in Alam et al. (2016). The gas chromatography–mass spectrometry (GC–MS) analysis was only qualitative due to unavailability of appropriate calibration standards, and the data were used to identify the VOCs in the samples.
4.1 Residence time
The residence time is one of the key parameters in Eq. (5) and therefore needs to be determined as accurately as possible. The geometry of the reactor and the total flow (sample + ozone) affect the residence time. Three methods were used to determine the residence time: (1) direct measurement using a double injection of acetone, (2) indirect measurement via determination of NO reactivity with O3, and (3) calculation using the flow rate and the internal volume of reactor (5990 cm3).
Method 1 is illustrated in Fig. 3: the reactor was connected to the PTR-MS and aliquots of 0.05 µL of acetone, in a flow of zero air, were injected simultaneously at the entrance and at the exit of the reactor. Acetone was used as a tracer for these experiments because it is easily ionized by H3O+ and does not undergo fragmentation, and consequently it is straightforward to detect by PTR-MS at its protonated mass (m∕z 59). The simplest way to determine the residence time is to measure the time lag between the detection of the first and the second acetone peak signals by the PTR-MS (“maxima” calculations in Fig. 3). However, the flow dynamics inside the reactor are complex and, as a consequence, there is no single value of residence time, but a distribution (Cazorla and Brune, 2010; Huang, 2016). Hence, the acetone signal from each injection can also be used to determine the mean residence time in the reactor (“means” calculations in Fig. 3): the residence times estimated using the means calculations are about 40 % larger than those estimated using the maxima calculations.
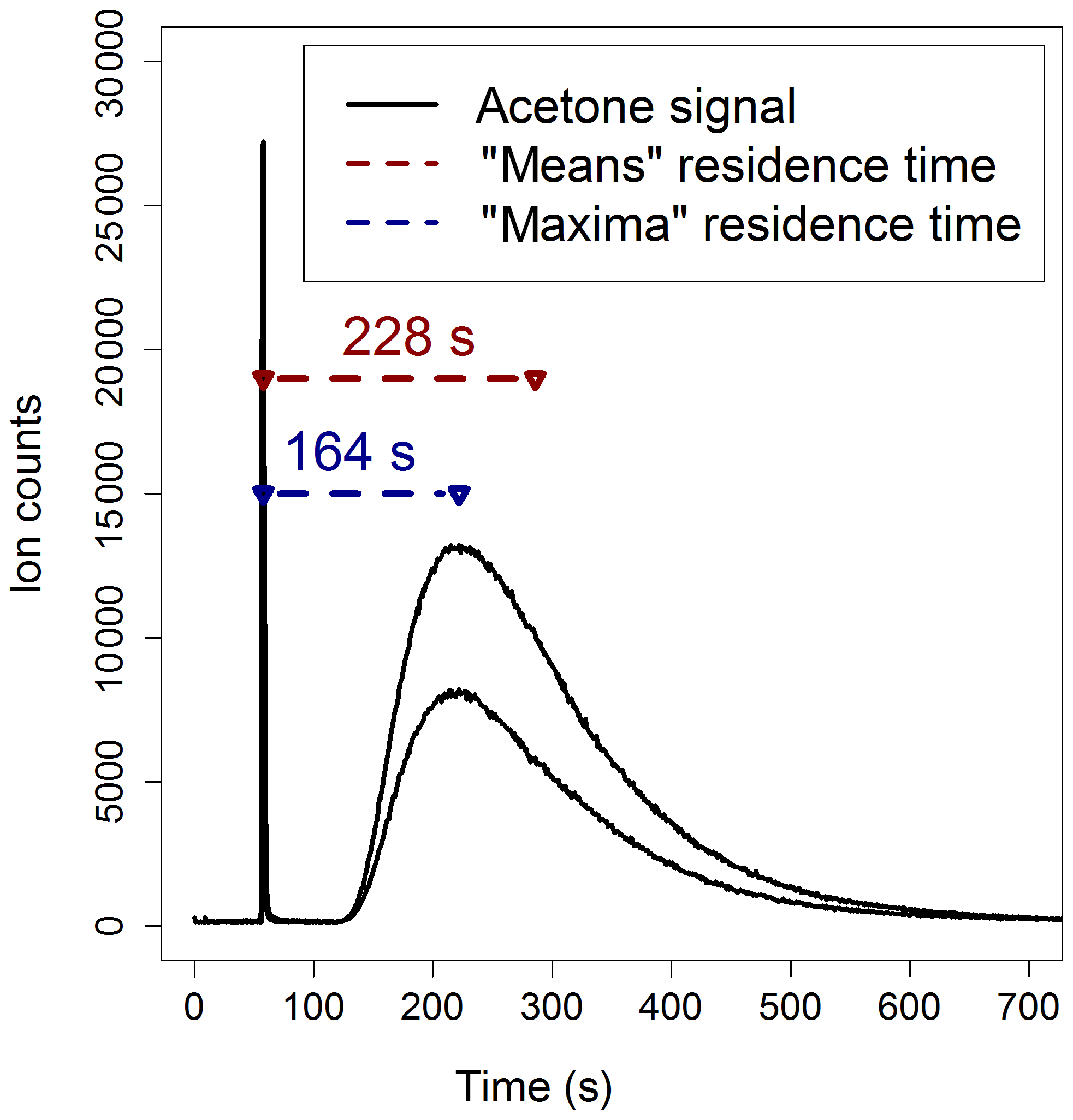
Figure 3Two acetone double-injection experiments used to estimate the residence time in the PTFE reactor for a flow rate of 1600 sccm. The “maxima” calculation uses the differences in time of the peak acetone signals; the “means” calculation uses the differences between the mean elapsed time of the two acetone signals.
Method 2 uses the TORS technique to measure the reactivity of O3 (∼20 ppb) with NO: the sample flow contained only ∼100 ppb of NO (diluted from a certified gas cylinder, 4.90±0.25 ppm in N2, by BOC UK) and, since the rate coefficient of NO+O3 is known ( at 298 K, with an uncertainty of 17 %; Atkinson et al., 2004), the only unknown variable in Eq. (5) was the reaction time t. Since under these conditions the chemical system deviates from pseudo-first-order conditions, the mean concentration of NO inside the reactor was used to analyse the experimental results. The averages of several experiments conducted at different flow rates are shown in Fig. 4a. Method 3 assumes perfect instant mixing and plug flow in the reactor: the calculation for a range of flow rates is shown in Fig. 4b.
