the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 27 May 2021
| 27 May 2021
Coupling a gas chromatograph simultaneously to a flame ionization detector and chemical ionization mass spectrometer for isomer-resolved measurements of particle-phase organic compounds
Chenyang Bi
Jordan E. Krechmer
Graham O. Frazier
Andrew T. Lambe
Megan S. Claflin
Brian M. Lerner
John T. Jayne
Douglas R. Worsnop
Manjula R. Canagaratna
Atmospheric oxidation products of volatile organic compounds consist of thousands of unique chemicals that have distinctly different physical and chemical properties depending on their detailed structures and functional groups. Measurement techniques that can achieve molecular characterizations with details down to functional groups (i.e., isomer-resolved resolution) are consequently necessary to provide understandings of differences of fate and transport within isomers produced in the oxidation process. We demonstrate a new instrument coupling the thermal desorption aerosol gas chromatograph (TAG), which enables the separation of isomers, with the high-resolution time-of-flight chemical ionization mass spectrometer (HR-ToF-CIMS), which has the capability of classifying unknown compounds by their molecular formulas, and the flame ionization detector (FID), which provides a near-universal response to organic compounds. The TAG-CIMS/FID is used to provide isomer-resolved measurements of samples from liquid standard injections and particle-phase organics generated in oxidation flow reactors. By coupling a TAG to a CIMS, the CIMS is enhanced with an additional dimension of information (resolution of individual molecules) at the cost of time resolution (i.e., one sample per hour instead of per minute). We found that isomers are prevalent in sample matrix with an average number of three to five isomers per formula depending on the precursors in the oxidation experiments. Additionally, a multi-reagent ionization mode is investigated in which both zero air and iodide are introduced as reagent ions, to examine the feasibility of extending the use of an individual CIMS to a broader range of analytes with still selective reagent ions. While this approach reduces iodide-adduct ions by a factor of 2, [M − H]− and [M + O2]− ions produced from lower-polarity compounds increase by a factor of 5 to 10, improving their detection by CIMS. The method expands the range of detected chemical species by using two chemical ionization reagents simultaneously, which is enabled by the pre-separation of analyte molecules before ionization.
- Article
(1441 KB) - Full-text XML
-
Supplement
(255 KB) - BibTeX
- EndNote





Atmospheric aerosols can impact climate by scattering light and absorbing radiation and thus changing earth's reflectivity directly or by impacting the formation of clouds (Seinfeld and Pandis, 2016). If inhaled, they may deposit into the alveoli region of lungs and transmit into the bloodstream to cause adverse health effects such as ischemic heart disease, cerebrovascular disease, and lung cancer (Burnett et al., 2014; Pope and Dockery, 2006). A significant fraction of submicrometer aerosol mass is organic, mostly formed through the oxidative conversion of gas-phase volatile organic compounds (VOCs) to lower-volatility oxygenated organic compounds that can condense to form secondary organic aerosol (SOA) (Goldstein and Galbally, 2007; Jimenez et al., 2009; Kroll and Seinfeld, 2008).
Atmospheric oxidation of volatile organic compounds produces thousands of unique chemicals that exist in a dynamic and complex mixture that spans a wide range of physicochemical properties (Atkinson and Arey, 2003; Goldstein and Galbally, 2007). The detailed structure and functional groups of each individual compound controls its properties (Kroll and Seinfeld, 2008; Nozière et al., 2015). Rate of reactions, partitioning between phases, and compound toxicity are all impacted by the functional groups present in a molecule (Arangio et al., 2016) and in some cases on its physical conformation (Atkinson, 2000; Lim and Ziemann, 2009). Characterization of a compound to the level of detail of its molecular structure (i.e., isomer-resolved composition) is therefore necessary to predict whether a compound will deposit to a particle or surface, react with an oxidant, or persist in the atmosphere and to understand its potential adverse impacts on public or ecosystem health.
A major advance in the online nontarget analysis of oxygenated organics in the atmosphere has been the development and widespread adoption of the high-resolution time-of-flight chemical ionization mass spectrometer (HR-ToF-CIMS). These systems directly sample atmospheric constituents with minimal pretreatment and classify them by their molecular formulas, providing unprecedented characterizations of thermally and/or chemically labile atmospheric constituents. Multiple negative-ion chemical ionization methods have been applied to atmospheric sampling, including acetate for the detection of carboxylic acids and some inorganic acids (Bertram et al., 2011; Brophy and Farmer, 2016; Veres et al., 2008), nitrate for the detection of highly oxidized organic matter (Ehn et al., 2010; Krechmer et al., 2015), and iodide for the detection of moderately and highly polar organic compounds (Aljawhary et al., 2013; Isaacman-Vanwertz et al., 2018; Lee et al., 2014; Lopez-Hilfiker et al., 2014; Riva et al., 2019). Iodide has seen particularly wide use due to its relatively general selectivity and straightforward chemistry, in which an adduct is formed between the analyte and iodide ion. Due to the high negative mass defect of iodine, iodide-adduct ions are separated from other isobaric ions in the mass spectrum by the so-called “iodide valley” in which few ions are present and so can be classified by their molecular formula with high confidence and low detection limits (Lee et al., 2014). Ions with positive mass defects (i.e., non-iodide adducts) are observed in iodide-CIMS mass spectra of atmospheric mixtures and may represent peroxy acids (Lee et al., 2014; Mielke and Osthoff, 2012), but these ions are generally poorly understood and frequently excluded from analyses due to their uncertain interpretation.
A major barrier to understanding most reagent ion chemistries is that isomers cannot be differentiated since analytes are classified by molecular formulas. Studying instrument response requires introducing analytes as known standards, so it is limited to commercially available or custom-synthesized compounds. Such compounds do not fully capture the composition of real-world atmospheres, which contain compounds that are not easily synthesized or available, such as transient and short-lived oxidation products. An improved approach would be the generation of oxidized products through the simulated atmospheric oxidation of atmospherically relevant precursors, but this approach has limited utility due to the formation of multiple isomers of each formula (Brophy and Farmer, 2016). The range of CIMS reagent ion chemistries that are practically useful is therefore limited to those, such as iodide, that yield distinguishable products, while other potentially promising techniques (e.g., acetate CIMS) have seen less use due to the complex ionization pathways. The lack of isomer resolution afforded by CIMS introduces several other limitations as well. The fate of a compound and its impacts on the environment are dependent on molecular structure, so they may not be fully predicted or captured by molecular formulas (Isaacman-VanWertz and Aumont, 2021). Furthermore, a formula may represent many isomers, but CIMS signals will preferentially represent isomers to which the ionization method is more sensitive. This may bias the interpretation and quantification of CIMS data, though the extent to which this issue has impacted the literature is unknown.
The resolution of individual components in a complex mixture is typically performed through some form of chromatography. While liquid chromatography (LC, e.g., Gómez-González et al., 2008), ion chromatography (e.g., VandenBoer et al., 2012), and gas chromatography (e.g., Hamilton, 2010; Schauer et al., 1996) have all been used in atmospheric measurements, we limit our discussion here to gas chromatography (GC), as it has been demonstrated as a field-deployable technique for the analysis of oxygenated organics, complementary with CIMS instrumentation (Isaacman-VanWertz et al., 2016; Thompson et al., 2017; Zhang et al., 2018). GC separates compounds in a complex mixture based upon their vapor pressure and polarity and has been deployed in the field for decades for the online measurement of low-polarity gas-phase components (Goldan et al., 2004; Goldstein et al., 1995; Helmig et al., 2007; Millet, 2005; Prinn et al., 2000; Vasquez et al., 2018). More recent developments on the field-deployable thermal desorption aerosol gas chromatograph (TAG) have enabled the GC analysis of semivolatile and particle-phase air samples through novel sampling cells, autonomous calibration via liquid injections of authentic standards, and derivatization of oxygenates for the analysis of oxygenated compounds (Isaacman et al., 2014; Williams et al., 2006; Zhao et al., 2013). Detection of analytes eluting from the GC may rely on a flame ionization detector (FID), which has near-universal response but provides no chemical information about an analyte (Kolb et al., 1977; Grob and Barry, 2004), or an electron ionization mass spectrometer (EI-MS), which provides identification of compounds with mass spectra available in existing libraries but requiring careful interpretation to identify those not in the libraries. These techniques have allowed for quantification of some individual isomers in SOA, but the substantial majority of analytes observed in GC analyses of SOA have no known structures or identification (Isaacman-VanWertz et al., 2016) due to a lack of commercially available standards of atmospheric oxidation products.
We demonstrate here a new instrument coupling the TAG, which enables the separation of isomers, with the HR-ToF-CIMS, which has the capability of classifying unknown compounds by their molecular formulas, and with the FID, which provides a near-universal response to all analytes for quantification with low uncertainty. The specific objectives of this work are to (1) describe the setup and operation of a TAG-CIMS/FID, (2) evaluate the presence of isomers in laboratory-generated SOA, (3) demonstrate the utility of this instrument for investigating ionization chemistries by resolving mass spectra of individual compounds separated from a complex mixture; and (4) expand the range of chemical species which can be measured by an individual CIMS using multiple reagent ions.
As shown in Fig. S1 in the Supplement, the TAG-CIMS/FID integrates the use of a GC instrument, TAG, with two detectors, HR-ToF-CIMS (Aerodyne Research Inc.) and FID (Agilent 7820A). The GC column effluent is split between the two detectors using a heated and passivated tee together with heated fused-silica transfer lines. The instrument allows for the collection of chromatographic signals generated by CIMS and FID simultaneously.
