the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 07 Jun 2021
| 07 Jun 2021
Measurement of iodine species and sulfuric acid using bromide chemical ionization mass spectrometers
Mingyi Wang
Henning Finkenzeller
Siddharth Iyer
Dexian Chen
Jiali Shen
Mario Simon
Victoria Hofbauer
Jasper Kirkby
Joachim Curtius
Norbert Maier
Theo Kurtén
Douglas R. Worsnop
Markku Kulmala
Matti Rissanen
Rainer Volkamer
Neil M. Donahue
Mikko Sipilä
Iodine species are important in the marine atmosphere for oxidation and new-particle formation. Understanding iodine chemistry and iodine new-particle formation requires high time resolution, high sensitivity, and simultaneous measurements of many iodine species. Here, we describe the application of a bromide chemical ionization mass spectrometer (Br-CIMS) to this task. During the iodine oxidation experiments in the Cosmics Leaving OUtdoor Droplets (CLOUD) chamber, we have measured gas-phase iodine species and sulfuric acid using two Br-CIMS, one coupled to a Multi-scheme chemical IONization inlet (Br-MION-CIMS) and the other to a Filter Inlet for Gasses and AEROsols inlet (Br-FIGAERO-CIMS). From offline calibrations and intercomparisons with other instruments, we have quantified the sensitivities of the Br-MION-CIMS to HOI, I2, and H2SO4 and obtained detection limits of 5.8 × 106, 3.8 × 105, and 2.0 × 105 molec. cm−3, respectively, for a 2 min integration time. From binding energy calculations, we estimate the detection limit for HIO3 to be 1.2 × 105 molec. cm−3, based on an assumption of maximum sensitivity. Detection limits in the Br-FIGAERO-CIMS are around 1 order of magnitude higher than those in the Br-MION-CIMS; for example, the detection limits for HOI and HIO3 are 3.3 × 107 and 5.1 × 106 molec. cm−3, respectively. Our comparisons of the performance of the MION inlet and the FIGAERO inlet show that bromide chemical ionization mass spectrometers using either atmospheric pressure or reduced pressure interfaces are well-matched to measuring iodine species and sulfuric acid in marine environments.
- Article
(1897 KB) - Full-text XML
-
Supplement
(57 KB) - BibTeX
- EndNote





Reactive iodine species are released into the atmosphere mainly by biological processes in marine environments (i.e., from macro- and micro-algae) (McFiggans et al., 2004), O3 deposition on the sea surface (Carpenter et al., 2013), and from the sea ice (Spolaor et al., 2013) and snowpack in the polar region (Raso et al., 2017). Once emitted, iodine species can modify atmospheric oxidative capacity via a chain of catalytic reactions with O3 that form iodine oxides, leading to about 20 %–28 % of O3 loss in the marine boundary layer (Prados-Roman et al., 2015; Sherwen et al., 2016). Through convection, reactive iodine species can be transported from the lower troposphere to the upper troposphere–lower stratosphere, causing one third of the iodine-induced ozone loss in the upper troposphere–lower stratosphere (Koenig et al., 2020). Another important effect of iodine species is their contribution to atmospheric new-particle formation. O'Dowd et al. (2002) showed that particles are produced from condensable iodine-containing vapors at a coastal location (Mace Head in Ireland). Recent studies have demonstrated that iodine oxoacids (iodous acid, HIO2, and iodic acid, HIO3) dominate the iodine cluster formation processes (He et al., 2021a, b) and drive the bursts of freshly formed particles in coastal regions (Sipilä et al., 2016). This process thereby may enhance cloud condensation nuclei formation, affecting climate both directly and indirectly (Saiz-Lopez et al., 2012; Simpson et al., 2015).
Understanding iodine chemistry and iodine-driven new-particle formation requires high time resolution, high sensitivity, and simultaneous measurements of iodine species. However, this has been a long-standing challenge due to their low abundance and short atmospheric lifetimes. Previous studies have achieved detection of relatively more abundant molecular iodine (I2), iodine monoxide (IO), and iodine dioxide (OIO) via optical spectroscopy, such as differential optical absorption (Leigh et al., 2010), cavity ring-down (Bitter et al., 2005), cavity-enhanced absorption (Vaughan et al., 2008), laser-induced fluorescence (Dillon et al., 2006), and resonance fluorescence (Gómez Martín et al., 2011). The spectroscopic techniques are invaluable; however, their very specificity limits them to the detection of a few iodine compounds, and they are less sensitive to other iodine species that have congested or broad absorption features, such as hypoiodous acid (HOI) and iodic acid (HIO3).
Another commonly used technique is a mass spectrometer; it has a fast response time and a low detection limit, but extra calibration efforts are needed for the quantification of the detection sensitivity. For example, photoionization (Gómez Martín et al., 2013) and a chemical ionization mass spectrometer (CIMS) have been employed to detect a suite of halogen species. Reagent ions used with CIMS include the following: SF for HCl and ClONO2 (Marcy et al., 2004); iodide (I−) for atmospheric chlorine and bromine species such as ClNO2, Cl2, ClO, BrO, and BrCl (Kercher et al., 2009; Lee et al., 2018; Tham et al., 2016); superoxide (O) for molecular iodine (I2) (Finley and Saltzman, 2008); and both nitrate (NO) (Sipilä et al., 2016) and protonated water (H3O+) (Pfeifer et al., 2020) for HIO3. The nitrate-CIMS and H3O+-CIMS suffer from the limited analyte affinity to the reagent ions. The iodide-CIMS can effectively measure chlorine and bromine species, but it is not suitable to detect iodine species due to the ambiguity in peak identification.
The bromide ion (Br−) exhibits an affinity to a wide spectrum of iodine containing species. A bromide chemical ionization mass spectrometer (Br-CIMS) has been routinely used to measure chlorine species (Lawler et al., 2011), HO2 radicals (Sanchez et al., 2016), organic vapors and sulfuric acid (Rissanen et al., 2019), and nitric acid (Wang et al., 2020). Like chlorine species, iodine species are known to cluster with bromide ions via halogen (or hydrogen) bonds; as such, here we explore using the Br-CIMS to measure gas-phase iodine species and sulfuric acid simultaneously at concentrations relevant to the marine boundary layer. In this study, we demonstrate the detection of various gas-phase inorganic iodine species with the Br-CIMS and explore the effect of relative humidity (RH) on that detection. We then quantify the sensitivities of several gas-phase iodine species via inter-method calibration, offline calibration, and quantum chemical calculations. Finally, we compare the performance of Br-CIMS coupled to a Multi-scheme chemical IONization inlet (Br-MION-CIMS) and Br-CIMS coupled to a Filter Inlet for Gasses and AEROsols inlet (Br-FIGAERO-CIMS) and show that both of them are well-suited for iodine species measurement in the marine boundary layer.