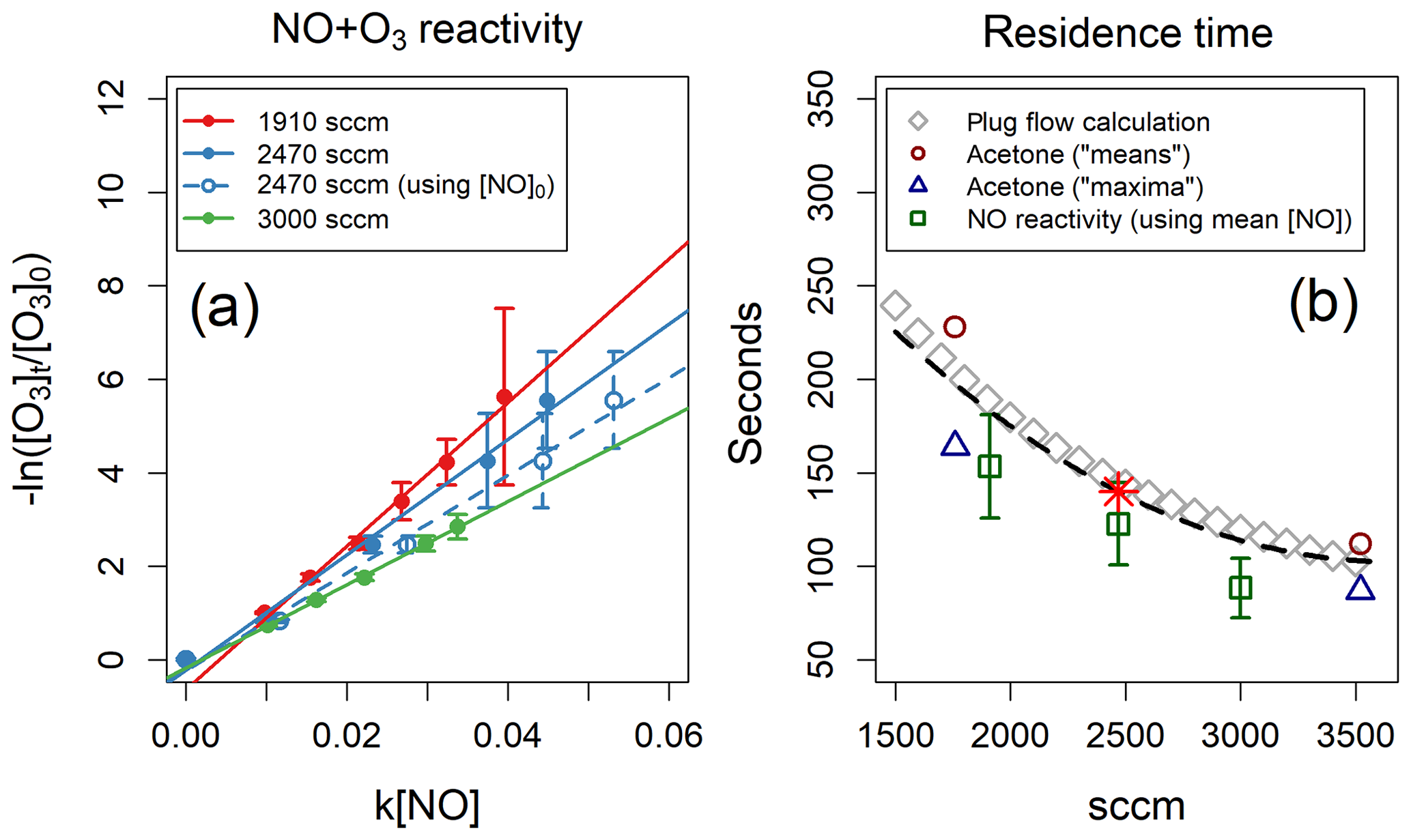
Figure 4(a) NO reactivity experiments analysed using the mean NO concentration inside the reactor. One experiment analysed using the initial NO concentration is also shown, for reference. (b) Residence times as a function of reactor flow – determined by three different methods – and polynomial fit to the three methods (black dashed line). The results of the acetone-injection method are taken from Fig. 3. The red star indicates the residence time used in this work (140 s for a reactor flow of 2470 sccm).
All three methods offer internally consistent results, within the respective uncertainties (Fig. 4b). Method 3 agrees well with the acetone-injection means calculation but overestimates the residence times determined with method 2 by ∼18 %. Method 2 relies on well-known kinetic parameters and implicitly takes into account the real distribution of flow paths through the reactor; it also provides a simple test of the TORS functionality using NO instead of a BVOC. In the experiments described in Sect. 5, we used a value of 140 s for the residence time, determined by fitting a second-degree polynomial to all three methods, as shown in Fig. 4b for a reactor flow of 2470 sccm (standard cubic centimetres per minute).
4.2 Ozone wall loss
One of the most important parameters and key uncertainties of the TORS technique is the ozone wall loss (Rwall), as discussed in Sect. 2.2. Other species can be lost on the reactor surface, but these are likely to have little or no impact on the determination of the ozone reactivity either because they do not react with O3 (e.g. multifunctional products of VOC oxidation) or because they are present at very low concentrations.
Measured is obtained by subtracting the loss of O3 on the reactor wall (Rwall) and, if present, to NO (RNO) from the total loss rate of O3 (Eq. 6). Small changes in the O3 wall loss could lead to significant variation in the final determination of ozone reactivity. It is therefore important to minimize this parameter in order to reduce the uncertainty of the measurement. In the design stage, several materials and sizes were tested under dry and humid conditions: (1) a glass cylinder (5×70 cm), (2) a quartz cylinder (9×100 cm), (3) a PFA coil (1.9×1524 cm), (4) a PTFE cylinder (9×100 cm). Based on these experiments, the large-diameter (o.d. =9 cm, i.d. =8.73 cm) PTFE cylinder showed the lowest ozone wall loss and minimal dependence on humidity.
The ozone wall loss was regularly determined during measurements by switching to a flow of zero air instead of the regular sample with a three-way Teflon valve (Fig. 2): in this operating mode there are no reactants (NO or VOCs) in the reactor and the measured ozone reactivity is equal to Rwall (Eq. 6). During each experiment and measurement period, multiple determinations of the ozone wall loss were made. Figure 5 shows the average Rwall determined during a series of laboratory experiments (Sect. 5.1 and 5.2), as a function of measured humidity, temperature, and time. Rwall showed a weak dependence on the relative humidity inside the reactor (, with R2=0.198). Ambient humidity is usually higher than the range shown in Fig. 5, but because of the dilution of the sample flow by the dry ozone flow before entering the reactor (Fig. 2), the relative humidity in the reactor is always less than 50 %. There was no clear dependence of Rwall on temperature, at least in the limited range experienced in the laboratory (14–24 ∘C). The ozone wall loss can be expected to vary with time, as the surface of the reactor is passivated and exposed to ambient air, but there was no obvious temporal trend over the 9-month period of these experiments (Fig. 5).
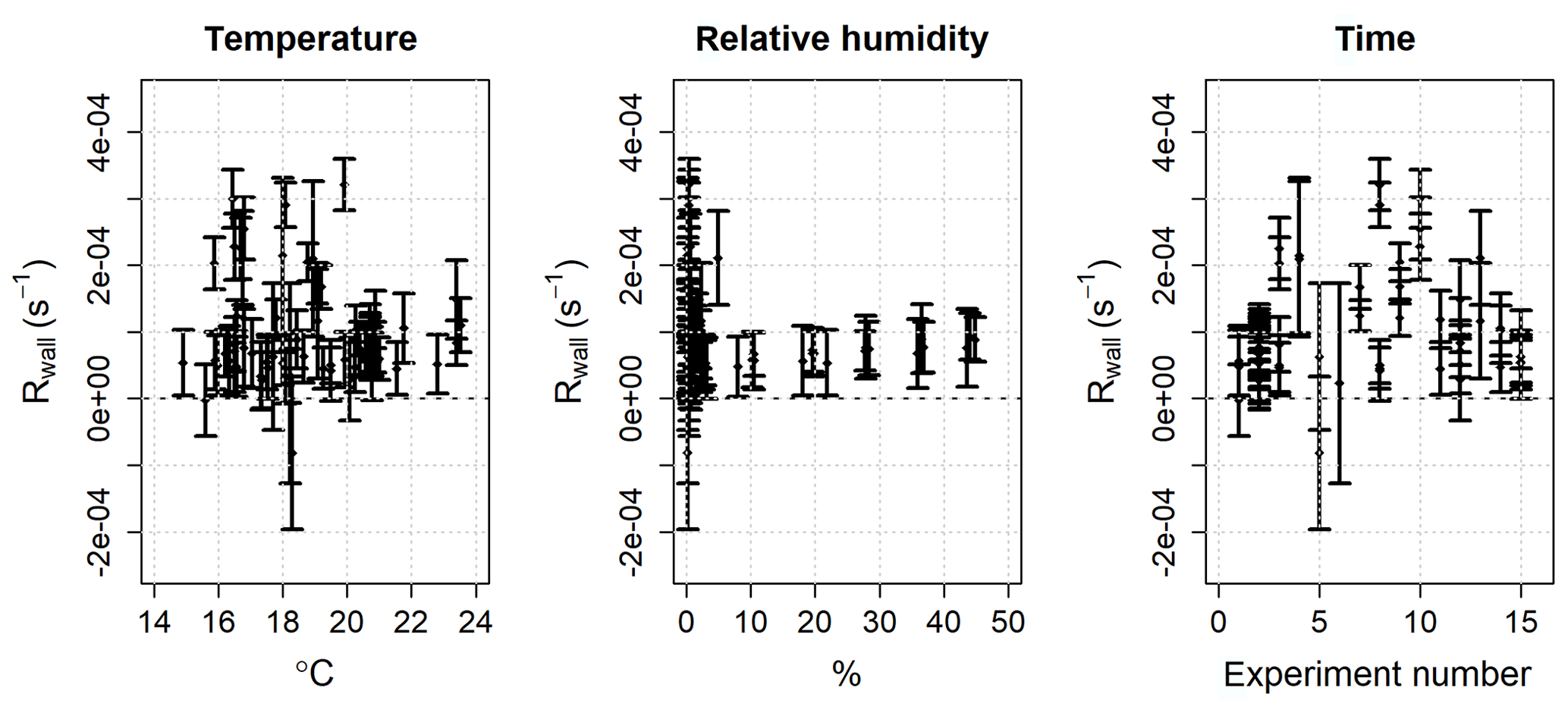
Figure 5Mean and 2σ standard deviation of the ozone wall loss (Rwall) in the PTFE reactor as a function of humidity, temperature, and time. The data are taken from 15 experiments, with multiple determinations of Rwall per experiment, over a 9-month period.