2.1 HR-ToF-CIMS
The HR-ToF-CIMS (Lee et al., 2014) uses a similar physical configuration as described by Isaacman-Vanwertz et al. (2018) with modification at the inlet for adapting the GC flow. The inlet is a heated metal interface which is kept at 225 ∘C and has a in. inner diameter bore-through center hole to allow for insertion of the fused-silica guard column into the ion–molecule region (IMR) which is kept at room temperature (∼ 20 ∘C) . The tip of the guard column extends 2 mm out of the heated cartridge to mix with the reagent ion flow inside the IMR.
Two different ionization modes are used in this study: iodide and multi-reagent ionization mode. The iodide ionization mode (Lee et al., 2014) is used in a similar configuration to those published in the past for a gas-phase iodide CIMS (Krechmer et al., 2016). Iodide ions are generated by passing a 2 slpm flow of humidified ultrahigh-purity (UHP) N2 over a permeation tube filled with methyl iodide and then through a radioactive source (Po-210, 10 mCi, NRD) into the IMR. The IMR pressure is maintained at 100 mbar. Besides the pure iodide ionization mode, the CIMS operated using a multi-reagent ionization mode to investigate the use of multiple reagent ions in the IMR. Instead of using pure nitrogen, the multi-reagent ionization mode uses a mixture of 1.91 slpm of UHP N2 and 0.09 slpm of ultra zero air as the methyl iodide sheath flow. This mixture is selected such that iodide ions (I−) accounted for ∼ 50 % of the total reagent ion count, with the rest comprised of air-based components as described later.
To obtain a smooth chromatographic peak, raw negative-ion spectra are typically acquired at a rate of 4 Hz without time averaging, which is higher than the rate typically used for direct air sampling by CI-ToF-MS (1 Hz and 1 min averaging). Data collection as fast as 20 Hz was tested and found to be viable for applications requiring higher data frequency but is not necessary for data collection described here. The ToF-MS is operated in “V mode” and achieves a mass resolution of ∼ 4100 at 212 and a mass accuracy of < 5 ppm, which enables the assignment of elemental composition to observed mass-to-charge values.
2.2 TAG
The TAG used in this study is a custom-built instrument similar to those commercially available through Aerodyne Research Inc. In brief, aerosols are collected with a sampling flow rate of 9 slpm through a nine-jet impactor with a 50 % collection cut-off point of 0.085 µm (Williams et al., 2006). The collection cell is equipped with a liquid injection port where chemical standards can be quantitatively added through an automatic liquid injection system (Isaacman et al., 2011). After a 5–15 min sample collection period, which is equivalent to 0.045–0.135 m3 of air sampled, samples are thermally desorbed in helium to the head of the GC column by heating the cell to 300 ∘C at a rate of 50 ∘C min−1 while the GC column is held at 50 ∘C with ∼ 3 sccm of helium carrier gas flow. Samples pass through a passivated metal manifold with restrictive capillaries to separate sample pressures from GC operating pressures without the use of in-line valves (Kreisberg et al., 2014). A polar GC column (MXT-WAX, 17 m × 0.25 mm × 0.25 µm, Restek) wrapped onto the temperature-controlled metal hub is used for the separation of oxygenated organic compounds. The GC analysis is conducted with 1 sccm of helium, and the temperature of the GC column is ramped to 250 ∘C at a rate of 10 ∘C min−1 and then held for 25 min. Though analysis focuses on oxygenated compounds to which the CIMS may exhibit sensitivity, we highlight that derivatization, which is typically used in the analysis of oxygenates by GC (Isaacman et al., 2014), is not employed here to minimize chemical alterations to the functionality of the analytes reaching the detectors. Although not observed with the injection of liquid standards, it is possible that skipping derivatization may bring up certain limitations of the instruments such as the decrease of the transmission efficiency and stability of the analytes (Isaacman et al., 2014).
2.3 TAG-CIMS/FID interface
The design of the TAG-CIMS/FID interface needs to allow for the efficient transfer of analytes from GC effluent to CIMS and FID. This interface is subject to three technical challenges: (1) the connections between capillary columns and fittings need to be leak-tight, (2) all components in the interfaces require proper heating to avoid cold spots and dead volume, and (3) the relative flow rates to CIMS and FID need to be controlled to maintain roughly equal split of flow. As shown in Fig. 1, a in. passivated (SilcoNert 2000, SilcoTek Corp.) metal cross piece (part no. ZX.5, VICI) is covered by a two-piece heated aluminum block. The four ports on this connector allow for splits from the GC to three detectors (FID, CIMS, optional third). Because the IMR pressure of the CIMS is at 100 mbar while the FID is at ambient pressure, the restriction to the CIMS side needs to be significantly higher than that of the FID side to maintain comparable flow rates. These restrictions are achieved using deactivated fused-silica guard columns which have an inner diameter of 0.075 and 0.25 mm and a length of 0.18 and 0.5 m to connect the cross with CIMS and FID, respectively. Because the change of temperature of GC column during a run cycle may potentially influence the split flow, we mitigate the impacts by maintaining the TAG-CIMS/FID interface at a constant temperature, 300 ∘C, that is 50 ∘C higher than the maximum temperature of the GC column. The guard column to FID passes through a narrow-bore metal tube wrapped in heater cable with fiberglass insulation and heated to 300 ∘C to prevent the condensation of analytes in the transfer lines. The temperature of the entrance capillary to the IMR of the CIMS is maintained at 225 ∘C in order to prevent the degradation of PTFE components of CIMS inlet; this leads to some peak broadening of low-volatility analytes in the CIMS data. With these dimensions and temperatures, the flow rate to FID is approximately one-third of total GC flow (0.3 sccm, measured using Sensidyne Gilibrator-2 at the inlet of FID) with the remainder to the CIMS (0.7 sccm). To further evaluate the stability of the split ratio of flow, test runs were conducted prior to the experiments to monitor the flow rate at the inlet of FID, variability in the flow split was found to be less than 10 % variation throughout a run cycle, which is stable enough to be quantitative.

Figure 1Schematic of the TAG CIMS/FID interface, with a photo inserted. Details of each instrument are simplified. Restrictive capillaries to each detector are used to balance flows to each detector due to differences in pressure (dimensions and temperatures shown).
2.4 Laboratory experiments
Data are collected from two sources: injection of liquid standards and laboratory-generated SOA through the oxidation of atmospherically relevant precursors. Liquid standards include 1,12-dodecanediol (Sigma-Aldrich, 99 % purity), undecanoic acid (AccuStandard, 100 % purity), eugenol (Sigma-Aldrich, 99 % purity), vanillin (Sigma-Aldrich, 99 % purity), vanillic acid (Sigma-Aldrich, 97 % purity), and oleic acid (Sigma-Aldrich, 99 % purity). Additionally, six commercially available liquid fragrance samples (MakingCosmetics Inc.) are also used to serve as representative complex mixtures. SOA was generated via gas-phase O3 and/or OH oxidation of limonene (Sigma-Aldrich, 97 % purity), 1,3,5-trimethylbenzene (Sigma-Aldrich, 98 % purity), and eucalyptol (Sigma-Aldrich, 99 % purity) in a potential aerosol mass (PAM) flow reactor (Lambe et al., 2011) and collected onto the TAG impactor cell at 9 L min−1 sampling flow rate and analyzed by the TAG-CIMS/FID. We hereafter label data from a given set of oxidation experiments system as “precursor–oxidant” (e.g., limonene–OH).
2.5 Data processing
CIMS and FID data are acquired as continuous time series throughout a single GC run. TAG sends a start signal to both CIMS and FID once the GC run starts and sends a digital signal to stop the data acquisition once a sample run is completed. For each run file, high-resolution (HR) mass spectrum peak fitting of CIMS data is conducted using the Tofware (Tofwerk, AG and Aerodyne Research Inc., version 3.1.2) toolkit developed for the Igor Pro 7 analysis software package (WaveMetrics, Inc.). HR fitting of iodide-adduct ions was used to generate a list of potential molecular formulas present within a given set of experiments. Once the formulas are extracted and assigned in CIMS, the high-resolution time-series data with the peak list are then imported into TERN, which is the freely available (https://sites.google.com/site/terninigor/, last access: 20 May 2021) Igor-based software tool for the quantification of chromatographic data (Isaacman-VanWertz et al., 2017). The retention times of peaks between CIMS and FID are aligned based on linear regression of retention times of internal standards (i.e., vanillin and 1,12-dodecanediol) and the most abundant compounds in chromatographic runs.
3.1 Chromatograms
Figure 2 shows an example comparison of chromatograms collected by the FID and CIMS for limonene–O3 SOA. The FID produces a total signal (y axis) as a function primarily of the mass of carbon combusted, so each analyte (i.e., chromatographic peak eluting at a given retention time) responds with similar mass-based sensitivity. However, the total chromatogram shown is the full extent of the data available for a given sample; there is no further separation by mass or other dimension. Co-eluting peaks that are not well resolved may not be able to be accurately integrated. In contrast, the CIMS analyte total ion count signal is the sum of all observed ions with the reagent ions removed, so while two peaks may not be chromatographically resolved (i.e., co-eluting total signal), they may be mass-resolved by producing signals on different ions. Additionally, many peaks that are small yet above the signal-to-noise ratio on their individual ions might not show up in the CIMS chromatogram of analyte total ion counts due to noise in the total counts. The results demonstrate that the responses from FID and CIMS have distinctly different patterns with the number of peaks, and the height of aligned peaks in the chromatograms of FID and CIMS differ significantly. Many early-eluting compounds (i.e., compounds having peaks with retention times less than 1500 s) that are observed by FID do not produce a clear signal in the CIMS. Compounds with retention time longer than 1500 s usually have peaks detected by both FID and CIMS chromatograms. Since the TAG-CIMS/FID interface and the capillary to the FID is held at 50 ∘C above the maximum column temperature, differences in the transfer of analytes to these two detectors should be negligible. Instead, these differences are due to the selectivity of the two detectors. FID is a near-universal detector, which is able to detect almost all organic compounds with relatively similar and predictable responses (Scanlon and Willis, 1985). The sensitivity of the iodide-CIMS may differ by orders of magnitude and is highest for compounds that contain multiple OH groups and can therefore more readily form an adduct with the iodide ion (Iyer et al., 2016). Since the TAG here used a polar (MXT-WAX) GC column that more preferably retains polar compounds, the early-eluting compounds are likely less polar and consequently less sensitive or not detected in the iodide-CIMS. Some early-eluting compounds may be present but have peaks too small to be visible due to the linear display of signal in Fig. 2. It is also possible that some of those peaks are thermally decomposed analytes which exhibit low sensitivity in CIMS since all thermal desorption instruments, including the TAG, are known to potentially cause thermal decomposition of samples (Isaacman-VanWertz et al., 2016; Stark et al., 2017).