2.1 The CLOUD facility
We conducted measurements and instrument intercomparison at the CERN Cosmics Leaving OUtdoor Droplets (CLOUD) facility, a 26.1 m3 electropolished stainless-steel chamber that enables new-particle formation experiments simulating the typical range of tropospheric conditions with scrupulous cleanliness and minimal contamination (Duplissy et al., 2016; Kirkby et al., 2011). The CLOUD chamber is mounted in a thermal housing, capable of keeping temperature constant in a range of −65 ∘C and +100 ∘C with ±0.1 ∘C precision (Dias et al., 2017) and relative humidity commonly between <0.5 % and 80 %. Photochemical processes are driven by different light sources, including four 200 W Hamamatsu Hg-Xe lamps with significant spectral irradiance between 250 and 450 nm and an array of 48 green light emitting diodes (LEDs) at 528 nm with adjustable optical power up to 153 W. Ion-induced nucleation under different ionization levels is simulated with a combination of electric fields (electrodes at ±30 kV at top and bottom of the chamber) which can be turned on to rapidly scavenge smaller ions, and a high-flux beam of 3.6 GeV pions (π+) which enhances ion production when turned on. Mixing is accelerated with magnetically coupled fans mounted at the top and bottom of the chamber. The characteristic gas mixing time in the chamber during experiments is a few minutes. The loss rate of condensable vapors onto the chamber wall is comparable to the condensation sink in pristine boundary layer environments (e.g., 2.2 × 10−3 s−1 for H2SO4 at 5 ∘C). To avoid a memory effect between different experiments, the chamber is periodically cleaned by rinsing the walls with ultra-pure water and heating to 100 ∘C for at least 24 h, ensuring extremely low contaminant levels of sulfuric acid (<5 × 104 cm−3) and total organics (<150 pptv, parts per trillion by volume) (Kirkby et al., 2016; Schnitzhofer et al., 2014). The CLOUD gas system is also built to the highest technical standards of cleanliness and performance.
The dry air supply for the chamber is provided by cryogenic nitrogen (Messer, 99.999 %) and cryogenic oxygen (Messer, 99.999 %) mixed at the atmospheric ratio of 79:21. Ultrapure water vapor, ozone, and other trace gases can be precisely added to attain desired mixing ratios at different levels. The total injection rate of the humidified air is fixed at 330 standard liters per minute (slpm) to compensate for the sampling consumption of various instruments. Molecular iodine (I2) is injected into the chamber by passing a flow of cryogenic nitrogen through a crystalline iodine (I2, Sigma-Aldrich, 99.999 % purity) reservoir, which is temperature-controlled at 10 ∘C, to achieve levels of 0.4 to 168 pptv in the chamber. The sulfinert-coated injection lines are temperature stabilized to minimize line conditioning effects. High intensity green light emitting diodes (LEDs) are used to photolyze molecular iodine into iodine atoms and initiate the subsequent oxidation reactions in the presence of ozone and water vapor. The 48 green LEDs (light sabre 4, LS4) are mounted in pairs (one upward facing, one downward facing) on a copper cooling bar within a long quartz jacket that protrudes into the chamber in the midplane. The maximum total optical power output is 153 W, centered on 528 nm. Actinic fluxes are regulated by controlling the number of LEDs used and the set point of individual LEDs. Light fluxes are monitored by two photodiodes and a spectrometer.
2.2 Br-MION-CIMS
We measured gas-phase iodine species with a bromide chemical ionization atmospheric pressure interface time-of-flight mass spectrometer (Junninen et al., 2010) coupled with a Multi-scheme chemical IONization inlet (Br-MION-CIMS) (Rissanen et al., 2019). The Br-MION inlet consists of an electrically grounded 24 mm inner diameter stainless steel flow tube attached to an ion source. For the CLOUD measurements, the length of the sampling inlet was ∼1.5 m and was designed to be in a laminar flow with a fixed total flow rate of 32 standard liters per minute (slpm). An ion filter, operated with positive and negative voltage, was placed at the front of the inlet to filter out any ions in the sample air prior to ion-molecule reaction chamber in the inlet. The reagent ions, bromide (Br−) and the bromide–water cluster (H2O ⋅ Br−), were produced by feeding 25 standard milliliters per minute (mlpm) of nitrogen (N2) flow through a saturator containing dibromomethane (CH2Br2; >99.0 %, Tokyo Chemical Industry) into the ion source, where the reagent was ionized by soft X-ray radiation. The resulting ions were then accelerated by a −2800 V ion accelerator array and focused by a −290 V ion deflector into the laminar sampling flow of the inlet via a 5 mm orifice. A small counter flow (∼40 mlpm) was applied through the orifice to prevent any mixing of the electrically neutral reagent vapor with the sampling flow. The details of the chemical ionization method, inlet design, setup, and operation have been described previously (He, 2017; Rissanen et al., 2019).
2.3 Br-FIGAERO-CIMS
We also measured both the gas- and particle-phase compositions via thermal desorption using a bromide chemical ionization time-of-flight mass spectrometer equipped with a Filter Inlet for Gases and AEROsols (Br-FIGAERO-CIMS) (Lopez-Hilfiker et al., 2014). FIGAERO is a manifold inlet for a CIMS with two operating modes. In the sampling mode, gases are directly sampled into a 150 mbar ion-molecule reactor using coaxial core sampling to minimize their wall losses in the sampling line. The total flow is maintained at 18.0 slpm and the core flow at 4.5 slpm; the CIMS samples at the center of the core flow with a flow rate at ∼1.6 slpm. Concurrently, particles are collected on a PTFE filter via a separate dedicated port with a flow rate of 6 slpm. In the desorption mode, the filter is automatically moved into a pure N2 gas stream flowing into the ion-molecule reactor, while the N2 is progressively heated upstream of the filter to evaporate the particles via temperature programmed desorption. Analytes are then chemically ionized by Br− and extracted into a mass spectrometer.
We optimize the adduct-ion signals in both the Br-MION-CIMS and Br-FIGAERO-CIMS by tuning the electric field strengths in the first two low-pressure stages of the mass spectrometer as weak as possible to minimize collision-induced cluster fragmentation while maintaining sufficient ion transmission. Optimization is achieved by maximizing the ratio of I2 ⋅ Br− Br− at a constant I2 concentration. We list relevant instrument specifications and operational conditions in Table S1. It should be noted that these values are specific to our instruments, can thus vary according to instrument parameters, and may not be applicable to other instruments.