Although there was no clear pattern with respect to the measured parameters, it is apparent that there was significant variability in the ozone wall loss and, therefore, it is necessary to measure Rwall often during an experiment and/or ambient measurements. The average standard deviation of Rwall was s−1 and the interquartile range was 0.5– s−1 (mean s−1, median s−1), which corresponds to the reactivity of 21–51 ppb of α-pinene (mean =41.5 ppb, median =30 ppb). The range of the measured ozone wall losses suggests that the limit of detection of TORS is of the order of a few tens of parts per billion of α-pinene or the equivalent concentration, in terms of reactivity, of other BVOCs (Table 1); the limit of detection was quantified with laboratory experiments using known concentrations of α-pinene, as described in Sect. 5.1.
4.3 TORS operation
The flows in the TORS instrument are constrained by several competing factors (Fig. 2): first, the total flow (sample + ozone) must be larger than the inlet flows of both O3 monitors, which are fixed at ∼1.4 slpm (standard litres per minute) each. Second, the residence time in the reactor must be long enough to allow the ozonolysis reactions to take place to a measurable extent, but short enough that the consumption of the reactants does not become significant, so that pseudo-first-order conditions are maintained (Sect. 2.2).
During the design phase of the TORS instrument several combinations of flow settings were experimented with, eventually settling on a sample flow of ∼2.3 slpm and an ozone flow – composed of a flow of zero air through the ozone lamp, plus a dilution flow to control the concentration of ozone, and a flow of cyclohexane as OH scrubber – of ∼1.5 slpm (Fig. 2). These settings result in a reactor flow rate of 2470 sccm, corresponding to a residence time of 140 s (Sect. 4.1). Since the sample flow is mixed with the ozone flow (Fig. 2), a correction factor needs to be applied to account for the dilution of the sample by the ozone flow. In the experiments and measurements described in Sect. 5, the mixing ratio of ozone at the entrance of the reactor was ∼120 ppb. The instrument settings, such as the residence time and the ozone concentration, can in principle be varied to adjust the sensitivity of TORS for environments with different values of .
Once the flows are set, the basic operation cycle of the TORS instrument consists of three main steps.
- 1.
“Bypass mode” to check the agreement between the two O3 monitors. The sample flow is substituted with an equal flow of zero air and the reactor is bypassed for a period of approximately 15 min. In this mode, both monitors measure the O3 concentration in zero air (i.e. with no reactants) before the entrance of the reactor, so that a correction factor can be derived if they differ. In this work, the difference between the O3 monitors was checked every day at the start and at the end of each experiment/measurement period and was typically ∼1 ppb.
- 2.
“Wall loss mode” to determine Rwall (Eq. 6). The sample flow is substituted with an equal flow of zero air inside the reactor for a period of approximately half an hour. The effect of the humidity change in the reactor within such a short period of time was found to be negligible. In this work, the wall loss was determined every 2–3 h.
- 3.
“Sampling mode”. The main operation mode of the instrument has a sample flow containing BVOCs and/or NO.
The rate coefficients required by Eq. (6) to retrieve the ozone reactivity are calculated using the actual temperature measured inside the reactor (Fig. 2) and ambient pressure. This procedure was followed during all the experiments and measurements described in Sect. 5.
The TORS instrument was tested in a series of experiments to evaluate its functionality and potential. The experiments were designed and conducted in order of increasing complexity from individual species in laboratory conditions to the complex BVOC mixture of a horticultural glasshouse. The TORS instrument was at first tested in the laboratory using pure α-pinene (Sect. 5.1), followed by emissions from small plants (Sect. 5.2). After these tests showed that the instrument was behaving as expected under controlled conditions, it was deployed in a glasshouse containing various aromatic plants to demonstrate that TORS can measure total ozone reactivity under quasi-ambient conditions (Sect. 5.3).
It must be noted that the rate coefficients of O3 with BVOCs span several orders of magnitude (Table 1). Therefore, for individual species in isolation, a measured ozone reactivity of (for example) s−1 corresponds equally to 1.9 ppm of camphene, 10 ppb of α-pinene, 75 ppb of isoprene, and 80 ppt of β-caryophyllene. This affects the interpretation of measurements by TORS, particularly if the composition of the sample is not known, as would be the case when taking ambient measurements.
5.1 Laboratory experiments
Several laboratory experiments were carried out using known concentrations of a selected biogenic compound. A thermostated diffusion tube and pure α-pinene (98 %, from Sigma-Aldrich) in zero air were used to provide a constant source of BVOC. The diffusion rate was controlled by varying the temperature of the diffusion tube and determined by regularly weighing it with a precision balance over a period of several weeks. The concentration of α-pinene was then calculated from the diffusion rate and confirmed via direct measurements by PTR-MS (Sect. 3.2), with an agreement of ∼14 %.
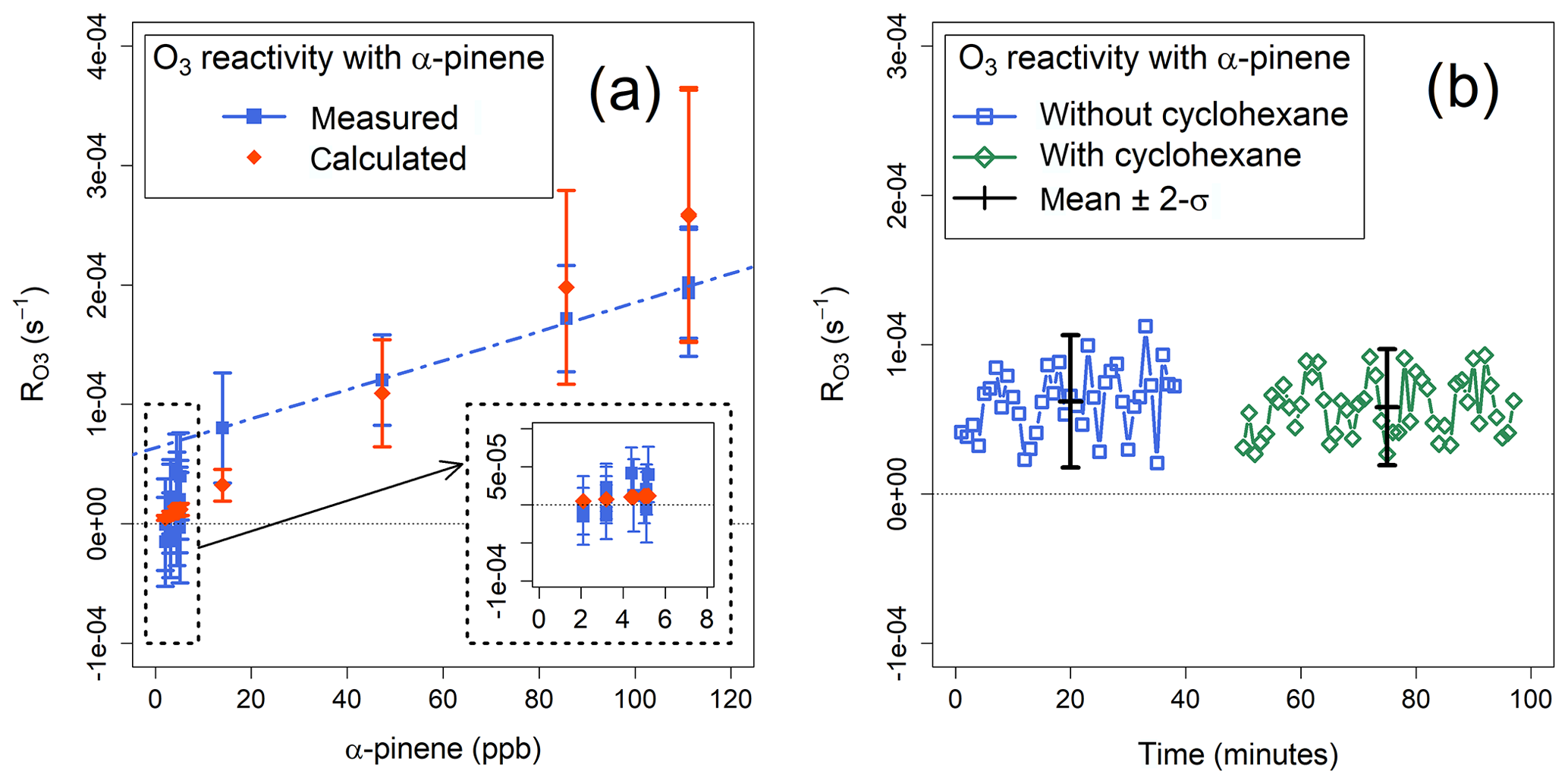
Figure 6(a) Measured and calculated mean ozone reactivities with α-pinene. The blue dashed line indicates the linear regression of the measured values for α-pinene mixing ratios >14 ppb. (b) Ozone reactivity with α-pinene, measured with and without cyclohexane as OH scrubber.