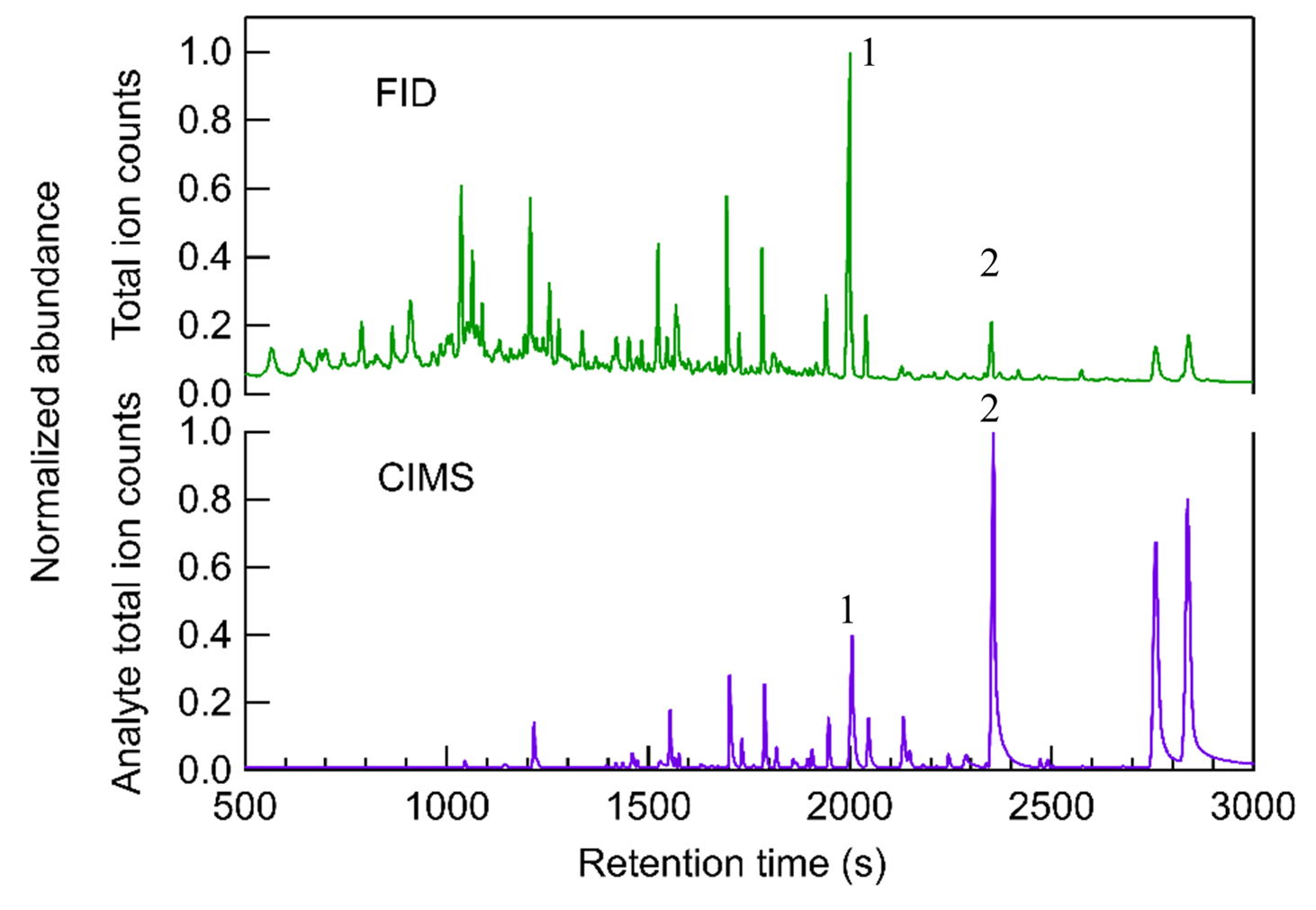
Figure 2Comparison of chromatograms between FID and CIMS from particle sample generated from limonene–O3 reaction. The time-series signals of CIMS are the analyte total ion counts, which are the summation of all ions with the reagent ions removed, while signals of FID are single channel. The signals of both CIMS and FID are normalized to the highest peak in the chromatogram.
The low abundance of early-eluting peaks in the CIMS chromatogram is an example of the value of the coupled CIMS/FID detector pair for investigating CIMS response. Although compounds having larger retention times can be found in both FID and CIMS, their abundance is significantly different between the two detectors. For example, analyte 1 with a retention time of 2000 s has the most abundant signals in FID while analyte 2 with a retention time of 2380 s is the largest peak in CIMS. Since FID sensitivity differs between compounds by less than a factor of two (Hurley et al., 2020), the peak area of FID approximates the relative mass of a compound in a sample matrix. However, comparing to the near-universal response of FID signals, the signals of iodide CIMS per unit mole of analytes may vary up to 5 orders of magnitude and highly depend on their enthalpies of binding with iodide (Iyer et al., 2016; Lopez-Hilfiker et al., 2016a). The two peaks highlighted provide an example of the variability of CIMS response: analyte 1 has a larger FID peak area, indicating a higher mass concentration in the sample mixture than analyte 2. However, since the CIMS peak area of analyte 1 is lower, it must be less sensitive than analyte 2 in an iodide CIMS. With the use of FID in addition to the CIMS detector, calibration of compounds in CIMS without using authentic standards can therefore theoretically be achieved. While implementation of this calibration approach is complex, here we provide a proof-of-concept quantitative analysis. Hurley et al. (2020) demonstrated that the number of moles of an analyte can be calculated from its FID peak area based on a calibration response factor to hydrocarbons, with a correction for oxygenation based on the chemical formula identified by CIMS (specifically, the FID response per carbon atom relative to maximum = −0.54 O C + 0.99, where O C is the oxygen-to-carbon ratio in the target analyte; Hurley et al., 2020). By applying the calibration approach for the two analytes in the example, analyte 1 (i.e., C10H14O3) is found to be roughly 4 times more abundant than analyte 2 (i.e., C9H14O3) on a per mole basis, but appear substantially lower in its CIMS signal due to a 10 times lower sensitivity. The detailed methods of quantification and determination of isomer sensitivity will be addressed in future work. This article focuses instead on the descriptions of technical hurdles overcome by TAG-CIMS/FID and its potential value in understanding existing and new ionization chemistries, as well as atmospheric systems.
3.2 Number of isomers per formula
We explore here the extent to which isomers are present in atmospherically relevant mixtures to assess the potential utility of a TAG-CIMS/FID (or, more generally, a GC-CIMS/FID). The number of isomers per molecular formula identified in the high mass resolution peak fitting can be obtained by counting the number of peaks in the chromatograms of a specific high-resolution of the iodide adduct (i.e., a specific molecular formula). These data should be interpreted cautiously. Overestimation may occur when large parent molecules decompose to isomers of a smaller formula during thermal desorption. Conversely, isomers may be undercounted if they fall below the threshold used to count them, which was selected here to be conservative. The limitations in these estimates will be discussed in detail later in this section, but these data nevertheless provide a useful, if uncertain, understanding of the prevalence of isomers in atmospherically relevant samples.
Figure 3a shows the cumulative number of isomers found in formulas identified in the PAM oxidation experiments. In this study, compounds with a peak height higher than 1.0 × 104 ions s−1 are taken into the isomer counts; the number of isomers is sensitive to the selection of this threshold so we set as a threshold the approximate level at which the chromatographic peak height clearly rises above the baseline by at least a factor of 50; in many cases, peaks are present below this threshold (e.g., Fig. S2, showing 12 isomers identified in (C9H12O4)I). Although 30 % of formulas in the oxidation experiments have only one isomer, a significant portion (34 %) of formulas have more than five isomers. For limonene–O3, limonene–OH, 1,3,5 trimethylbenzene–OH, and eucalyptol–OH reactions, the median numbers of isomers per formula are 2.0, 4.0, 3.5, and 5.0; the average numbers of isomers per formula are 3.7, 4.9, 5.1, and 5.3; and the maximum numbers of isomers per formula are 14, 19, 19, and 17, respectively. The results indicate that isomers are prevalent in sample matrix with an average number of three to five isomers per formula depending on the precursors in the oxidation experiments. We compare these data to previously published studies using isomer-resolved analyses of SOA (Fig. 3b). Ambient measurements from a range of environments show a qualitatively similar distribution of isomers per formula, though with somewhat lower averages of 2.7, 2.1, and 1.9, based on data collected from ambient air in a ponderosa pine forest in California measured by two-dimensional GC without derivatization (Worton et al., 2017), southeastern USA by two-dimensional GC with derivatization (Zhang et al., 2018), and combined datasets from SOA formation measured by liquid chromatography (Bryant et al., 2020; Dixon, 2020). These data were collected by different instruments (two-dimensional GC with filter samples analyzed by thermal desorption and derivatization, two-dimensional GC with filter samples analyzed by thermal desorption, and liquid chromatography, respectively) that are likely not sensitive to the same compounds. Another possible reason for the lower number of isomers might be that the use of two-dimensional GC can limit the range of compounds detected since those analytes have to be such that they make it through two consecutive capillary columns. Together, the published data and that collected by TAG-CIMS/FID support the conclusion that isomers are abundant for molecular formulas with 10 or less carbon number in particle-phase samples. Those isomers may have significantly different physical and chemical properties that impact the formation, transport, and toxicity of SOA, and the distribution of isomers could vary temporally or spatially. The isomer-resolved classification of SOA components provided by TAG-CIMS/FID therefore provides valuable understandings of the oxidative process. Although the molecular structure of each isomer cannot be recognized directly in the chromatograph and the specific functional groups within the formula remain unknown, the chromatographic separation of the TAG-CIMS provides some comparison of polarity, which is dependent on chemical structures, for isomers within a formula.