2.4 CE-DOAS
For the quantitative measurement of gas-phase molecular iodine (I2), we deployed a cavity-enhanced differential optical absorption spectroscopy instrument (CE-DOAS) (Meinen et al., 2010). CE-DOAS determines concentrations of trace gases from the strength of differential spectral features in a reference spectrum. The overall accuracy for the I2 time series is estimated to be 20 %, never better than the detection limit (3σ precision), resulting from the uncertainty in cross sections and the stability of the baseline. It is thus an absolute method and does not depend on an instrument-specific detection efficiency. To maximize the measurement sensitivity towards I2, we used a setup optimized for the green wavelength range (508–554 nm), where I2 exhibits strong differential absorption features. The measurement light is provided by a green light emitting diode (LED Engin). Spectral dispersion is established with a Czerny–Turner grating spectrometer (Princeton Instruments Acton 150), resulting in an optical resolution of 0.73 nm full width at half maximum at 546 nm. Intensities are monitored with a charge-coupled device (CCD) detector (Princeton Instruments PIXIS400B) cooled to −70 ∘C. Highly reflective mirrors (Advanced Thin Films) enhance the 1 m mirror separation to an effective optical path length of 15–23 km. The effective spectral mirror reflectivity was established by comparing light intensity spectra in the presence of N2 and He (Washenfelder et al., 2008). The abundance of trace gases is then determined by comparing spectra of chamber air relative to reference spectra recorded with ultrapure N2 without I2. Chamber air is drawn into the cavity with a constant flow rate of 1 slpm. Variations in the sampling flow did not result in changes in measured I2 concentrations, indicating that photolysis from the measurement light within the instrument was negligible. The following absorbers were included in the fit: I2 (Spietz et al., 2006), NO2 (Vandaele et al., 1998), H2O (Rothman et al., 2010), O2–O2 collision-induced absorption (Thalman and Volkamer, 2013), and a polynomial of sixth order. The setup allowed a 1 min detection limit of 25 pptv, or 8 pptv for integration times of 10 min, respectively. Periodic automated recordings of N2 reference spectra were recorded to ensure baseline stability. The optical path length at the time of measurement was continuously confirmed for consistency by the measurement of the O2–O2 collision-induced absorption and H2O column in the same analysis window. The overall systematic accuracy for the I2 time series is estimated to be 20 %, never better than the detection limit, resulting from the uncertainty in cross sections and the stability of the baseline.
2.5 Offline calibration setup
2.5.1 I2 permeation device
We used an iodine permeation tube (VICI Metronics) as a source for offline laboratory calibration. The permeation tube was encased within an electronically controlled heating mantle (80–140 (±2) ∘C) to allow for adjustable yet steady iodine permeation rates. The heating device (for holding the permeation tube) was made from a stainless steel tube ( in., 1.27 cm, outer diameter, OD) with a length of 25 cm, encased within an electronically controlled heating mantle. The configuration of the permeation device has been described in Tham et al. (2021). The iodine permeation device was run continuously for at least 72 h before any calibration experiments to ensure that a complete equilibrium was reached in the system. We then confirmed the robustness of the permeation device by the constant I2 signal measured with Br-MION-CIMS for over 24 h.
To determine the permeation rate of I2, we trapped iodine in n-hexane at cryogenic temperatures in an all-glass apparatus, following the method described in Chance et al. (2010). We initially filled the absorption glass vessel with 20 mL of n-hexane (99.95 %, Merck) and then weighed it to determine the combined mass. We then immersed the absorption vessel into a wide-necked Dewar vessel, filled with an acetone and dry ice mixture (at ∘C). After temperature equilibration, the I2 molecules, carried by 50 mlpm N2 flow from the permeation device, were bubbled through the absorption vessel. After a continuous collection for 5 h, we removed the absorption apparatus from the cooling mixture and allowed it to warm to room temperature prior to disassembling the setup to prevent any losses of iodine on the tip of the inlet capillary. The absorption vessel was then reweighed; the mass compared with that prior to absorption was less than 2 %, indicating a negligible loss during the trapping process. The I2 and n-hexane sample solutions were stored at 4 ∘C for 14 h before being subjected to analysis.
We determined the I2 concentration of the samples using a UV–vis spectrophotometer (Shimadzu Model UV2450) at a wavelength of 522 nm. We established a calibration curve via a set of I2 solutions ranging from 270 to 5300 nmol, diluted with n-hexane from a freshly prepared stock solution (0.5 g L−1). Repetition of the same analysis after 2 and 7 d yielded identical results, confirming that the sample solutions were stable at 4 ∘C. As an alternative analytical approach, we also quantified the I2 concentration in the sample solutions using an inductively coupled plasma mass spectrometer (ICP-MS; Agilent 7800). Before being introduced to the ICP-MS, the sample solutions were treated with NaHSO3 water solution (0.100 M), accomplishing efficient hexane-to-water extraction and simultaneous reduction of iodine to iodide (Schwehr et al., 2005) (Agilent Clinical Sample Preparation Guide (v3), ref. ISO 17294-2). The ICP-MS results were in good agreement (within 20 % discrepancies) with those from the UV–vis spectrophotometry.
We conducted the I2 trapping and quantification experiments in triplicate with satisfactory reproducibility (standard deviation <10 %). The calculated iodine permeation rate at 50 mlpm N2 flow and 140 ∘C oven temperature is 278 ± 12 ng min−1 (mean ± standard deviation). This result was used as the benchmark to estimate temperature-corrected permeation rates according to the formula provided by the permeation tube vendor (VICI Metronics). We checked the validity of the temperature-corrected values by conducting a second iodine absorption experiment in which the iodine permeation tube was kept at 130 ∘C with an N2 flow rate of 50 mlpm, and the determined permeation rate agreed within 10 % of the calculated value.
2.5.2 Cl2 permeation device
We used a commercial chlorine permeation tube (VICI Metronics) as a source for offline calibration. We passed a 20 mlpm high-purity nitrogen (99.999 %) flow at room temperature through a 25 cm long stainless-steel tube (” O.D.) containing the permeation tube. We quantified the permeation rate of Cl2 following a procedure described in a previous study (Finley and Saltzman, 2008). The output of 20 mlpm flow was bubbled into a buffered aqueous potassium iodide solution (2.0 % KI – – prepared in 1.00 mM aqueous phosphate buffer, pH = 7.0) filled in an all-glass two-stage serial absorption apparatus (stage 1 = 100 mL; stage 2 = 50 mL) for 3 h and kept at room temperature. The Cl2 oxidized the iodide (I−) into iodine (I2) once contacting with the KI absorption solution, and the I2 further reacted with the excess KI present in the absorption solution to form I, which can be quantified by UV–vis spectrophotometry. We analyzed the resulting sample solutions with an UV–vis spectrophotometer (Shimadzu Model UV-1800) using 1 cm quartz cells at 352 nm, corresponding to the I. We detected no I in the second stage absorption solution, indicating that all the chlorine was quantitatively trapped and rapidly converted to I within the first absorption unit. The samples were quantified relative to I standards in the range of 5 to 68 × 10−6 M, prepared by dilution of a stock obtained by dissolving 174 mg iodine in 200 mL of a solution containing 2 % KI in 1.00 mM aqueous phosphate buffer, pH 7.0. From this calibration curve, we calculated a molar absorptivity of 26 800 L mol−1 cm−1, which is consistent with the values reported in the literature (Finley and Saltzman, 2008; Kazantseva et al., 2002). Samples and standard solutions were re-analyzed after being stored in the dark at room temperature for 24 h, and the results were within 3 % of those obtained with the fresh solutions. We repeated the absorption experiment, and the calculated chlorine permeation rate at room temperature was 764 ± 74 ng Cl2 min−1 (mean ± standard deviation).