The values of measured during several experiments were compared with the values calculated using the known concentrations of α-pinene in the sample (Eq. 6). The agreement between the calculated and measured ozone reactivity for α-pinene mixing ratios larger than 40 ppb was about 25 % – within the combined uncertainties of the instrument and of the α-pinene +O3 rate coefficient (41 %; Atkinson et al., 2004) (Fig. 6a). At mixing ratios below 10 ppb of α-pinene, the measured reactivities cannot be statistically distinguished from each other and from zero; in fact, the corresponding reactivity ( s−1) is of the same magnitude as the average standard deviation of Rwall (Sect. 4.2). Measured corresponding to concentrations of α-pinene >14 ppb are linearly correlated (r2=0.993) with a slope of , corresponding to the sensitivity of the instrument, and an intercept of s−1 (Fig. 6a).
Based on these experiments, the TORS detection limit, for a residence time of 140 s, can be estimated between and s−1, corresponding to ozone reactivities equivalent to 20–40 ppb of α-pinene (Table 1). These values are consistent with the estimates based on the range of measured Rwall, as discussed in Sect. 4.2; the actual detection limit for a given set of measurements depends on the magnitude of the ozone wall loss, which can vary significantly (Fig. 5). These values are also comparable to the detection limit of the instrument described by Matsumoto (2014) of s−1 for a residence time of 57 s. From Eqs. (5) and (6), the total uncertainty of TORS can be estimated by propagating the uncertainties in the determination of the residence time (related to the uncertainty in the concentration of the NO cylinder and in the rate coefficient of the NO+O3 reaction, Sect. 4.1), the uncertainty of the ozone monitors (Sect. 3.1), and the median variability of Rwall during individual experiments (Sect. 4.2) at ∼32 %.
Some experiments were conducted without adding cyclohexane to the ozone flow (Fig. 2) to verify the effect of the OH scrubber, as discussed in Sect. 2.3. Ozonolysis of BVOCs is known to generate OH radicals with different yields (Rickard et al., 1999; Johnson and Marston, 2008), which may lead to consumption of BVOCs by OH in the reactor, thus causing underestimation of . However, the experiments where the OH scrubber was used did not show substantially different results from those where it was not used (Fig. 6b). Measured with ∼30 ppb of α-pinene was s−1 with cyclohexane and s−1 without cyclohexane: the corresponding p value was 0.394, indicating that the difference between the two measurements is not statistically significant. The difference between the ozone reactivities determined with and without OH scrubber was 6 %–7 %, in agreement with the modelling results (Sect. 2.3). While this is less than the precision of the ozone monitors (Sect. 3.1), we note that, in principle, the comparison of total ozone reactivity measurements with and without an OH scrubber can yield additional information on the speciation of the VOC mixture in the sample.
5.2 Plant experiments
Laboratory experiments were carried out using small aromatic plants in a controlled environment to test the TORS instrument under more realistic conditions. Three plants of lemon thyme (Thymus citriodorus) were enclosed in a Teflon bag (Adtech Polymer Engineering Ltd, UK) with an approximate volume of 0.1 m3, filled with a continuous flow of zero air. A halogen lamp (ROK 120 W) was located over the bag and a temperature–humidity probe (Vaisala HMP110) was inserted into the bag, together with a small fan (RS Components) to ensure homogeneous conditions. The natural release of water vapour via transpiration and evaporation by the plants caused the humidity in the bag to rise over the course of the experiment, but relative humidity remained below 50 % inside the TORS reactor due to dilution with the dry ozone flow (Fig. 2) and therefore it did not affect the loss of O3 on the reactor wall, as discussed in Sect. 4.2. The PTR-MS was connected to the bag to identify and quantify the BVOCs that constitute the plant emissions (Sect. 3.2). The TORS instrument and the PTR-MS sampled continuously from the Teflon bag during the experiment, which had a duration of about 48 h.
Figure 7 shows the ozone reactivity measurements of one lemon thyme experiment, together with the reactivity calculated using the BVOC measurements by PTR-MS. The interquartile range of measured was 3– s−1 for the first experiment and 6– s−1 for the second experiment, with mean values of and s−1, respectively. Measured ozone reactivity increased by about a factor of 2 when the lamp was turned on, due to increased emissions of all BVOCs and, in particular, of the more reactive ones (i.e. monoterpenes and sesquiterpenes, Table 1). This is because when the lamp was switched on, the temperature inside the bag increased (by ∼10 ∘C, Fig. 7), as well as the light. Isoprene emissions are controlled by both light and temperature, but monoterpenes and sesquiterpenes emissions are mostly controlled by temperature and have an exponential response to temperature (Duhl et al., 2008; Guenther et al., 2012; Hellén et al., 2018). Therefore, the emissions of these more reactive compounds tend to increase faster than those of isoprene when temperature rises quickly.
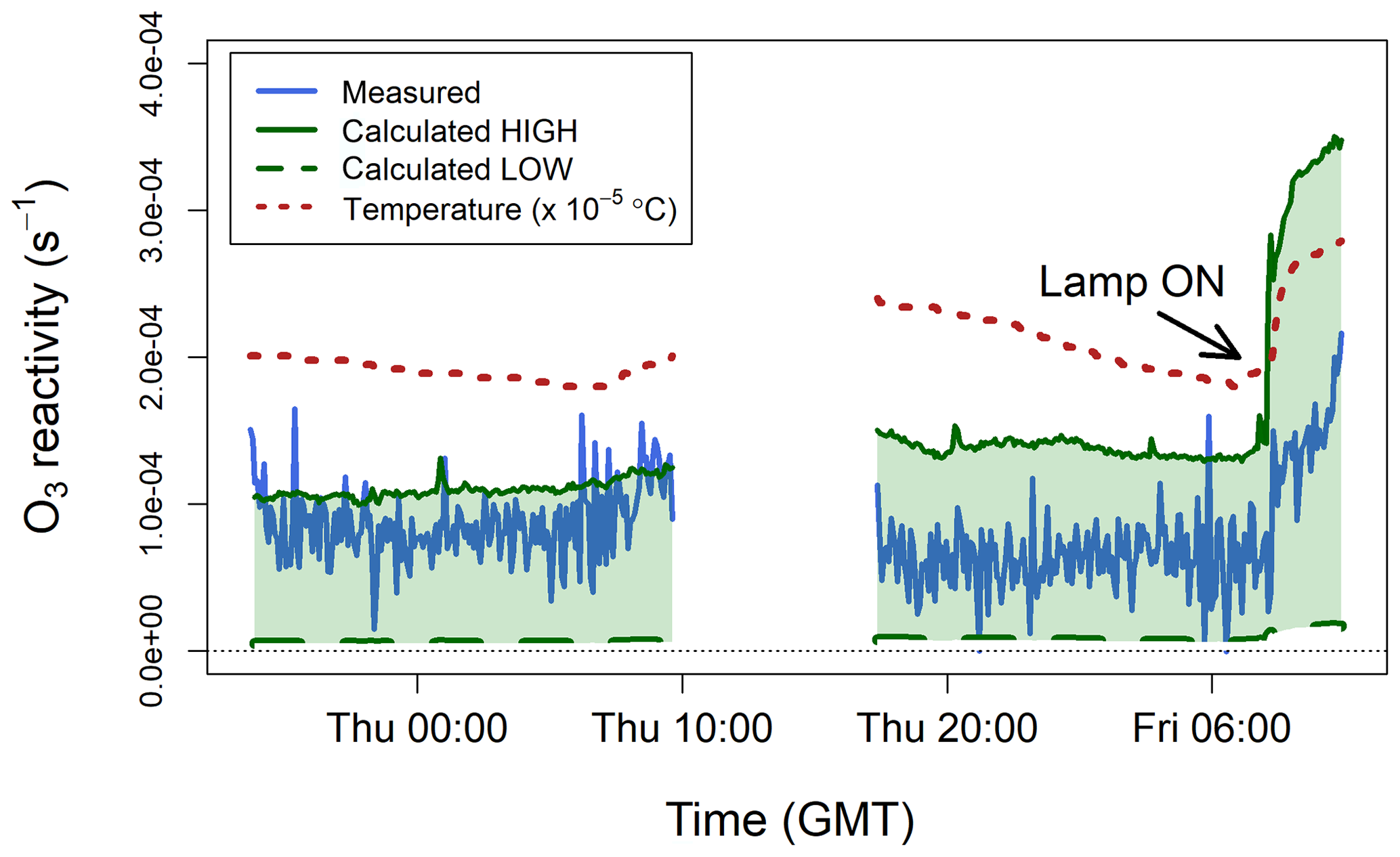
Figure 7Measured ozone reactivity () from three plants of lemon thyme (Thymus citriodorus) in a Teflon bag, compared to the ozone reactivity calculated from BVOC measurements by PTR-MS. The temperature measured inside the bag is also shown. All data are averaged to 5 min.