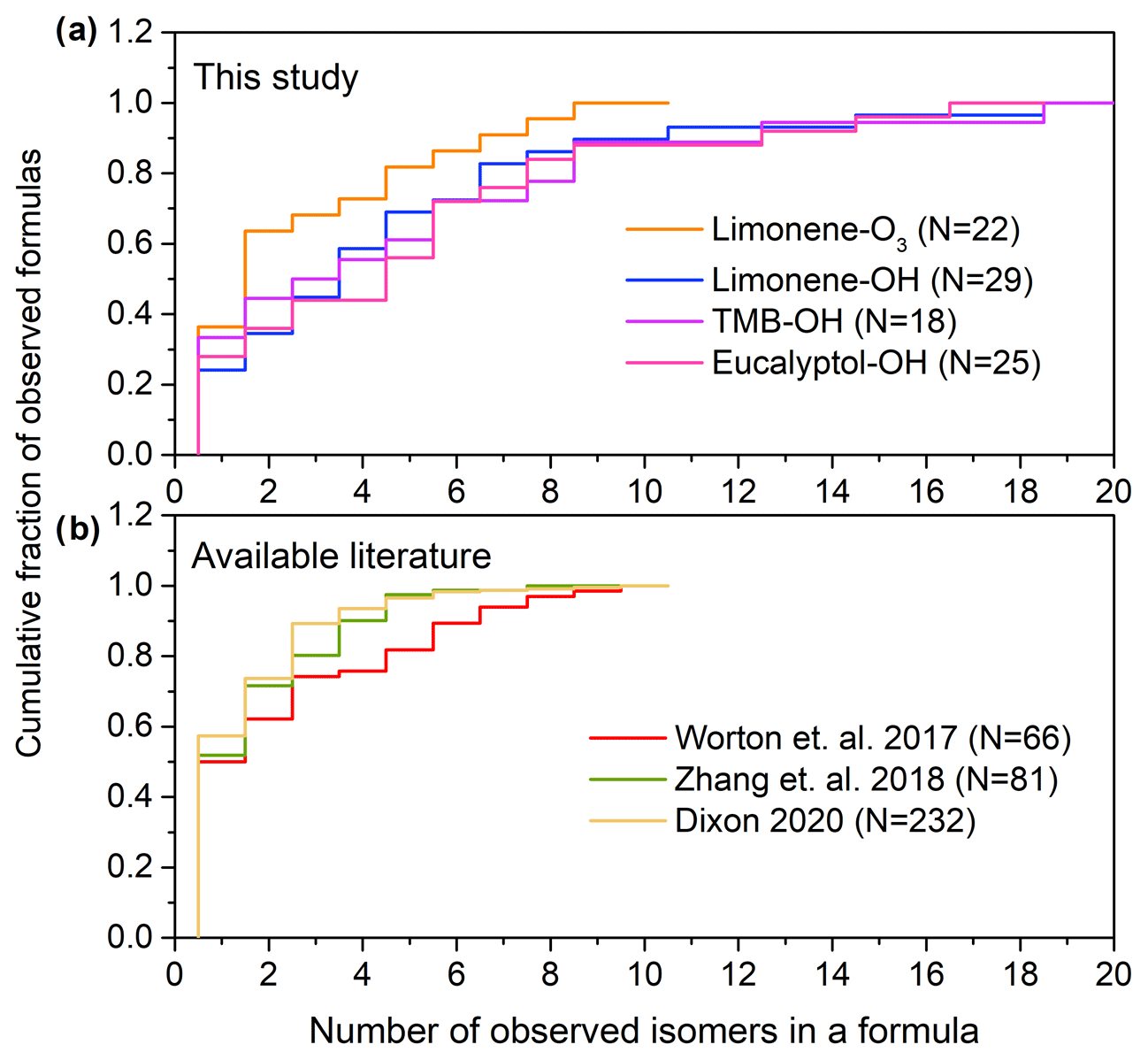
Figure 3Number of isomers per chemical formula in (a) observed in this work in oxidation experiments and (b) available literature. Data in (b) are from ambient atmospheres in a ponderosa pine forest in California measured by two-dimensional GC without derivatization (Worton et al., 2017), southeastern USA by two-dimensional GC with derivatization (Zhang et al., 2018), and combined datasets from SOA formation measured by liquid chromatography (Bryant et al., 2019; Dixon, 2020). The number, N, of formulas observed in each dataset is provided in the legend.
There are certain limitations for the analysis of the number of isomers per formula in this study. The number of isomers is probably underestimated due to exclusion of peak heights less than 1.0 × 104 ions s−1. Furthermore, isomers may vary in their sensitivity, with isomers having less polar functional groups possibly not detected by CIMS. Additionally, the use of a GC column, which is selective towards a certain range of volatility and polarity of compounds, which limits the detection of compounds. Conversely, thermal desorption within TAG may fragment larger accretion products to form analytes not present in the original sample (Buchholz et al., 2020; Isaacman-VanWertz et al., 2016; Lopez-Hilfiker et al., 2016b; Stark et al., 2017) or may reverse particle-phase oligomerization reactions (Claflin and Ziemann, 2019). These fragments may not represent the actual molecular composition of SOA, though they nevertheless may provide insight into the formation mechanisms of SOA (Isaacman-VanWertz et al., 2016). Consequently, the potential multiple fragments from one parent compound may result in an overestimation of the number of isomers. We note, however, that similar numbers of isomers are observed when using liquid chromatography (Fig. 3b), which does not involve thermal desorption. Given these uncertainties, we believe that the results presented are not a floor or a ceiling on the number of isomers in the atmosphere but a step toward understanding a poorly constrained problem. In any case, the number of isomers observed by any single instrument is expected to be underestimated as no instrument is capable of measuring all atmospheric components with molecular speciation. The number of isomers shown in Fig. 3 is therefore likely more illustrative as an example than quantitative, demonstrating the general ubiquity of isomers in the atmosphere. This issue raises significant questions the atmospheric measurement community should address as to how isomers differ in their sources, physicochemical properties, instrument sensitivities, and atmospheric transformations (Isaacman-VanWertz and Aumont, 2021).
3.3 Exploring new chemistries: multi-reagent ionization
Unlike direct air sampling by CIMS, in which the mass spectrum at a given time point is the summation of all analytes, the mass spectra of TAG-CIMS/FID analytes are separated in time by chromatography. Consequently, if a chromatographic peak of a compound is well resolved in CIMS, all signals detected from the ionization of a single analyte are observed at the same chromatographic retention time and unambiguously assignable to that specific compound, including iodide adducts, products of adduct declustering, fragments (generally not from iodide clustering), and any ions produced by simultaneous alternate chemistry with other ions present in the atmospheric pressure interface (e.g., air). This provides a clean mass spectrum for each chromatographically well-resolved analyte and is particularly useful when analytes are in a complex mixture (Fig. S3). Consequently, this technique shows a significant advantage for understanding ionization chemistry. Previous work has demonstrated that coupling a GC interface to a NO+ CIMS can determine the products ion distributions for VOCs (Koss et al., 2016). This instrument complements this previous work by examining less volatile and more oxidized compounds, as well as other CIMS chemistries. This technique is particularly interesting in the context of iodide CIMS chemistry, as it allows us to explore ions with positive mass defects (i.e., non-adduct ions), which are not particularly well understood (Lee et al., 2014). We demonstrate the capability of the technique by showing the chromatograms of a complex sample containing a mixture of liquid chemical standards and six commercially available fragrances in Fig. 4. Three known liquid standards introduced in the mixture – undecanoic acid, vanillin, and 1,12-dodecanediol – are observed in iodide ionization mode using their iodide-adduct ion, i.e., [M + I]− (Fig. 4a). However, while this mode is nominally dominated by ionization by the iodide ion (Fig. 4b), other ions are observed in the mass spectrum of these standards, including the deprotonated form of vanillin (i.e., [M − H]−) and its nitrite adduct (i.e., [M + NO2]−), indicating that the iodide ionization mode used in these experiments includes declustering of iodide adducts for low-polarity analytes and/or side reactions. The CIMS voltage settings used in this study were not optimized to minimize declustering of lower-polarity compounds like vanillin, leading to spectra of these compounds in which the iodide adduct significantly is significantly less dominant than the deprotonated form, even in iodide ionization mode. However, it is a benefit of the GC-CIMS approach that a clean mass spectrum is obtained for each analyte within this complex mixture and is therefore a pathway toward better understanding iodide adduct chemistry and co-existing side reactions. Other than the introduced standards, which were selected in part due to their tendency to form iodide adducts, few other major analytes are observed, despite the co-introduction of multiple commercially available fragrances known to contain many organic constituents.