2.5.3 HOI calibrator
We produced a continuous HOI source via the reaction of I2 and hydroxyl radicals (OH) in a setup similar to the sulfuric acid (H2SO4) calibrator (Kürten et al., 2012). The OH was generated by photolyzing H2O with a mercury (Hg) lamp at 184.9 nm, whose calibrated intensity was used to estimate the OH concentration. We tested the system by removing the I2 or OH source from the calibrator, upon which HOI production was undetectable, confirming that any single reactant did not produce HOI. A numerical model was constructed to predict the mean HOI concentration entering the Br-MION-CIMS, which is analogous to the model used for H2SO4 calibration (Kürten et al., 2012). We only included the formation pathway of I2+ OH to HOI in the model for simplicity; the other pathway of IO + HO2 was considered minor as its reaction rate is about an order of magnitude slower than that of I2 + OH. Furthermore, IO is likely at negligible concentration in the calibration device due to the absence of O3 for IO formation.
2.6 Quantum chemical calculations
We used quantum chemical calculations to estimate the cluster formation enthalpy of halogen-containing species and bromide ions. The initial conformational sampling was performed using the Spartan'14 program. The cluster geometry was then optimized using density function theory methods at the ωB97X-D/aug-cc-pVTZ-PP level of theory (Chai and Head-Gordon, 2008; Kendall et al., 1992). Iodine pseudo-potential definitions were taken from the Environmental Molecular Sciences Laboratory (EMSL) basis set library (Feller, 1996). Calculations were carried out using the Gaussian 09 program (Frisch et al., 2010). An additional coupled-cluster single-point energy correction was carried out on the lowest energy geometry to calculate the final cluster formation enthalpy. The coupled-cluster calculation was performed at the DLPNO-CCSD(T)/def2-QZVPP level using the ORCA program version 4.0.0.2 (Neese, 2012; Riplinger and Neese, 2013). In Table 1 we present calculated cluster formation enthalpies based on the optimized geometries.
Table 1Cluster formation enthalpies of different species with bromide ions. The cluster geometries are optimized at the ωB97X-D/aug-cc-pVTZ-PP level at 298.15 K. The enthalpies are calculated at the DLPNO-CCSD(T)/def2-QZVPP//ωB97xD/aug-cc-pVTZ-PP level at 298.15 K.
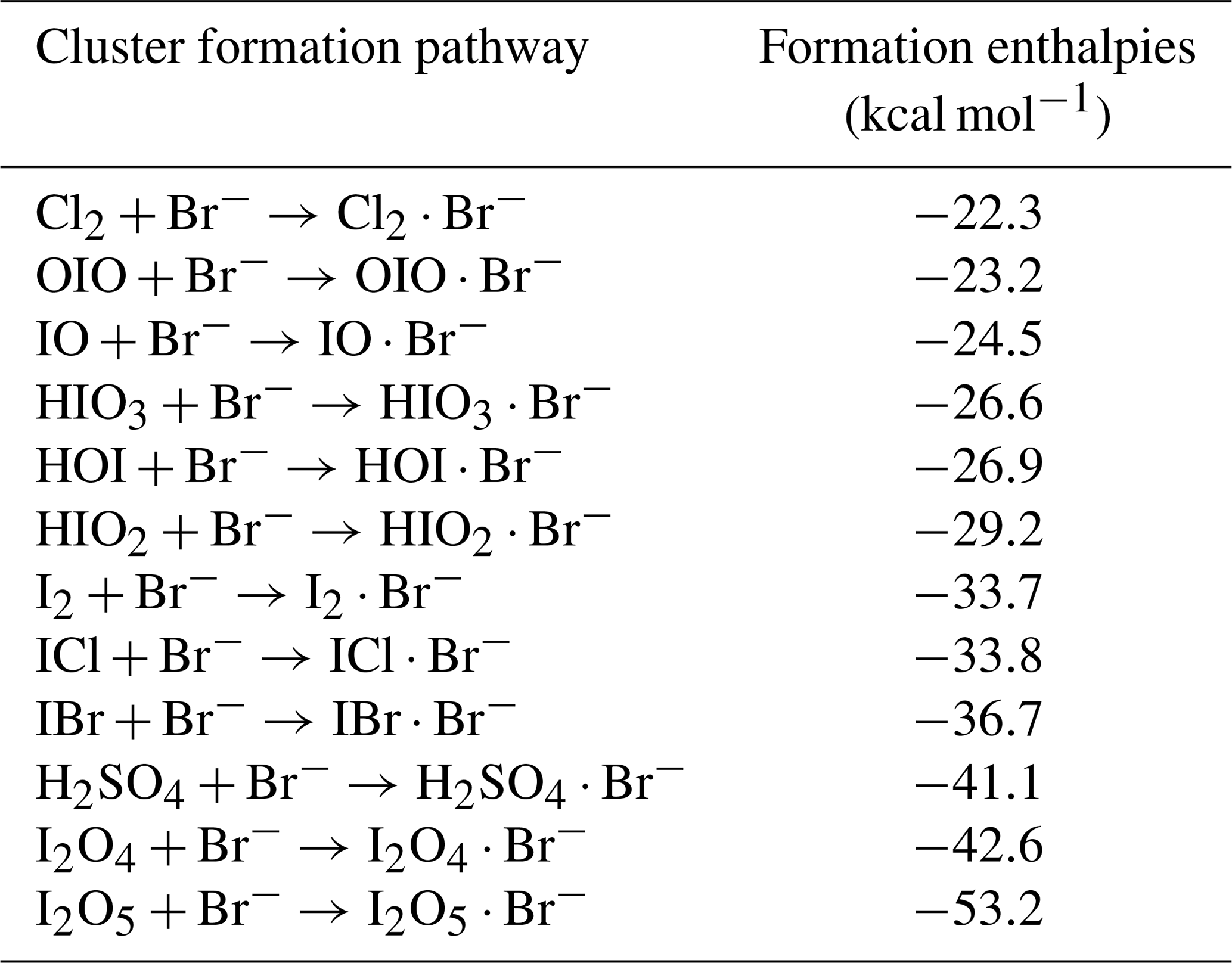
3.1 Detection of gas-phase inorganic species by Br-MION-CIMS
We show in Fig. 1 the selected inorganic species observed with the Br-MION-CIMS during an iodine oxidation experiment in the CLOUD chamber. The peak identities are indicated in the labels. Observed species include I2 and its various oxidation products. There are also a few other halogen-containing inorganic species such as Cl2, ICl, and IBr, likely coming from the impurities in the iodine source. Non-halogen inorganic species such as H2SO4 can also react with bromide ions and are detected. Due to the large negative mass defect of the bromine and iodine atoms, and the high resolution (∼10 000 Th Th−1) of the mass spectrometer, the peaks can be unambiguously distinguished and identified in the mass spectrum. As shown in the lower panel of Fig. 1, most of the iodine-containing species appear as a single peak in the unit mass range, except for HIO2 ⋅ 79Br− (), which overlaps with the reagent ion cluster (79Br281Br)− ().