To calculate from the PTR-MS measurements using Eq. (6), a number of assumptions have to be made. The only BVOC that the proton-transfer-reaction technique can uniquely identify is isoprene. All monoterpenes and sesquiterpenes have the same molecular weight (136.24 and 204.36 g mol−1, respectively) and therefore are very difficult to distinguish from each other using a soft ionization technique (de Gouw and Warneke, 2007). Thus, the PTR-MS instrument effectively reports the sum of monoterpenes and the sum of sesquiterpenes. To account for this problem, estimated low and high limits were calculated. Lemon thyme is an evergreen broadleaf plant, whose main emissions (besides isoprene) are α-pinene, β-pinene, and β-ocimene (monoterpenes) and β-caryophyllene and α-farnesene (sesquiterpenes) (Fares et al., 2011; Guenther et al., 2012). The lower-limit estimate was calculated assuming that the measured monoterpene signal was solely due to β-pinene and that the measured sesquiterpene signal was solely due to α-farnesene. The higher-limit estimate was calculated assuming that the measured monoterpene signal was solely due to β-ocimene and that the measured sesquiterpene signal was solely due to β-caryophyllene. This provides a range of which likely includes that of the particular BVOC mixture emitted by the lemon thyme plants (Table 1). The calculated low and high limit estimates are compared to the TORS measurements in Fig. 7. The TORS measured reactivities were within the range of these estimates and followed the same pattern, with higher values when the light was on and the temperature higher.
5.3 Glasshouse experiments
In order to evaluate the TORS technique under quasi-ambient conditions, the instrument was deployed in a horticultural glasshouse containing a range of aromatic plants. The glasshouse is a similar environment to ambient and was subject to a continuous inflow of ambient air, but, being a semi-enclosed system, the concentrations of BVOCs emitted from the plants are higher and the concentrations of NO are lower than the external environment, resulting in a stronger signal. The glasshouse is located at the Winterbourne House and Garden (https://www.winterbourne.org.uk/, last access: 1 March 2020), adjacent to the University of Birmingham campus, and has an approximate volume of 200 m3. The following plants were inside the glasshouse during the sampling period: fringed “French” lavender (Lavandula dentata var. candicans), lemon verbena (Aloysia triphylla), scented-leaf geraniums (Pelargonium x citriodorum “Prince of Orange”, Pelargonium Radula), and several varieties of the Citrus genus (Citrus x limon, Citrus x latifolia “Tahiti”, Citrus reticulata “Clementine”).
The TORS instrument was set up in a similar way as in the plant experiments (Sect. 5.2), with regular determinations of the ozone wall loss using a flow of zero air instead of the ambient flow. The ozone wall loss during the measurement period varied between and s−1 (first and third quartiles), with mean values between and s−1. The measurements were taken over a period of 2 weeks in early June 2018; during this period the weather was dry (mean RH =57 %) with temperatures reaching a maximum of 39 ∘C inside the glasshouse (mean =15 ∘C). Cyclohexane was used as OH scrubber only during the second week of measurements. The PTR-MS was not available at the glasshouse, but two air samples were taken on two different days using adsorption tubes and qualitatively analysed by GC–MS (Sect. 3.2). The GC data were used to determine the most important monoterpenes and sesquiterpenes in the air inside the glasshouse, based on their relative abundance.
The total ozone reactivity measurements made in the glasshouse are shown in Fig. 8. For the period without cyclohexane the interquartile range was 1.9– s−1 with a mean value of s−1. For the period with cyclohexane the interquartile range was 2.0– s−1 with a mean value of s−1. Taking into account the natural variability of plant emissions, these numbers suggest that the use of an OH scrubber does not significantly change the TORS measurements, in keeping with the laboratory experiments (Sect. 5.1) and the model results of the chemistry inside the reactor (Sect. 2.3).
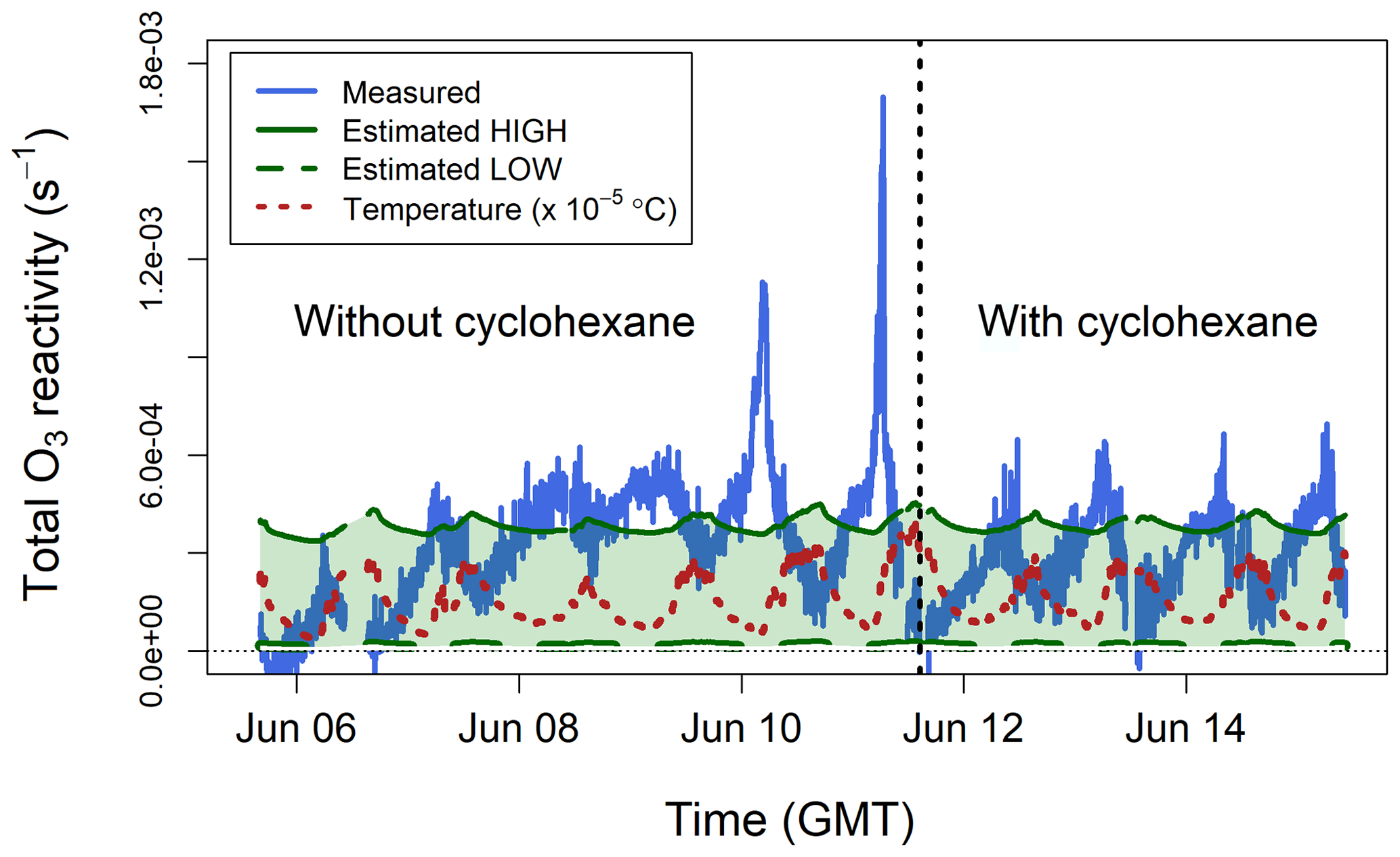
Figure 8Total ozone reactivity () measured in the Winterbourne House and Garden glasshouse compared to the ozone reactivity calculated from estimated concentrations of BVOCs. The temperature measured inside the glasshouse is also shown. All data are averaged to 5 min.