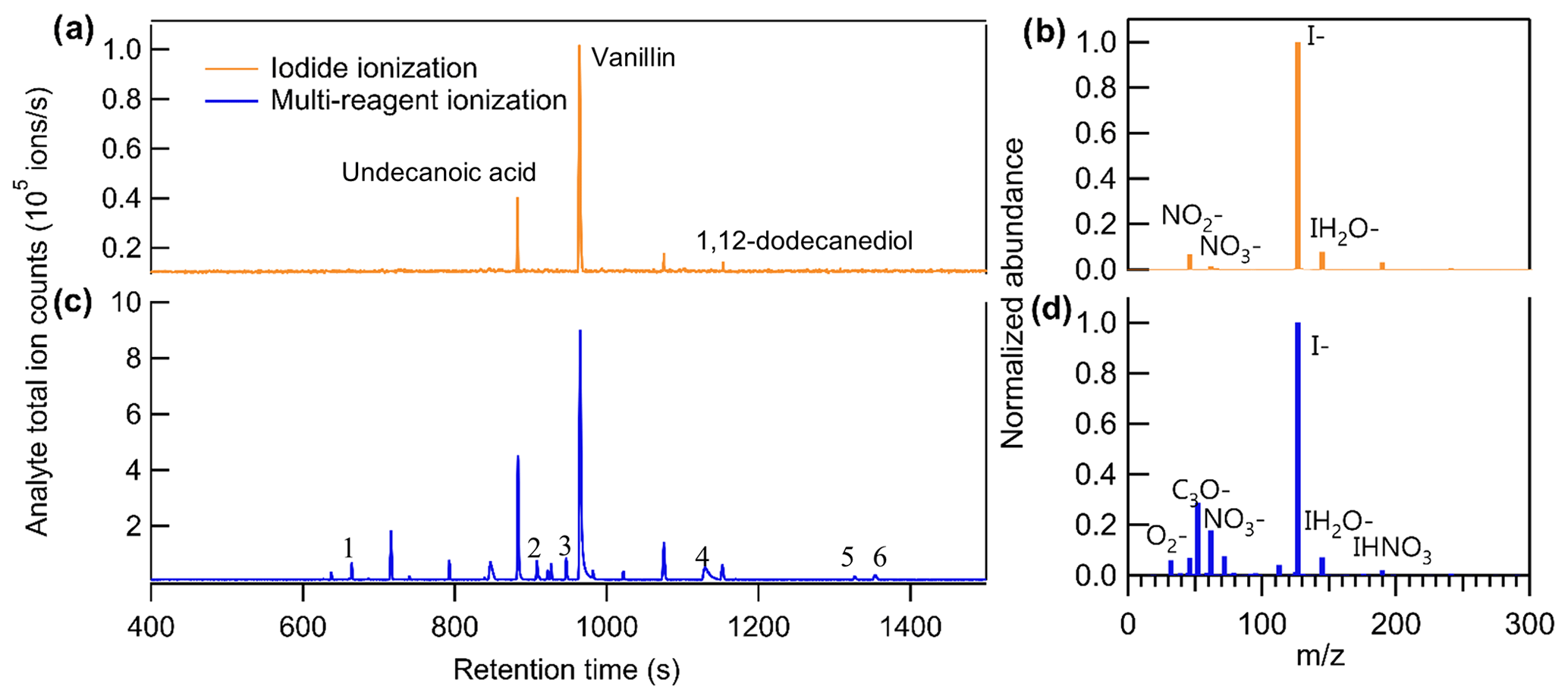
Figure 4Comparison of chromatograms of analyte total ion counts between (a) iodide ionization and (c) multi-reagent ionization. Comparison of background mass spectra between (b) iodide ionization and (d) multi-reagent ionization. The sample introduced in this run is the mixture of liquid chemical standards and six commercially available fragrances.
Using GC-CIMS not only enables the elucidation of different ionization pathways in the CI source and enables separation of interferences in the quantification but might also be useful for exploiting these co-existing chemistries to yield additional information. While chemical ionization intrinsically offers selectivity for ease of analysis, selectivity is also negatively limiting (Munson and Field, 1966). Thus, under certain circumstances it may be useful to use multiple reagent ions to detect different classes of compounds using separate, but still soft, ionization methods. Other ions like the deprotonated form of the analyte, [M − H]− spectra might be better suited for the identification and quantification of some analytes. The deprotonated ions are believed to be produced through the reaction with O present in the IMR (Dzidic et al., 1975; Hunt et al., 1975). It is reported that the presence of O, which is commonly found in atmospheric pressure ion sources such as electrospray ionization (ESI) (Hassan et al., 2017), atmospheric pressure chemical ionization (APCI) (McEwen and Larsen, 2009), atmospheric pressure photoionization (APPI) (Song et al., 2007), and direct analysis in real time (DART) (Cody et al., 2005), may result in the deprotonated molecules through oxidative ionization. Therefore, using multiple reagent ions including I− and O, it is possible that low-polarity compounds tend to be ionized through proton abstraction by O, while compounds with high polarity can still form iodide adducts. Although we did not find carbonate (as CO and CO) in the mass spectrum in this study, we caution that those ions, like O, can also deprotonate molecules and may interfere with the quantification of the deprotonated ions.
To explore the feasibility of purposefully introducing multiple simultaneous chemistries (e.g., deprotonation reactions in addition to iodide-adduct formation) to extend the utility of a CIMS with isomer resolution, the CIMS was operated in a multi-reagent ionization mode by adding 100 sccm flow (i.e., 5 %) of ultra zero air to the 2 slpm flow of N2 for the gas supply of the methyl iodide permeation tube. Figure 4b and d show the background ions under the two modes. With no sample introduced into the system (i.e., pure helium as GC effluent), the total ion counts are 2.4 × 106 and 1.4 × 106 ions s−1 and the I− ion counts are 1.8 × 106 and 0.7 × 106 ions s−1 for iodide and multi-reagent ionization, respectively. In other words, by mixing the reagent ion flow with 5 % air, the I− ion reduced by half, while the abundance of additional reagent ions such as O and NO increased by approximately an order of magnitude.
As shown in Fig. 4c, for compounds that can be detected by iodide ionization, the total number of ions produced by an analyte increased by a factor of 5 to 10 after switching to multi-reagent ionization mode (note that the scale of y axis in Fig. 4c is a factor of 10 higher than that in Fig. 4a). This increase in ions is observed to occur almost entirely through the addition of new chemical pathways. To examine increases in abundance of non-adduct ions in multi-reagent ionization, all identified ions are plotted as a function of their exact mass and mass defect for iodide ionization (Fig. 5a) and multi-reagent ionization (Fig. 5b) with the marker area representing the background-subtracted ion abundance. Analysis is limited to only ions that exhibit a chromatographic peak above the level of detection (taken as 10 times signal-to-noise ratio in the chromatographic baseline) and with ion abundance higher than 1 % of the maximum signal across both systems. The results show that despite slight decreases in their abundance, nearly all of the iodide-adduct ions (green markers within the dashed circle in Fig. 5a) are still present after switching to multi-reagent ionization mode. However, signals of non-iodide adduct ions observed in iodide ionization are enhanced significantly, even for lower-polarity compounds that exhibited non-iodide adduct ionization pathways in iodide ionization mode. Multi-reagent ionization also generates many new non-adduct ions. While shown summarily as mass defect plots, it is important to remember that all ions are not observed simultaneously but rather elute as chromatographic peaks comprised of some subset of ions. Figure 5 consequently demonstrates that by using multi-reagent ionization, identification of compounds with iodide-adduct signals can be maintained, while additional analytes are accessed through these new ionization pathways, as demonstrated by the increase in peaks observed in Fig. 4c. Enhancement of these side reactions expands formula identifications to compounds that do not strongly form iodide adducts in this instrument, due either to inherent chemical limitations (e.g., low polarity) or instrument operating conditions (e.g., adduct declustering). For example, six peaks labeled in Fig. 4c are not detected as iodide adducts but for which formulas can be assigned using [M − H]− and [M + O2]− as identifiers, i.e., 1: C15H24O, 2: C9H10O3, 3: C12H24O2, 4: C16H32O2, 5: C18H34O2, and 6: C18H36O2. The results suggest that the instrument selectivity to other classes of compounds can be enlarged by bringing in O as an additional reagent ion, without significantly suppressing the iodide ionization pathway. In other words, the sensitivity of compounds that tend to be ionized by O or other side reactions are significantly enhanced in multi-ionization CIMS with only minor decreases in the sensitivity of compounds typically observed by an iodide-CIMS. As long as individual analytes enter the CIMS at separate times, as in the case of chromatography, combining multiple ionization chemistries can provide additional information or selectivity.

Figure 5High-resolution mass defect spectrum obtained for liquid mixture samples in (a) iodide ionization mode and (b) multi-reagent ionization mode. The area of markers is proportional to the ion abundance.
A reasonable objection to multi-reagent ionization is that the complexity of adding up signals in multiple ionization chemistry with variable sensitivities may prohibit reasonable CIMS quantification. However, using CIMS for identification of unknowns by formula or other chemical information is valuable on its own, and quantification of many components is achievable using the FID channel of this instrument. This technique is likely only useful when analytes are individually resolved (i.e., isomer resolution), as the resulting mass spectrum of the complete complex mixture would be otherwise too difficult to interpret. We demonstrate here an example of exploring new reagent chemistries: simultaneously using multiple reagent ions is only made possible by the GC separation of analytes but expands the information provided by this instrument. An in-depth understanding of the competition between reagent chemistries in a multi-reagent system is beyond the scope of this article.
We couple a thermal desorption aerosol gas chromatograph with a chemical ionization mass spectrometer as a technique for isomer-resolved analysis of particle-phase organics in the air. The GC column effluent is also split and directed to a FID, which provides a near-universal response to carbon-containing analytes, to calibrate the compounds identified by CIMS. We demonstrate that the TAG-CIMS/FID can measure compounds from liquid injections as well as compounds in SOA generated in an oxidation flow reactor. By coupling a TAG to a CIMS, the CIMS is enhanced with an additional dimension of information (resolution of individual molecules) at the cost of time resolution (i.e., one sample per hour instead of per minute). This trade-off may be valuable in ambient atmospheres, as the number of isomers per formula, although subject to the positive bias during thermal decomposition and the negative bias due to the vapor pressure limit of the GC column, is observed in ambient samples and oxidation experiments to be typically 2 to 5 and as high as 10 to 20, depending on the instrument and the environment.