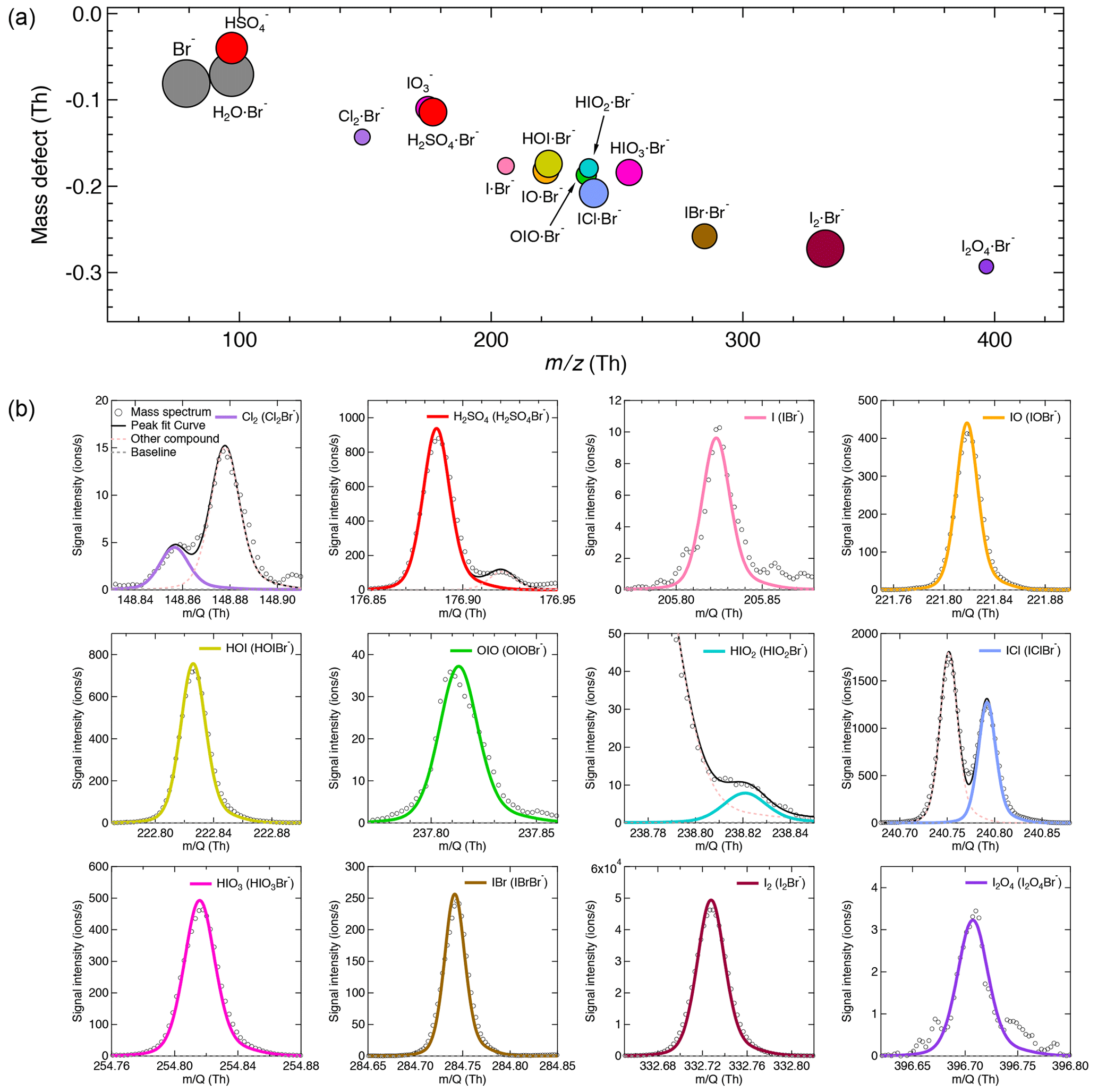
Figure 1Gas-phase inorganic species measured with the Br-MION-CIMS. (a) Mass defect (difference of exact mass to integer mass) versus of gas-phase halogen species and sulfuric acid during an iodine oxidation experiment at 69 % relative humidity and −10 ∘C (conducted on 5 October 2018). Ions shown here are either clustered with or formed via proton transfer to a bromide ion. The area of the markers is proportional to the logarithm of the signal (counts per second). (b) The high-resolution single peak fits for species in the mass defect plot in the upper panel. Species in both panels are color coded in the same style.
The iodine oxidation experiments were conducted under experimental conditions typically found in the high-latitude marine boundary layer, with a temperature of −10 ∘C and a relative humidity of 69 %. As illustrated in Fig. 2, a typical experiment started with illumination of the chamber at constant I2 (∼60 pptv) using the green light to photolytically produce I atoms. The subsequent reactions of I and ∼40 ppbv O3 led to the formation of various oxidized iodine species within a few minutes. The most prominent species we observed from these experiments were IO, HOI, and HIO3, with lower but significant levels of OIO, HIO2, and I2O4. Among these iodine oxides, IO rose the most rapidly; this is consistent with the first-generation production of IO from the I + O3 reaction. After a few steps of radical reactions, OIO, HIO2, and HIO3 reached steady state almost simultaneously. The only observed iodine oxide dimer was I2O4 in this event, while I2O2, I2O3, and I2O5 were below the detection limit of both mass spectrometers. A noticeable dip in the HIO3 traces a few minutes after the onset of the reactions is likely due to the participation of HIO3 in new-particle formation, resulting in an extra loss term and a lower steady-state concentration. When we turned off the green light, the production of I radicals stopped, and iodine species decayed away.