In the absence of BVOC measurements, an estimate of the ozone reactivity was calculated using the qualitative information obtained from the GC–MS analysis of the adsorption tubes and from the emission factors by Guenther et al. (2012): broadleaf evergreen plants emit isoprene, monoterpenes, and sesquiterpenes in a proportion of approximately . However, Fares et al. (2011) found that Citrus plants, several types of which were present in the glasshouse, emit more monoterpenes than isoprene. Since most monoterpenes are more reactive with ozone than isoprene (Table 1), the estimates of discussed below are relatively insensitive to the actual isoprene concentration. Analysis of the adsorption tubes showed that the most important monoterpenes were limonene, β-pinene, camphene, and myrcene, and the most important sesquiterpenes were longifolene and farnesene. Based on these results, a high value was estimated assuming 100 ppb of isoprene, 25 ppb of myrcene, and 5 ppb of α-farnesene. A low was estimated assuming 50 ppb of isoprene, 5 ppb of camphene, and 1 ppb of longifolene. The estimated low and high values were of the same magnitude as the TORS measurements (Fig. 8).
In contrast to the laboratory experiments (Sect. 5.1 and 5.2), which were performed using zero air, the ozone reactivity measurements in the glasshouse were affected by ambient NO (Reaction R5). Measurements of NO were not available inside the glasshouse, so it is not possible to definitively quantify the contribution of RNO to the total ozone reactivity measurements shown in Fig. 8. The average total ozone reactivity showed a clear diel pattern, with maximum values of about s−1 observed around 06:00 GMT (approximately 1 h after dawn), and was anticorrelated with ambient temperature (Fig. 8). BVOC emissions are driven by both light and temperature and are therefore higher during the day (Fares et al., 2011; Hellén et al., 2018); likewise, NO concentrations are higher during the day, due to traffic emissions. Therefore, it may be expected that measured total ozone reactivity (from both BVOCs and NO) is higher during the daylight hours, which was in fact observed during the laboratory experiments with the lemon thyme plants (Fig. 7). Under typical ambient conditions, BVOCs react during the day with OH radicals at a faster rate than they react with O3 (Atkinson et al., 2006; Johnson and Marston, 2008), and, as a result, ozone reactivity may be expected to peak in the early morning, when the NO and BVOC emissions start increasing, but the concentration of OH is still too low to compete with O3 for BVOC removal (Hellén et al., 2018). In the quasi-ambient conditions of the glasshouse, OH formation is also possible – but may be influenced by environment-specific factors, such as heterogeneous production of HONO – and may affect the diurnal variability of BVOCs in a complex manner including the behaviour shown in Fig. 8.
An instrument to measure total ozone reactivity, the Total Ozone Reactivity System (TORS), was developed, characterized, and tested under controlled conditions in the laboratory; both individual compounds and small plants were used. The instrument was deployed inside a horticultural glasshouse containing a range of aromatic plants to evaluate its functionality under quasi-ambient conditions.
The TORS instrument was able to measure O3 reactivities with BVOCs () of 4.5– s−1 or more – with a residence time of 140 s, an averaging time of 5 min, and an estimated total uncertainty of ∼32 %. These values correspond to 20–40 ppb of α-pinene, 150–300 ppb of isoprene, or 160–320 ppt of β-caryophyllene. These mixing ratios are larger than typical ambient levels, but they can be observed in BVOC-rich forested environments and in enclosure studies (Duhl et al., 2008; Bouvier-Brown et al., 2009; Kammer et al., 2018); they can also be easily reproduced in laboratory and environmental chamber experiments. An OH scrubber (cyclohexane) was used to remove the OH radicals formed by the ozonolysis of BVOCs; however, simulations of the chemistry inside the TORS reactor using a Master Chemical Mechanism (MCM v3.3.1) box model found that the formation of OH from BVOC+O3 reactions affected the measurements of by <6 %, under the conditions used during the experiments.
Further work will aim to improve the stability of the signal and to reduce the signal noise and the detection limit. This may require using ozone monitors with higher precision and/or a more stable O3 generator, as well as a detailed exploration of the various parameters affecting the TORS technique: gas flows, residence time, relative humidity, OH scrubber levels, and ozone concentrations. Moreover, our experimental data indicate that accurate measurements of NOx are always required to be able to interpret the TORS observations. With these improvements and proper supporting measurements, the detection limit and the uncertainty of TORS can be improved and the technique will be able to make measurements under a wider range of conditions, to advance understanding of the role of natural emissions in the ozone budget and the oxidative capacity of the atmosphere.
The original data are available from the authors upon request.
RS, LJK, and WJB designed the instrument and the experiments. LRC and MSA provided the measurements of volatile organic compounds. All authors contributed to the manuscript.
The authors declare that they have no conflict of interest.
We would like to thank Daniel Blenkhorn, Dewi Komalasari, Vasileios Matthaios, Daniel Rooney, and Karn Vohra for their help with some of the experiments and the University of Birmingham Biosciences Workshop for their continuous assistance. Special thanks are due to the staff of the Winterbourne House and Garden for their support and to Jun Matsumoto for his advice and kind hospitality.
This research has been supported by the Natural Environment Research Council (grant no. NE/P003524/1) and the UoB-Waseda Research Collaborationn Fund.
This paper was edited by Hendrik Fuchs and reviewed by two anonymous referees.