A key advantage of coupling a TAG to a CIMS is in characterizing the reagent–analyte reactions occurring in the IMR of a CIMS by resolving mass spectra of individual analytes that might not be commercially available. While the iodide-adduct ions do exist in the mass spectrum of individual analytes, we also observe high abundance of non-adduct ions such as [M − H]− and [M + O2]−. Although such high abundance of [M − H]− may be partially resulted by the tuning-driven declustering of low-polarity adduct ions, the observed non-adduct ions likely account for many ions in the non-adduct region of the iodide valley. By separating analytes chromatographically, these non-adduct ions can be used for the identification of some compounds. These non-iodide ionization pathways can be further enhanced by the intentional introduction of multiple reagent ions.
A multi-reagent ionization mode is investigated in which both zero air and iodide are introduced as reagent ions, to examine the feasibility of extending the use of an individual CIMS for detection of a broader range of analytes. While this approach reduces iodide-adduct ions by a factor of 2, [M − H]− and [M + O2]− ions produced from less polar compounds increase by a factor of 5 to 10, improving their detection by CIMS. The method expands the range of chemical species that can be measured by CIMS without losing the advantage of identifying chemical formula using the iodide adducts. This novel multi-reagent approach is made possible by combining GC and CIMS detection together with co-measurements from FID. The advantage of simultaneously measuring FID signals for isomer-resolved quantification of iodide-CIMS sensitivity will be discussed in more detail in a forthcoming paper. Thus, taken together, the GC-CIMS/FID instrument is not only inherently valuable for its resolution of isomers in complex atmospheric samples but also for its ability to characterize and calibrate known CIMS chemistries and to investigate novel and complex chemistries.
All raw and processed data collected as part of this project are available upon request.
The supplement related to this article is available online at: https://doi.org/10.5194/amt-14-3895-2021-supplement.
CB led hardware design, instrumentation, data collection, and data analysis under the guidance of GIVW. GIVW, JEK, BML, and MRC contributed to the development of the theory of the described approach. GIVW, GOF, JEK, JTJ, and DRW contributed to hardware design and instrumentation. JEK, WX, ATL, and MSC contributed to data collection. CB prepared the article with contributions by all authors.
Jordan E. Krechmer, Wen Xu, Andrew T. Lambe, Megan S. Claflin, Brian M. Lerner, John T. Jayne, Douglas R. Worsnop, and Manjula R. Canagaratna are employed by Aerodyne Research Inc., which commercializes TAG and CIMS instruments for geoscience research.
We would like to thank David Worton, Haofei Zhang, Allen Goldstein, Jacqueline Hamilton, and William Dixon for sharing their data, which is shown in Fig. 3.
This research has been supported by the Alfred P. Sloan Foundation (grant no. P-2018-11129).
This paper was edited by Mingjin Tang and reviewed by two anonymous referees.
Aljawhary, D., Lee, A. K. Y., and Abbatt, J. P. D.: High-resolution chemical ionization mass spectrometry (ToF-CIMS): application to study SOA composition and processing, Atmos. Meas. Tech., 6, 3211–3224, https://doi.org/10.5194/amt-6-3211-2013, 2013.
Arangio, A. M., Tong, H., Socorro, J., Pöschl, U., and Shiraiwa, M.: Quantification of environmentally persistent free radicals and reactive oxygen species in atmospheric aerosol particles, Atmos. Chem. Phys., 16, 13105–13119, https://doi.org/10.5194/acp-16-13105-2016, 2016.
Atkinson, R.: Atmospheric chemistry of vocs and nox, Atmos. Environ., 34, 2063–2101, https://doi.org/10.1016/S1352-2310(99)00460-4, 2000.
Atkinson, R. and Arey, J.: Atmospheric degradation of volatile organic compounds, Chem. Rev., 103, 4605–4638, https://doi.org/10.1021/cr0206420, 2003.
Bertram, T. H., Kimmel, J. R., Crisp, T. A., Ryder, O. S., Yatavelli, R. L. N., Thornton, J. A., Cubison, M. J., Gonin, M., and Worsnop, D. R.: A field-deployable, chemical ionization time-of-flight mass spectrometer, Atmos. Meas. Tech., 4, 1471–1479, https://doi.org/10.5194/amt-4-1471-2011, 2011.
Brophy, P. and Farmer, D. K.: Clustering, methodology, and mechanistic insights into acetate chemical ionization using high-resolution time-of-flight mass spectrometry, Atmos. Meas. Tech., 9, 3969–3986, https://doi.org/10.5194/amt-9-3969-2016, 2016.
Bryant, D. J., Dixon, W. J., Hopkins, J. R., Dunmore, R. E., Pereira, K. L., Shaw, M., Squires, F. A., Bannan, T. J., Mehra, A., Worrall, S. D., Bacak, A., Coe, H., Percival, C. J., Whalley, L. K., Heard, D. E., Slater, E. J., Ouyang, B., Cui, T., Surratt, J. D., Liu, D., Shi, Z., Harrison, R., Sun, Y., Xu, W., Lewis, A. C., Lee, J. D., Rickard, A. R., and Hamilton, J. F.: Strong anthropogenic control of secondary organic aerosol formation from isoprene in Beijing, Atmos. Chem. Phys., 20, 7531–7552, https://doi.org/10.5194/acp-20-7531-2020, 2020.
Buchholz, A., Ylisirniö, A., Huang, W., Mohr, C., Canagaratna, M., Worsnop, D. R., Schobesberger, S., and Virtanen, A.: Deconvolution of FIGAERO–CIMS thermal desorption profiles using positive matrix factorisation to identify chemical and physical processes during particle evaporation, Atmos. Chem. Phys., 20, 7693–7716, https://doi.org/10.5194/acp-20-7693-2020, 2020.
Burnett, R. T., Pope, C. A., Ezzati, M., Olives, C., Lim, S. S., Mehta, S., Shin, H. H., Singh, G., Hubbell, B., Brauer, M., Anderson, H. R., Smith, K. R., Balmes, J. R., Bruce, N. G., Kan, H., Laden, F., Prüss-Ustün, A., Turner, M. C., Gapstur, S. M., Diver, W. R., and Cohen, A.: An integrated risk function for estimating the global burden of disease attributable to ambient fine particulate matter exposure, Environ. Health Perspect., 122, 397–403, https://doi.org/10.1289/ehp.1307049, 2014.
Claflin, M. S. and Ziemann, P. J.: Thermal desorption behavior of hemiacetal, acetal, ether, and ester oligomers, Aerosol Sci. Technol., 53, 473–484, https://doi.org/10.1080/02786826.2019.1576853, 2019.
Cody, R. B., Laramée, J. A., and Durst, H. D.: Versatile new ion source for the analysis of materials in open air under ambient conditions, Anal. Chem., 77, 2297–2302, https://doi.org/10.1021/ac050162j, 2005.
Dixon, W.: Quantitative analysis of secondary organic aerosol using a high-resolution mass spectral database, PhD dissertation, University of York, York, UK, 2020.
Dzidic, I., Carroll, D. I., Stillwell, R. N., and Horning, E. C.: Atmospheric pressure ionization (api) mass spectrometry: formation of phenoxide ions from chlorinated aromatic compounds, Anal. Chem., 47, 1308–1312, https://doi.org/10.1021/ac60358a077, 1975.
Ehn, M., Junninen, H., Petäjä, T., Kurtén, T., Kerminen, V.-M., Schobesberger, S., Manninen, H. E., Ortega, I. K., Vehkamäki, H., Kulmala, M., and Worsnop, D. R.: Composition and temporal behavior of ambient ions in the boreal forest, Atmos. Chem. Phys., 10, 8513–8530, https://doi.org/10.5194/acp-10-8513-2010, 2010.
Goldan, P. D., Kuster, W. C., Williams, E., Murphy, P. C., Fehsenfeld, F. C., and Meagher, J.: Nonmethane hydrocarbon and oxy hydrocarbon measurements during the 2002 new england air quality study, J. Geophys. Res.-Atmos., 109, 1–14, https://doi.org/10.1029/2003JD004455, 2004.
Goldstein, A. H. and Galbally, I. E.: Known and unexplored organic constituents in the earth's atmosphere, Environ. Sci. Technol., 41, 1514–1521, https://doi.org/10.1021/es072476p, 2007.
Goldstein, A. H., Daube, B. C., Munger, J. W., and Wofsy, S. C.: Automated in-situ monitoring of atmospheric non-methane hydrocarbon concentrations and gradients, J. Atmos. Chem., 21, 43–59, https://doi.org/10.1007/BF00712437, 1995.
Gómez-González, Y., Surratt, J. D., Cuyckens, F., Szmigielski, R., Vermeylen, R., Jaoui, M., Lewandowski, M., Offenberg, J. H., Kleindienst, T. E., Edney, E. O., Blockhuys, F., Van Alsenoy, C., Maenhaut, W., and Claeys, M.: Characterization of organosulfates from the photooxidation of isoprene and unsaturated fatty acids in ambient aerosol using liquid chromatography/(-) electrospray ionization mass spectrometry, J. Mass Spectrom., 43, 371–382, https://doi.org/10.1002/jms.1329, 2008.
Grob, R. L. and Barry, E. F. Modern practice of gas chromatography, 4th edition, John Wiley & Sons, Inc., https://doi.org/10.1002/0471651141, ISBN 978-0-471-22983-4, 2004.
Hamilton, J. F.: Using comprehensive two-dimensional gas chromatography to study the atmosphere, J. Chromatogr. Sci., 48, 274–282, https://doi.org/10.1093/chromsci/48.4.274, 2010.