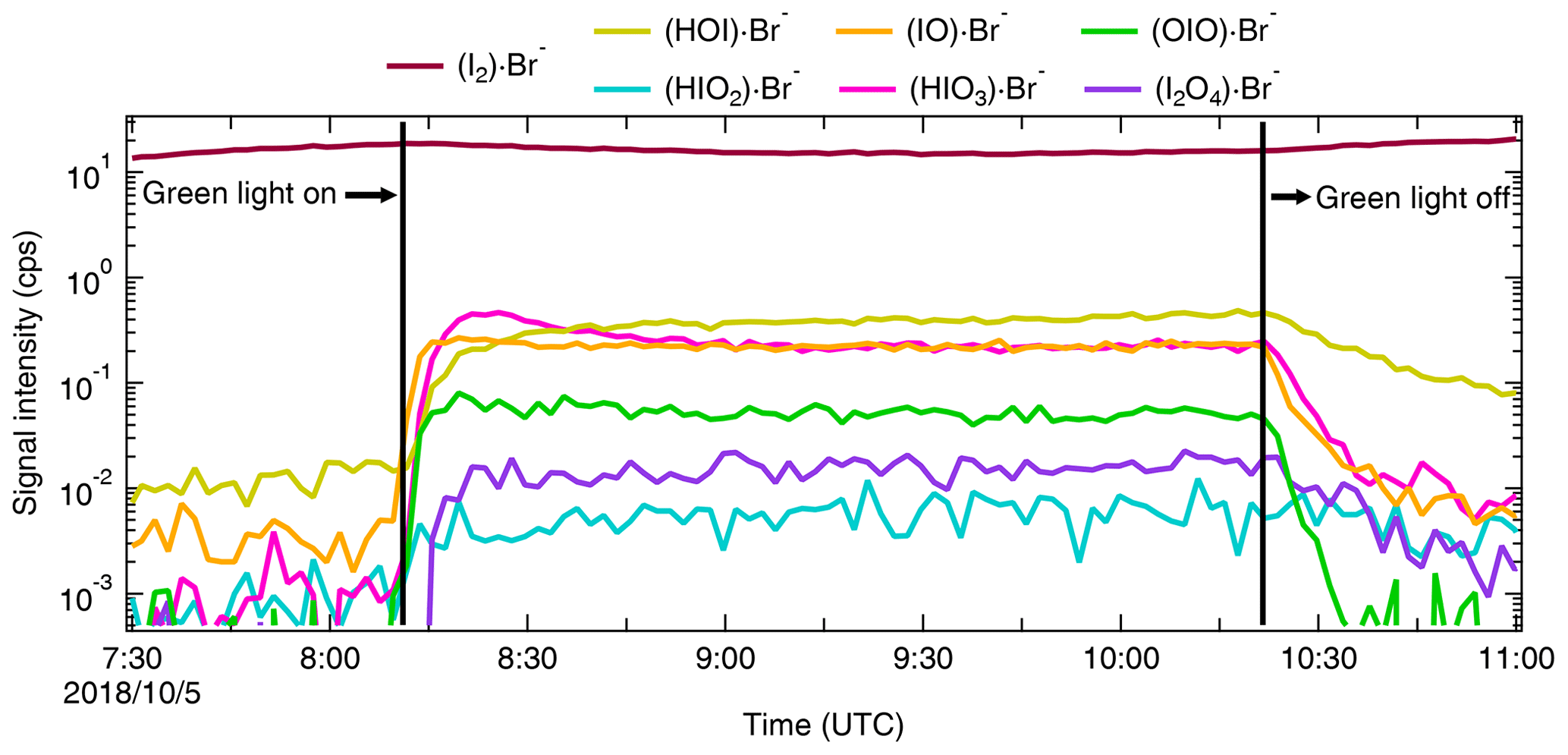
Figure 2Evolution of selected iodine species during a typical run. The experiment was performed at 60 pptv I2, 40 ppbv O3, 69 % relative humidity, and −10 ∘C. The oxidized iodine species start to appear soon after switching on the green light at 08:11, 5 October 2018. The I atom production was halted at 10:21, 5 October 2018 by switching off the green light, and the concentration of oxidized iodine species decayed away afterwards. All species are color coded in the same way as in Fig. 1.
3.2 Relative humidity dependence
Water molecules can cluster with I− to form H2O ⋅ I− in the iodide CIMS. This enhances the instrument sensitivities for small molecules (i.e., chlorine and bromine) and reduces them for large molecules (i.e., oxygenated organics) (Lee et al., 2014). To investigate the role of water concentration in the sensitivity of the Br-MION-CIMS, we varied the relative humidity (RH) from 40 % to 80 % at a constant temperature of −10 ∘C. We show in Fig. 3 the correlation of I2 time series from the Br-MION-CIMS and the CE-DOAS throughout the experiment.
Figure 3Signal normalization methods for the Br-MION-CIMS. Normalized I2 ⋅ Br− signal intensity for variable I2 concentrations, color coded by relative humidity (orange: 35 %–45 % relative humidity, red: 70 %–80 % relative humidity). The charger ions in the ion source of Br-MION-CIMS are Br− and H2O ⋅ Br− (both 79Br and 81Br). Their abundance depends both on the instrument tuning and the absolute humidity of the sampled flow. The normalization of the I2 ⋅ Br− signal by only Br− (a) or H2O ⋅ Br− (b) does not compensate for the humidity effect. Using the sum of Br− and H2O ⋅ Br− (c) for normalization yields a tight correlation to the true I2 as measured by CE-DOAS, independent of the humidity. The black dashed line indicates the fitted linear calibration.
Chemical ionization relies on an ion-molecule reaction to transfer charge from a reagent ion to an analyte, forming either a product ion or a charged cluster between the analyte and the reagent ion with a rate coefficient, kIM. This occurs in an ion-molecule reactor with a fixed flow rate and thus reaction time, dt, and ideally under pseudo-first-order conditions in which a small fraction of the analyte is ionized and the reagent ion concentration ([Ion]) remains constant. Under these (linear) conditions the fraction of analyte that is ionized is kIM × [Ion] × dt. However, the primary ion source strength can vary with time, and so we normalize the analyte signal by reagent ion signal to account for those small variations in analyte signal.
During the RH transition, the ratio of the two reagent ions, Br− and H2O ⋅ Br−, changed in the Br-MION-CIMS. As shown in Fig. 3a and b, using either reagent ion alone for I2 normalization results in discrepancies in recovered I2 concentrations at different RHs. However, if we use the sum of these two reagent ions (Br H2O ⋅ Br−) for normalization, the humidity effect vanishes, as shown in Fig. 3c. Separately, during the I2 calibration using a permeation tube (Sect. 2.5.1 and Sect. 3.3.2), the detection of I2 molecules is robust and independent of RH between 20 % and 40 % at 25 ∘C as long as a proper normalization method is used for the Br-MION-CIMS. Furthermore, we have also carried out the HOI calibration at 25 ∘C (Sect. 2.5.3 and Sect. 3.3.2) and used the same normalization method. During the calibration, we varied the water content in the calibrator to vary OH concentrations. A good correlation between the modeled HOI concentrations and the measured HOI signals also indicates that the different H2O concentrations in the system do not affect the HOI detection (Fig. 4d). This assertion may also be applicable to molecules such as iodine monochloride (ICl) and iodine monobromide (IBr), which share similar chemical and physical properties with I2; however, further confirmation is needed for other species such as oxygenated organics.