Alam, M. S., Camredon, M., Rickard, A. R., Carr, T., Wyche, K. P., Hornsby, K. E., Monks, P. S., and Bloss, W. J.: Total radical yields from tropospheric ethene ozonolysis, Phys. Chem. Chem. Phys., 13, 11002–11015, https://doi.org/10.1039/c0cp02342f, 2011. a, b, c
Alam, M. S., Stark, C., and Harrison, R. M.: Using variable ionization energy time-of-flight mass spectrometry with comprehensive GCxGC to identify isomeric species, Anal. Chem., 88, 4211–4220, https://doi.org/10.1021/acs.analchem.5b03122, 2016. a
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., and Troe, J.: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume I – gas phase reactions of Ox, HOx, NOx and SOx species, Atmos. Chem. Phys., 4, 1461–1738, https://doi.org/10.5194/acp-4-1461-2004, 2004. a, b, c
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., Troe, J., and IUPAC Subcommittee: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume II – gas phase reactions of organic species, Atmos. Chem. Phys., 6, 3625–4055, https://doi.org/10.5194/acp-6-3625-2006, 2006. a, b
Bell, N., Heard, D. E., Pilling, M. J., and Tomlin, A. S.: Atmospheric lifetime as a probe of radical chemistry in the boundary layer, Atmos. Environ., 37, 2193–2205, https://doi.org/10.1016/S1352-2310(03)00157-2, 2003. a
Bouvier-Brown, N. C., Holzinger, R., Palitzsch, K., and Goldstein, A. H.: Large emissions of sesquiterpenes and methyl chavicol quantified from branch enclosure measurements, Atmos. Environ., 43, 389–401, https://doi.org/10.1016/j.atmosenv.2008.08.039, 2009. a, b
Cazorla, M. and Brune, W. H.: Measurement of Ozone Production Sensor, Atmos. Meas. Tech., 3, 545–555, https://doi.org/10.5194/amt-3-545-2010, 2010. a
de Gouw, J. and Warneke, C.: Measurements of volatile organic compounds in the Earth's atmosphere using proton-transfer-reaction mass spectrometry, Mass Spectrom. Rev., 26, 223–257, https://doi.org/10.1002/mas.20119, 2007. a, b
Di Carlo, P., Brune, W. H., Martinez, M., Harder, H., Lesher, R., Ren, X., Thornberry, T., Carroll, M. A., Young, V., Shepson, P. B., Riemer, D., Apel, E., and Campbell, C.: Missing OH reactivity in a forest: evidence for unknown reactive biogenic VOCs, Science, 304, 722–725, https://doi.org/10.1126/science.1094392, 2004. a, b
Dubé, W. P., Brown, S. S., Osthoff, H. D., Nunley, M. R., Ciciora, S. J., Paris, M. W., McLaughlin, R. J., and Ravishankara, A. R.: Aircraft instrument for simultaneous, in situ measurement of NO3 and N2O5 via pulsed cavity ring-down spectroscopy, Rev. Sci. Instrum., 77, 034101, https://doi.org/10.1063/1.2176058, 2006. a
Duhl, T. R., Helmig, D., and Guenther, A.: Sesquiterpene emissions from vegetation: a review, Biogeosciences, 5, 761–777, https://doi.org/10.5194/bg-5-761-2008, 2008. a, b, c
Edwards, S. J., Lewis, A. C., Andrews, S. J., Lidster, R. T., Hamilton, J. F., and Rhodes, C. N.: A compact comprehensive two-dimensional gas chromatography (GCxGC) approach for the analysis of biogenic VOCs, Anal. Methods, 5, 141–150, https://doi.org/10.1039/c2ay25710f, 2013. a
Fares, S., Gentner, D. R., Park, J.-H., Ormeno, E., Karlik, J., and Goldstein, A. H.: Biogenic emissions from citrus species in California, Atmos. Environ., 45, 4557–4568, https://doi.org/10.1016/j.atmosenv.2011.05.066, 2011. a, b, c
Fuchs, H., Novelli, A., Rolletter, M., Hofzumahaus, A., Pfannerstill, E. Y., Kessel, S., Edtbauer, A., Williams, J., Michoud, V., Dusanter, S., Locoge, N., Zannoni, N., Gros, V., Truong, F., Sarda-Esteve, R., Cryer, D. R., Brumby, C. A., Whalley, L. K., Stone, D., Seakins, P. W., Heard, D. E., Schoemaecker, C., Blocquet, M., Coudert, S., Batut, S., Fittschen, C., Thames, A. B., Brune, W. H., Ernest, C., Harder, H., Muller, J. B. A., Elste, T., Kubistin, D., Andres, S., Bohn, B., Hohaus, T., Holland, F., Li, X., Rohrer, F., Kiendler-Scharr, A., Tillmann, R., Wegener, R., Yu, Z., Zou, Q., and Wahner, A.: Comparison of OH reactivity measurements in the atmospheric simulation chamber SAPHIR, Atmos. Meas. Tech., 10, 4023–4053, https://doi.org/10.5194/amt-10-4023-2017, 2017. a
Glasius, M. and Goldstein, A. H.: Recent discoveries and future challenges in atmospheric organic chemistry, Environ. Sci. Technol., 50, 2754–2764, https://doi.org/10.1021/acs.est.5b05105, 2016. a, b, c, d, e
Goldstein, A. H. and Galbally, I. E.: Known and unexplored organic constituents in the Earth's atmosphere, Environ. Sci. Technol., 41, 1514–1521, https://doi.org/10.1021/es072476p, 2007. a
Griffith, S. M., Hansen, R. F., Dusanter, S., Stevens, P. S., Alaghmand, M., Bertman, S. B., Carroll, M. A., Erickson, M., Galloway, M., Grossberg, N., Hottle, J., Hou, J., Jobson, B. T., Kammrath, A., Keutsch, F. N., Lefer, B. L., Mielke, L. H., O'Brien, A., Shepson, P. B., Thurlow, M., Wallace, W., Zhang, N., and Zhou, X. L.: OH and HO2 radical chemistry during PROPHET 2008 and CABINEX 2009 – Part 1: Measurements and model comparison, Atmos. Chem. Phys., 13, 5403–5423, https://doi.org/10.5194/acp-13-5403-2013, 2013. a, b
Guenther, A. B., Jiang, X., Heald, C. L., Sakulyanontvittaya, T., Duhl, T., Emmons, L. K., and Wang, X.: The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 5, 1471–1492, https://doi.org/10.5194/gmd-5-1471-2012, 2012. a, b, c, d, e, f
Hellén, H., Praplan, A. P., Tykkä, T., Ylivinkka, I., Vakkari, V., Bäck, J., Petäjä, T., Kulmala, M., and Hakola, H.: Long-term measurements of volatile organic compounds highlight the importance of sesquiterpenes for the atmospheric chemistry of a boreal forest, Atmos. Chem. Phys., 18, 13839–13863, https://doi.org/10.5194/acp-18-13839-2018, 2018. a, b, c, d
Holzinger, R., Acton, W. J. F., Bloss, W. J., Breitenlechner, M., Crilley, L. R., Dusanter, S., Gonin, M., Gros, V., Keutsch, F. N., Kiendler-Scharr, A., Kramer, L. J., Krechmer, J. E., Languille, B., Locoge, N., Lopez-Hilfiker, F., Materić, D., Moreno, S., Nemitz, E., Quéléver, L. L. J., Sarda Esteve, R., Sauvage, S., Schallhart, S., Sommariva, R., Tillmann, R., Wedel, S., Worton, D. R., Xu, K., and Zaytsev, A.: Validity and limitations of simple reaction kinetics to calculate concentrations of organic compounds from ion counts in PTR-MS, Atmos. Meas. Tech., 12, 6193–6208, https://doi.org/10.5194/amt-12-6193-2019, 2019. a, b
Huang, H.: Development of an instrument for the in situ measurement of atmospheric ozone production rates, PhD thesis, School of Geography, Earth and Environmental Sciences, University of Birmingham, UK, 2016. a
Ingham, T., Goddard, A., Whalley, L. K., Furneaux, K. L., Edwards, P. M., Seal, C. P., Self, D. E., Johnson, G. P., Read, K. A., Lee, J. D., and Heard, D. E.: A flow-tube based laser-induced fluorescence instrument to measure OH reactivity in the troposphere, Atmos. Meas. Tech., 2, 465–477, https://doi.org/10.5194/amt-2-465-2009, 2009. a
Johnson, D. and Marston, G.: The gas-phase ozonolysis of unsaturated volatile organic compounds in the troposphere, Chem. Soc. Rev., 37, 699–716, https://doi.org/10.1039/b704260b, 2008. a, b, c, d, e, f, g
Kammer, J., Perraudin, E., Flaud, P.-M., Lamaud, E., Bonnefond, J. M., and Villenave, E.: Observation of nighttime new particle formation over the French Landes forest, Sci. Total Environ., 621, 1084–1092, https://doi.org/10.1016/j.scitotenv.2017.10.118, 2018. a
Kim, S., Karl, T., Helmig, D., Daly, R., Rasmussen, R., and Guenther, A.: Measurement of atmospheric sesquiterpenes by proton transfer reaction-mass spectrometry (PTR-MS), Atmos. Meas. Tech., 2, 99–112, https://doi.org/10.5194/amt-2-99-2009, 2009. a, b
Kim, S., Karl, T., Guenther, A., Tyndall, G., Orlando, J., Harley, P., Rasmussen, R., and Apel, E.: Emissions and ambient distributions of Biogenic Volatile Organic Compounds (BVOC) in a ponderosa pine ecosystem: interpretation of PTR-MS mass spectra, Atmos. Chem. Phys., 10, 1759–1771, https://doi.org/10.5194/acp-10-1759-2010, 2010. a