Hassan, I., Pavlov, J., Errabelli, R., and Attygalle, A. B.: Oxidative ionization under certain negative-ion mass spectrometric conditions, J. Am. Soc. Mass Spectrom., 28, 270–277, https://doi.org/10.1007/s13361-016-1527-5, 2017.
Helmig, D., Ortega, J., Duhl, T., Tanner, D., Guenther, A., Harley, P., Wiedinmyer, C., Milford, J., and Sakulyanontvittaya, T.: Sesquiterpene emissions from pine trees – identifications, emission rates and flux estimates for the contiguous united states, Environ. Sci. Technol., 41, 1545–1553, https://doi.org/10.1021/es0618907, 2007.
Hunt, D., Harvey, M., and Russell, J.: Oxygen as a reagent gas for the analysis of 2,3,7,8-tetrachlorodibenzo-p-dioxin by negative ion chemical ionization mass spectrometry, J. Chem. Soc. Chem. Comm., 151–152, 1975.
Hurley, J. F., Kreisberg, N. M., Stump, B., Bi, C., Kumar, P., Hering, S. V., Keady, P., and Isaacman-VanWertz, G.: A new approach for measuring the carbon and oxygen content of atmospherically relevant compounds and mixtures, Atmos. Meas. Tech., 13, 4911–4925, https://doi.org/10.5194/amt-13-4911-2020, 2020.
Isaacman, G., Kreisberg, N. M., Worton, D. R., Hering, S. V., and Goldstein, A. H.: A versatile and reproducible automatic injection system for liquid standard introduction: application to in-situ calibration, Atmos. Meas. Tech., 4, 1937–1942, https://doi.org/10.5194/amt-4-1937-2011, 2011.
Isaacman, G., Kreisberg, N. M., Yee, L. D., Worton, D. R., Chan, A. W. H., Moss, J. A., Hering, S. V., and Goldstein, A. H.: Online derivatization for hourly measurements of gas- and particle-phase semi-volatile oxygenated organic compounds by thermal desorption aerosol gas chromatography (SV-TAG), Atmos. Meas. Tech., 7, 4417–4429, https://doi.org/10.5194/amt-7-4417-2014, 2014.
Isaacman-VanWertz, G. and Aumont, B.: Impact of organic molecular structure on the estimation of atmospherically relevant physicochemical parameters, Atmos. Chem. Phys., 21, 6541–6563, https://doi.org/10.5194/acp-21-6541-2021, 2021.
Isaacman-VanWertz, G., Yee, L. D., Kreisberg, N. M., Wernis, R., Moss, J. A., Hering, S. V., De Sá, S. S., Martin, S. T., Alexander, M. L., Palm, B. B., Hu, W., Campuzano-Jost, P., Day, D. A., Jimenez, J. L., Riva, M., Surratt, J. D., Viegas, J., Manzi, A., Edgerton, E., Baumann, K., Souza, R., Artaxo, P., and Goldstein, A. H.: Ambient gas-particle partitioning of tracers for biogenic oxidation, Environ. Sci. Technol., 50, 9952–9962, https://doi.org/10.1021/acs.est.6b01674, 2016.
Isaacman-VanWertz, G., Sueper, D. T., Aikin, K. C., Lerner, B. M., Gilman, J. B., de Gouw, J. A., Worsnop, D. R., and Goldstein, A. H.: Automated single-ion peak fitting as an efficient approach for analyzing complex chromatographic data, J. Chromatogr. A, 1529, 81–92, https://doi.org/10.1016/j.chroma.2017.11.005, 2017.
Isaacman-Vanwertz, G., Massoli, P., O'Brien, R., Lim, C., Franklin, J. P., Moss, J. A., Hunter, J. F., Nowak, J. B., Canagaratna, M. R., Misztal, P. K., Arata, C., Roscioli, J. R., Herndon, S. T., Onasch, T. B., Lambe, A. T., Jayne, J. T., Su, L., Knopf, D. A., Goldstein, A. H., Worsnop, D. R., and Kroll, J. H.: Chemical evolution of atmospheric organic carbon over multiple generations of oxidation, Nat. Chem., 10, 462–468, https://doi.org/10.1038/s41557-018-0002-2, 2018.
Iyer, S., Lopez-Hilfiker, F., Lee, B. H., Thornton, J. A., and Kurtén, T.: Modeling the detection of organic and inorganic compounds using iodide-based chemical ionization, J. Phys. Chem. A, 120, 576–587, https://doi.org/10.1021/acs.jpca.5b09837, 2016.
Jimenez, J. L., Canagaratna, M. R., Donahue, N. M., Prevot, A. S. H., Zhang, Q., Kroll, J. H., DeCarlo, P. F., Allan, J. D., Coe, H., Ng, N. L., Aiken, A. C., Docherty, K. S., Ulbrich, I. M., Grieshop, A. P., Robinson, A. L., Duplissy, J., Smith, J. D., Wilson, K. R., Lanz, V. A., Hueglin, C., Sun, Y. L., Tian, J., Laaksonen, A., Raatikainen, T., Rautiainen, J., Vaattovaara, P., Ehn, M., Kulmala, M., Tomlinson, J. M., Collins, D. R., Cubison, M. J., Dunlea, E. J., Huffman, J. A., Onasch, T. B., Alfarra, M. R., Williams, P. I., Bower, K., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Salcedo, D., Cottrell, L., Griffin, R., Takami, A., Miyoshi, T., Hatakeyama, S., Shimono, A., Sun, J. Y., Zhang, Y. M., Dzepina, K., Kimmel, J. R., Sueper, D., Jayne, J. T., Herndon, S. C., Trimborn, A. M., Williams, L. R., Wood, E. C., Middlebrook, A. M., Kolb, C. E., Baltensperger, U., and Worsnop, D. R.: Evolution of organic aerosols in the atmosphere, Science, 326, 1525–1529, https://doi.org/10.1126/science.1180353, 2009.
Kolb, B., Auer, M., and Pospisil, P.: Ionization detector for gas chromatography with switchable selectivity for carbon, nitrogen and phosphorus, J. Chromatogr. A, 134, 65–71, https://doi.org/10.1016/S0021-9673(00)82570-4, 1977.
Koss, A. R., Warneke, C., Yuan, B., Coggon, M. M., Veres, P. R., and de Gouw, J. A.: Evaluation of NO+ reagent ion chemistry for online measurements of atmospheric volatile organic compounds, Atmos. Meas. Tech., 9, 2909–2925, https://doi.org/10.5194/amt-9-2909-2016, 2016.
Krechmer, J. E., Coggon, M. M., Massoli, P., Nguyen, T. B., Crounse, J. D., Hu, W., Day, D. A., Tyndall, G. S., Henze, D. K., Rivera-Rios, J. C., Nowak, J. B., Kimmel, J. R., Mauldin, R. L., Stark, H., Jayne, J. T., Sipilä, M., Junninen, H., St. Clair, J. M., Zhang, X., Feiner, P. A., Zhang, L., Miller, D. O., Brune, W. H., Keutsch, F. N., Wennberg, P. O., Seinfeld, J. H., Worsnop, D. R., Jimenez, J. L., and Canagaratna, M. R.: Formation of low volatility organic compounds and secondary organic aerosol from isoprene hydroxyhydroperoxide low-no oxidation, Environ. Sci. Technol., 49, 10330–10339, https://doi.org/10.1021/acs.est.5b02031, 2015.
Krechmer, J. E., Pagonis, D., Ziemann, P. J., and Jimenez, J. L.: Quantification of gas-wall partitioning in teflon environmental chambers using rapid bursts of low-volatility oxidized species generated in situ, Environ. Sci. Technol., 50, 5757–5765, https://doi.org/10.1021/acs.est.6b00606, 2016.
Kreisberg, N. M., Worton, D. R., Zhao, Y., Isaacman, G., Goldstein, A. H., and Hering, S. V.: Development of an automated high-temperature valveless injection system for online gas chromatography, Atmos. Meas. Tech., 7, 4431–4444, https://doi.org/10.5194/amt-7-4431-2014, 2014.
Kroll, J. H. and Seinfeld, J. H.: Chemistry of secondary organic aerosol: formation and evolution of low-volatility organics in the atmosphere, Atmos. Environ., 42, 3593–3624, https://doi.org/10.1016/j.atmosenv.2008.01.003, 2008.
Lambe, A. T., Onasch, T. B., Massoli, P., Croasdale, D. R., Wright, J. P., Ahern, A. T., Williams, L. R., Worsnop, D. R., Brune, W. H., and Davidovits, P.: Laboratory studies of the chemical composition and cloud condensation nuclei (CCN) activity of secondary organic aerosol (SOA) and oxidized primary organic aerosol (OPOA), Atmos. Chem. Phys., 11, 8913–8928, https://doi.org/10.5194/acp-11-8913-2011, 2011.
Lee, B. H., Lopez-Hilfiker, F. D., Mohr, C., Kurtén, T., Worsnop, D. R., and Thornton, J. A.: An iodide-adduct high-resolution time-of-flight chemical-ionization mass spectrometer: application to atmospheric inorganic and organic compounds, Environ. Sci. Technol., 48, 6309–6317, https://doi.org/10.1021/es500362a, 2014.
Lim, Y. B. and Ziemann, P. J.: Effects of molecular structure on aerosol yields from oh radical-initiated reactions of linear, branched, and cyclic alkanes in the presence of no x, Environ. Sci. Technol., 43, 2328–2334, https://doi.org/10.1021/es803389s, 2009.
Lopez-Hilfiker, F. D., Mohr, C., Ehn, M., Rubach, F., Kleist, E., Wildt, J., Mentel, Th. F., Lutz, A., Hallquist, M., Worsnop, D., and Thornton, J. A.: A novel method for online analysis of gas and particle composition: description and evaluation of a Filter Inlet for Gases and AEROsols (FIGAERO), Atmos. Meas. Tech., 7, 983–1001, https://doi.org/10.5194/amt-7-983-2014, 2014.