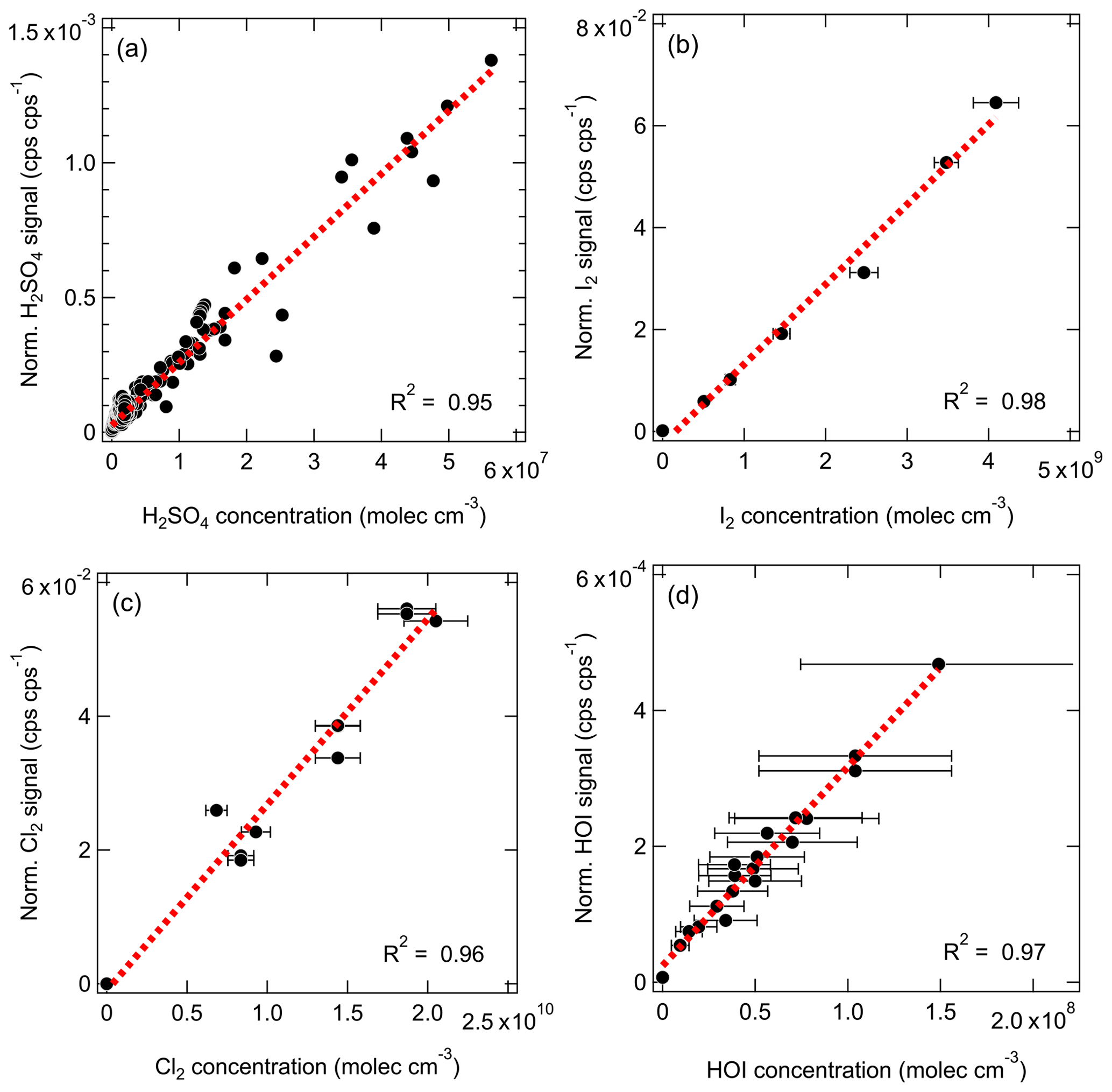
Figure 4The normalized signals (cps cps−1; cps signifies counts per second) vs. the absolute concentrations (molec. cm−3) measured with the Br-MION-CIMS for (a) H2SO4, (b) I2, (c) Cl2, and (d) HOI. The dashed red lines are the linear fits. The overall 3σ accuracy of % on [H2SO4] is not shown here.
3.3 Quantification of gas-phase inorganic species
Inter-method calibrations and offline calibrations were carried out to quantify the sensitivities of the Br-MION-CIMS to selected calibrants. For the inter-method calibrations, we used the CLOUD chamber as a stable source of I2 and H2SO4 and intercompared the signals in the Br-MION-CIMS with the CE-DOAS and a pre-calibrated nitrate-CIMS, respectively. For the offline calibrations, we carried out the experiments separately in a laboratory at the University of Helsinki, using permeation tubes to quantify I2 and Cl2 and a calibrator to quantify HOI.
3.3.1 Inter-method calibrations at CLOUD
I2 calibration using the CE-DOAS
As shown in Fig. 3, we use the accurate I2 time series measured with the CE-DOAS to calibrate normalized I2 signals in the Br-MION-CIMS. The I2 concentrations used for the calibration span approximately 2 orders of magnitude, reaching up to 4.6 × 1010 molec. cm−3. A linear fit, limited to I2 concentrations smaller than 1010 molec. cm−3, establishes the calibration factor as follows:
For this range of concentrations, which are typically encountered in the atmosphere, the calibrated Br-MION-CIMS time series agrees within error with the CE-DOAS measurement (1σ accuracy 20 %, detection limit 25 pptv for 1 min data). Deviations between both time series are generally smaller than 10 % (25 and 75 percentile 0.88 and 1.03, respectively). These small differences may be explained by incomplete homogeneity of iodine concentrations in the chamber and the different sampling positions of CE-DOAS and Br-MION-CIMS.
H2SO4 calibration using a nitrate-CIMS
We derive the H2SO4 calibration coefficient for the Br-MION-CIMS using the absolute H2SO4 concentrations measured with a pre-calibrated nitrate-CIMS. The calibration protocol of H2SO4 in the nitrate-CIMS has been described in detail previously (Kürten et al., 2012). The H2SO4 time series used for the inter-method calibration covers a wide concentration range from less than 5.0 × 104 (detection limit of the nitrate-CIMS) to 6.0 × 107 molec. cm−3. For Br-MION-CIMS, although both HSO and H2SO4 ⋅ Br− appear as distinct peaks for sulfuric acid, we only use the normalized H2SO4 ⋅ 79Br− for the intercalibration, as HSO () has substantial interference from the reagent ion H2O ⋅ 79Br− (). We show in Fig. 4a the linear fit.
The H2SO4 calibration coefficient is thus 4.1 × 1010 molec. cm−3 per normalized signal (cps cps−1; cps signifies counts per second), and the correlation coefficient between the two H2SO4 traces is 0.95. The systematic 3σ accuracy is % for H2SO4 calibration using a nitrate-CIMS; detailed accuracy estimation has been discussed previously (Stolzenburg et al., 2020).