Kovacs, T. A. and Brune, W. H.: Total OH loss rate measurement, J. Atmos. Chem., 39, 105–122, https://doi.org/10.1023/a:1010614113786, 2001. a
Lewis, A. C., Carslaw, N., Marriott, P. J., Kinghorn, R. M., Morrison, P., Lee, A. L., Bartle, K. D., and Pilling, M. J.: A larger pool of ozone-forming carbon compounds in urban atmospheres, Nature, 405, 778–781, https://doi.org/10.1038/35015540, 2000. a, b
Liebmann, J. M., Muller, J. B. A., Kubistin, D., Claude, A., Holla, R., Plass-Dülmer, C., Lelieveld, J., and Crowley, J. N.: Direct measurements of NO3 reactivity in and above the boundary layer of a mountaintop site: identification of reactive trace gases and comparison with OH reactivity, Atmos. Chem. Phys., 18, 12045–12059, https://doi.org/10.5194/acp-18-12045-2018, 2018. a
Lou, S., Holland, F., Rohrer, F., Lu, K., Bohn, B., Brauers, T., Chang, C. C., Fuchs, H., Häseler, R., Kita, K., Kondo, Y., Li, X., Shao, M., Zeng, L., Wahner, A., Zhang, Y., Wang, W., and Hofzumahaus, A.: Atmospheric OH reactivities in the Pearl River Delta – China in summer 2006: measurement and model results, Atmos. Chem. Phys., 10, 11243–11260, https://doi.org/10.5194/acp-10-11243-2010, 2010. a
Matsumoto, J.: Comprehensive analyzer for biogenic volatile organic compounds detected as total ozone reactivity, in: AGU 2011 Fall Meeting, San Francisco, USA, 5–9 December 2011, Abstract id. A51A-0232, 2011. a
Matsumoto, J.: Measuring biogenic volatile organic compounds (BVOCs) from vegetation in terms of ozone reactivity, Aerosol Air Qual. Res., 14, 197–206, https://doi.org/10.4209/aaqr.2012.10.0275, 2014. a, b, c, d, e
Matsumoto, J.: Temperature dependence of rate constant for the gas-phase reaction of ozone with linalool, Chem. Lett., 45, 1102–1104, https://doi.org/10.1246/cl.160500, 2016. a, b
Miyazaki, K., Nakashima, Y., Schoemaecker, C., Fittschen, C., and Kajii, Y.: A laser-flash photolysis and laser-induced fluorescence detection technique for measuring total HO2 reactivity in ambient air, Rev. Sci. Instrum., 84, 076106, https://doi.org/10.1063/1.4812634, 2013. a
Monks, P. S., Archibald, A. T., Colette, A., Cooper, O., Coyle, M., Derwent, R., Fowler, D., Granier, C., Law, K. S., Mills, G. E., Stevenson, D. S., Tarasova, O., Thouret, V., von Schneidemesser, E., Sommariva, R., Wild, O., and Williams, M. L.: Tropospheric ozone and its precursors from the urban to the global scale from air quality to short-lived climate forcer, Atmos. Chem. Phys., 15, 8889–8973, https://doi.org/10.5194/acp-15-8889-2015, 2015. a, b, c
Pankow, J. F., Luo, W., Melnychenko, A. N., Barsanti, K. C., Isabelle, L. M., Chen, C., Guenther, A. B., and Rosenstiel, T. N.: Volatilizable Biogenic Organic Compounds (VBOCs) with two dimensional Gas Chromatography-Time of Flight Mass Spectrometry (GC × GC-TOFMS): sampling methods, VBOC complexity, and chromatographic retention data, Atmos. Meas. Tech., 5, 345–361, https://doi.org/10.5194/amt-5-345-2012, 2012. a
Park, J., Guenther, A. B., and Helmig, D.: Ozone reactivity of biogenic volatile organic compound (BVOC) emissions during the Southeast Oxidant and Aerosol Study (SOAS), in: AGU 2013 Fall Meeting, San Francisco, USA, 9–13 December 2013, Abstract id. A13A-0172, 2013. a
Paulson, S. E., Chung, M. Y., and Hasson, A. S.: OH radical formation from the gas-phase reaction of ozone with terminal alkenes and the relationship between structure and mechanism, J. Phys. Chem. A, 103, 8125–8138, https://doi.org/10.1021/jp991995e, 1999. a
Pollmann, J., Ortega, J., and Helmig, D.: Analysis of atmospheric sesquiterpenes: sampling losses and mitigation of ozone interferences, Environ. Sci. Technol., 39, 9620–9629, https://doi.org/10.1021/es050440w, 2005. a
Rickard, A. R., Johnson, D., McGill, C. D., and Marston, G.: OH yields in the gas-phase reactions of ozone with alkenes, J. Phys. Chem. A, 103, 7656–7664, https://doi.org/10.1021/jp9916992, 1999. a, b
Sanchez, D., Jeong, D., Seco, R., Wrangham, I., Park, J.-H., Brune, W. H., Koss, A., Gilman, J., de Gouw, J., Misztal, P., Goldstein, A., Baumann, K., Wennberg, P. O., Keutsch, F. N., Guenther, A., and Kim, S.: Intercomparison of OH and OH reactivity measurements in a high isoprene and low NO environment during the Southern Oxidant and Aerosol Study (SOAS), Atmos. Environ., 174, 227–236, https://doi.org/10.1016/j.atmosenv.2017.10.056, 2018. a
Saunders, S. M., Jenkin, M. E., Derwent, R. G., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): tropospheric degradation of non-aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 161–180, https://doi.org/10.5194/acp-3-161-2003, 2003. a
Simpson, W. R., Brown, S. S., Saiz-Lopez, A., Thornton, J. A., and von Glasow, R.: Tropospheric halogen chemistry: sources, cycling, and impacts, Chem. Rev., 115, 4035–4062, https://doi.org/10.1021/cr5006638, 2015. a
Sinha, V., Williams, J., Lelieveld, J., Ruuskanen, T. M., Kajos, M. K., Patokoski, J., Hellen, H., Hakola, H., Mogensen, D., Boy, M., Rinne, J., and Kulmala, M.: OH reactivity measurements within a boreal forest: evidence for unknown reactive emissions, Environ. Sci. Technol., 44, 6614–6620, https://doi.org/10.1021/es101780b, 2010. a, b
Sobanski, N., Tang, M. J., Thieser, J., Schuster, G., Pöhler, D., Fischer, H., Song, W., Sauvage, C., Williams, J., Fachinger, J., Berkes, F., Hoor, P., Platt, U., Lelieveld, J., and Crowley, J. N.: Chemical and meteorological influences on the lifetime of NO3 at a semi-rural mountain site during PARADE, Atmos. Chem. Phys., 16, 4867–4883, https://doi.org/10.5194/acp-16-4867-2016, 2016. a
Sommariva, R., Cox, S., Martin, C., Borońska, K., Young, J., Jimack, P. K., Pilling, M. J., Matthaios, V. N., Nelson, B. S., Newland, M. J., Panagi, M., Bloss, W. J., Monks, P. S., and Rickard, A. R.: AtChem (version 1), an open-source box model for the Master Chemical Mechanism, Geosci. Model Dev., 13, 169–183, https://doi.org/10.5194/gmd-13-169-2020, 2020. a
Sulzer, P., Hartungen, E., Hanel, G., Feil, S., Winkler, K., Mutschlechner, P., Haidacher, S., Schottkowsky, R., Gunsch, D., Seehauser, H., Striednig, M., Jürschik, S., Breiev, K., Lanza, M., Herbig, J., Märk, L., Märk, T. D., and Jordan, A.: A proton transfer reaction-quadrupole interface time-of-flight mass spectrometer (PTR-QiTOF): high speed due to extreme sensitivity, Int. J. Mass Spectrom., 368, 1–5, https://doi.org/10.1016/j.ijms.2014.05.004, 2014. a
Tani, A., Hayward, S., and Hewitt, C. N.: Measurement of monoterpenes and related compounds by proton transfer reaction-mass spectrometry (PTR-MS), Int. J. Mass Spectrom., 223-224, 561–578, https://doi.org/10.1016/s1387-3806(02)00880-1, 2003. a
Whalley, L. K., Edwards, P. M., Furneaux, K. L., Goddard, A., Ingham, T., Evans, M. J., Stone, D., Hopkins, J. R., Jones, C. E., Karunaharan, A., Lee, J. D., Lewis, A. C., Monks, P. S., Moller, S. J., and Heard, D. E.: Quantifying the magnitude of a missing hydroxyl radical source in a tropical rainforest, Atmos. Chem. Phys., 11, 7223–7233, https://doi.org/10.5194/acp-11-7223-2011,1, 2011. a, b
Whalley, L. K., Stone, D., Bandy, B., Dunmore, R., Hamilton, J. F., Hopkins, J., Lee, J. D., Lewis, A. C., and Heard, D. E.: Atmospheric OH reactivity in central London: observations, model predictions and estimates of in situ ozone production, Atmos. Chem. Phys., 16, 2109–2122, https://doi.org/10.5194/acp-16-2109-2016, 2016. a
Williams, J.: Organic trace gases in the atmosphere: an overview, Environ. Chem., 1, 125–136, https://doi.org/10.1071/EN04057, 2004. a
Total Ozone Reactivity Systemor TORS – that measures the total loss of ozone in the troposphere. The objective of the TORS instrument is to provide an estimate of the organic compounds emitted by plants which are not measured and thus to improve our understanding of the ozone budget.