Lopez-Hilfiker, F. D., Iyer, S., Mohr, C., Lee, B. H., D'Ambro, E. L., Kurtén, T., and Thornton, J. A.: Constraining the sensitivity of iodide adduct chemical ionization mass spectrometry to multifunctional organic molecules using the collision limit and thermodynamic stability of iodide ion adducts, Atmos. Meas. Tech., 9, 1505–1512, https://doi.org/10.5194/amt-9-1505-2016, 2016a.
Lopez-Hilfiker, F. D., Mohr, C., D'Ambro, E. L., Lutz, A., Riedel, T. P., Gaston, C. J., Iyer, S., Zhang, Z., Gold, A., Surratt, J. D., Lee, B. H., Kurten, T., Hu, W. W., Jimenez, J., Hallquist, M., and Thornton, J. A.: Molecular composition and volatility of organic aerosol in the southeastern u.s.: implications for iepox derived soa, Environ. Sci. Technol., 50, 2200–2209, https://doi.org/10.1021/acs.est.5b04769, 2016b.
McEwen, C. N. and Larsen, B. S.: Ionization mechanisms related to negative ion appi, apci, and dart, J. Am. Soc. Mass Spectrom., 20, 1518–1521, https://doi.org/10.1016/j.jasms.2009.04.010, 2009.
Mielke, L. H. and Osthoff, H. D.: On quantitative measurements of peroxycarboxylic nitric anhydride mixing ratios by thermal dissociation chemical ionization mass spectrometry, Int. J. Mass Spectrom., 310, 1–9, https://doi.org/10.1016/j.ijms.2011.10.005, 2012.
Millet, D. B.: Atmospheric volatile organic compound measurements during the pittsburgh air quality study: results, interpretation, and quantification of primary and secondary contributions, J. Geophys. Res., 110, D07S07, https://doi.org/10.1029/2004JD004601, 2005.
Munson, M. S. B. and Field, F. H.: Chemical ionization mass spectrometry. i. general introduction, J. Am. Chem. Soc., 88, 2621–2630, https://doi.org/10.1021/ja00964a001, 1966.
Nozière, B., Kalberer, M., Claeys, M., Allan, J., D'Anna, B., Decesari, S., Finessi, E., Glasius, M., Grgić, I., Hamilton, J. F., Hoffmann, T., Iinuma, Y., Jaoui, M., Kahnt, A., Kampf, C. J., Kourtchev, I., Maenhaut, W., Marsden, N., Saarikoski, S., Schnelle-Kreis, J., Surratt, J. D., Szidat, S., Szmigielski, R., and Wisthaler, A.: The molecular identification of organic compounds in the atmosphere: state of the art and challenges, Chem. Rev., 115, 3919–3983, https://doi.org/10.1021/cr5003485, 2015.
Pope, C. A. and Dockery, D. W.: Health effects of fine particulate air pollution: lines that connect, J. Air Waste Manag. Assoc., 56, 709–742, https://doi.org/10.1080/10473289.2006.10464485, 2006.
Prinn, R. G., Weiss, R. F., Simmonds, P. J. F. P. G., Cunnold, D. M., Alyea, F. N., Doherty, S. O., Salameh, P., Miller, B. R., Huang, J., Wang, R. H. J., Hartley, D. E., Harth, C., Steele, L. P., Sturrock, G., Midgley, P. M., and Mcculloch, A.: A history of chemically and radiatively important gases in air deduced from ale/gage/agage, J. Geophys. Res., 105, 17751–17792, 2000.
Riva, M., Heikkinen, L., Bell, D. M., Peräkylä, O., Zha, Q., Schallhart, S., Rissanen, M. P., Imre, D., Petäjä, T., Thornton, J. A., Zelenyuk, A., and Ehn, M.: Chemical transformations in monoterpene-derived organic aerosol enhanced by inorganic composition, npj Clim. Atmos. Sci., 2, 1–9, https://doi.org/10.1038/s41612-018-0058-0, 2019.
Scanlon, J. T. and Willis, D. E.: Calculation of flame ionization detector relative response factors using the effective carbon number concept, J. Chromatogr. Sci., 23, 333–340, 1985.
Schauer, J. J., Rogge, W. F., Hildemann, L. M., Mazurek, M. A., Cass, G. R., and Simoneit, B. R. T.: Source apportionment of airborne particulate matter using organic compounds as tracers, Atmos. Environ., 30, 3837–3855, https://doi.org/10.1016/1352-2310(96)00085-4, 1996.
Seinfeld, J. H. and Pandis, S. N.: Atmospheric chemistry and physics: from air pollution to climate change, 3rd edition, available from: https://www.wiley.com/en-us/Atmospheric+Chemistry+and+Physics:+From+Air+Pollution+to+Climate+Change,+3rd+Edition-p-9781118947401 (last access: 11 December 2019), 2016.
Song, L., Wellman, A. D., Yao, H., and Bartmess, J. E.: Negative ion-atmospheric pressure photoionization: electron capture, dissociative electron capture, proton transfer, and anion attachment, J. Am. Soc. Mass Spectrom., 18, 1789–1798, https://doi.org/10.1016/j.jasms.2007.07.015, 2007.
Stark, H., Yatavelli, R. L. N., Thompson, S. L., Kang, H., Krechmer, J. E., Kimmel, J. R., Palm, B. B., Hu, W., Hayes, P. L., Day, D. A., Campuzano-Jost, P., Canagaratna, M. R., Jayne, J. T., Worsnop, D. R., and Jimenez, J. L.: Impact of thermal decomposition on thermal desorption instruments: advantage of thermogram analysis for quantifying volatility distributions of organic species, Environ. Sci. Technol., 51, 8491–8500, https://doi.org/10.1021/acs.est.7b00160, 2017.
Thompson, S. L., Yatavelli, R. L. N., Stark, H., Kimmel, J. R., Krechmer, J. E., Day, D. A., Hu, W., Isaacman-VanWertz, G., Yee, L., Goldstein, A. H., Khan, M. A. H., Holzinger, R., Kreisberg, N., Lopez-Hilfiker, F. D., Mohr, C., Thornton, J. A., Jayne, J. T., Canagaratna, M., Worsnop, D. R., and Jimenez, J. L.: Field intercomparison of the gas/particle partitioning of oxygenated organics during the southern oxidant and aerosol study (soas) in 2013, Aerosol Sci. Technol., 51, 30–56, https://doi.org/10.1080/02786826.2016.1254719, 2017.
VandenBoer, T. C., Markovic, M. Z., Petroff, A., Czar, M. F., Borduas, N., and Murphy, J. G.: Ion chromatographic separation and quantitation of alkyl methylamines and ethylamines in atmospheric gas and particulate matter using preconcentration and suppressed conductivity detection, J. Chromatogr. A, 1252, 74–83, https://doi.org/10.1016/j.chroma.2012.06.062, 2012.
Vasquez, K. T., Allen, H. M., Crounse, J. D., Praske, E., Xu, L., Noelscher, A. C., and Wennberg, P. O.: Low-pressure gas chromatography with chemical ionization mass spectrometry for quantification of multifunctional organic compounds in the atmosphere, Atmos. Meas. Tech., 11, 6815–6832, https://doi.org/10.5194/amt-11-6815-2018, 2018.
Veres, P., Roberts, J. M., Warneke, C., Welsh-Bon, D., Zahniser, M., Herndon, S., Fall, R., and de Gouw, J.: Development of negative-ion proton-transfer chemical-ionization mass spectrometry (ni-pt-cims) for the measurement of gas-phase organic acids in the atmosphere, Int. J. Mass Spectrom., 274, 48–55, https://doi.org/10.1016/j.ijms.2008.04.032, 2008.
Williams, B., Goldstein, A., Kreisberg, N., and Hering, S.: An in-situ instrument for speciated organic composition of atmospheric aerosols: thermal desorption aerosol gc/ms-fid (tag), Aerosol Sci. Technol., 40, 627–638, https://doi.org/10.1080/02786820600754631, 2006.
Worton, D. R., Decker, M., Isaacman-VanWertz, G., Chan, A. W. H., Wilson, K. R., and Goldstein, A. H.: Improved molecular level identification of organic compounds using comprehensive two-dimensional chromatography, dual ionization energies and high resolution mass spectrometry, Analyst, 142, 2395–2403, https://doi.org/10.1039/c7an00625j, 2017.
Zhang, H., Yee, L. D., Lee, B. H., Curtis, M. P., Worton, D. R., Isaacman-VanWertz, G., Offenberg, J. H., Lewandowski, M., Kleindienst, T. E., Beaver, M. R., Holder, A. L., Lonneman, W. A., Docherty, K. S., Jaoui, M., Pye, H. O. T., Hu, W., Day, D. A., Campuzano-Jost, P., Jimenez, J. L., Guo, H., Weber, R. J., De Gouw, J., Koss, A. R., Edgerton, E. S., Brune, W., Mohr, C., Lopez-Hilfiker, F. D., Lutz, A., Kreisberg, N. M., Spielman, S. R., Hering, S. V., Wilson, K. R., Thornton, J. A., and Goldstein, A. H.: Monoterpenes are the largest source of summertime organic aerosol in the southeastern united states, Proc. Natl. Acad. Sci. USA, 115, 2038–2043, https://doi.org/10.1073/pnas.1717513115, 2018.
Zhao, Y., Kreisberg, N. M., Worton, D. R., Teng, A. P., Hering, S. V., and Goldstein, A. H.: Development of an in situ thermal desorption gas chromatography instrument for quantifying atmospheric semi-volatile organic compounds, Aerosol Sci. Technol., 47, 258–266, https://doi.org/10.1080/02786826.2012.747673, 2013.