3.3.2 Offline laboratory calibrations for I2, Cl2, and HOI
For the I2 calibration, we diluted the I2 flow to seven different values and measured the flow with the Br-MION-CIMS. We repeated the calibration five times; we show the data along with a linear fit between the I2 concentration and normalized I2 signal in Fig. 4b. The slope of the line gives a calibration coefficient of 6.3 × 1010 molec. cm−3 per normalized signal (cps cps−1), with R2 of 0.98 and an overall 1σ accuracy of ±45 %.
For the Cl2 calibration, the Cl2 permeation source was run continuously for 12 h prior to calibration experiments to ensure complete system equilibrium. A two-stage dilution system similar to the setup of Gallagher et al. (1997) was set up for diluting the output of the Cl2 permeation device. The 20 mlpm of N2 stream emerging from the Cl2 permeation device (operated at room temperature) was diluted in a stream of 6 slpm of dry N2. Then, a small fraction of this mixture (50 to 300 mlpm) was further mixed with the total flow of 25 slpm of N2 (20 slpm dry N2+5 slpm humidified N2) before being sampled by the Br-MION-CIMS. The calibration coefficient for Cl2 was determined to be 3.5 × 1011 molec. cm−3 per normalized signal (cps cps−1) from three separate calibration experiments (Fig. 4c), with an 1σ accuracy of 30 %.
As for the HOI calibration, we produced a range of HOI concentrations by varying I2 and OH concentrations in the calibrator. We show in Fig. 4d the linear correlation between the modeled HOI concentrations and measured HOI signals. The slope of the fit corresponds to a calibration coefficient of 3.3 × 1011 molec. cm−3 per normalized signal (cps cps−1), with an overall 1σ accuracy of 55 %. The good correlation (R2=0.97) including various H2O levels also indicates that H2O concentrations did not affect the HOI detection.
3.3.3 Connecting sensitivity to binding enthalpy
Beyond the species for which we carried out calibrations, there are many more, especially iodine species, that cannot be directly calibrated due to a lack of authentic standards or generation methods. However, the sensitivity of an iodide-CIMS towards analytes can be predicted by the cluster binding enthalpy, calculated by relatively simple quantum chemical methods (Iyer et al., 2016). This holds for the Br-CIMS as well. In the instrument, ion clusters, formed from reactions between analytes and reagent ions, are guided and focused by ion optics during transmission to the detector. The electric forces applied to the clusters enhance their collision energies with carrier gas molecules. If sufficient energy is transferred during the collision, cluster fragmentation may occur, affecting the instrument sensitivity for the analytes (Passananti et al., 2019). However, clusters with higher binding enthalpy will be more easily preserved and detected. Analytes that bind to the reagent ions with enthalpies higher than a critical level are likely detected at maximum sensitivity (kinetic-limited detection) by the instrument. For example, the calculated critical enthalpy is 25 kcal mol−1 for the iodide-CIMS used in Iyer et al. (2016) and Lopez-Hilfiker et al. (2016), calculated at the DLPNO-CCSD(T)/def2-QZVPP//PBE/aug-cc-pVTZ-PP level of theory.
For the bromide chemical ionization, there are two types of fragmentation pathways:
-
reversion to the original form of Br− and analyte,
-
proton transfer from the analyte to Br−,
where the X−H is the hydrogen bond donor. An analyte should be detected at the maximum sensitivity when the dissociation enthalpy for the first pathway is either (a) much higher than the critical enthalpy (dissociation of X−H ⋅ Br− to X−H and Br− does not occur) or (b) lower than the critical enthalpy but much higher than that of the second pathway (dissociation of X−H ⋅ Br− to X−H and Br− may occur, but dissociation to X− and HBr is the dominant pathway). Whether the enthalpy for the second pathway is higher than the critical enthalpy does not directly affect the sensitivity as both X−H ⋅ Br− and X− can be measured and counted. The sensitivity toward X−H would be reduced only when the first reversion pathway occurs to a non-negligible extent. Taking H2SO4 as an example, the dissociation enthalpies for the first and second pathways are 41.1 and 27.9 kcal mol−1, respectively. If some of the H2SO4 ⋅ Br− dissociate, they preferably become HSO and are detectable by the Br-CIMS. Thus, H2SO4 can be detected at the maximum sensitivity.
While we were unable to experimentally establish a correlation between sensitivities and binding enthalpies due to limited quantifiable halogen species, we can predict the tentative critical enthalpy as the binding enthalpy of a species that is likely detected at the maximum sensitivity. We list the cluster formation enthalpies for a selection of halogen-containing species in Table 1 and the corresponding cluster dissociation enthalpies in Table 2. Among all the calibration coefficients listed in Table 3, H2SO4 and I2 have the lowest calibration coefficients (highest sensitivities). Thereby, we conclude that both H2SO4 and I2 are detected at the maximum sensitivity, suggesting a critical enthalpy not higher than 33.7 kcal mol−1. We can then infer the sensitivity for other species that are difficult to calibrate by comparing their binding enthalpies to those of the benchmark species. For example, ICl and IBr should have the maximum sensitivity since the dissociation enthalpies for ICl ⋅ Br− and IBr ⋅ Br− are both much higher than 33.7 kcal mol−1 (Table 2). Although HIO3 ⋅ Br− has a lower dissociation enthalpy than the critical value, the favored dissociation pathway is proton transfer (the second pathway); HIO3 can thus be considered as a maximum sensitivity species detectable as IO ions after proton transfer. This is consistent with the fact that both HIO3 ⋅ Br− and IO are detected in Fig. 1, as is the case with H2SO4. We thus assume that HIO3 has a kinetic calibration coefficient of 4.1 × 1010 molec. cm−3 cps cps−1, the value for H2SO4. However, the lowest dissociation enthalpies of HOI ⋅ Br− and Cl2 ⋅ Br− are 26.9 and 22.3 kcal mol−1, respectively, consistent with their higher calibration coefficients of 3.3 × 1011 and 3.5 × 1011 molec. cm−3 cps cps−1. The dissociation enthalpies for IO ⋅ Br−, OIO ⋅ Br−, and HIO2 ⋅ Br− are 24.5, 23.2, and 29.2 kcal mol−1, respectively. We would expect that their sensitives are lower than the maximum sensitivity. Since the dissociation enthalpies for IO ⋅ Br− and OIO ⋅ Br− are between those of HOI ⋅ Br− and Cl2 ⋅ Br−, a similar calibration coefficient may be applied, but direct calibrations are more preferable. We note that when transferring the calibration factor from one species to another, the diffusivity difference should be accounted for since it affects the inlet line loss. This factor is not considered in the cluster enthalpy calculations.
Table 2Fragmentation reaction enthalpies of different species with bromide ions. The cluster geometries are optimized at the ωB97X-D/aug-cc-pVTZ-PP level at 298.15 K. The enthalpies are calculated at the DLPNO-CCSD(T)/def2-QZVPP//ωB97xD/aug-cc-pVTZ-PP level at 298.15 K.

Table 3Calibration coefficients for selected species (“n/a” stands for “not available”).
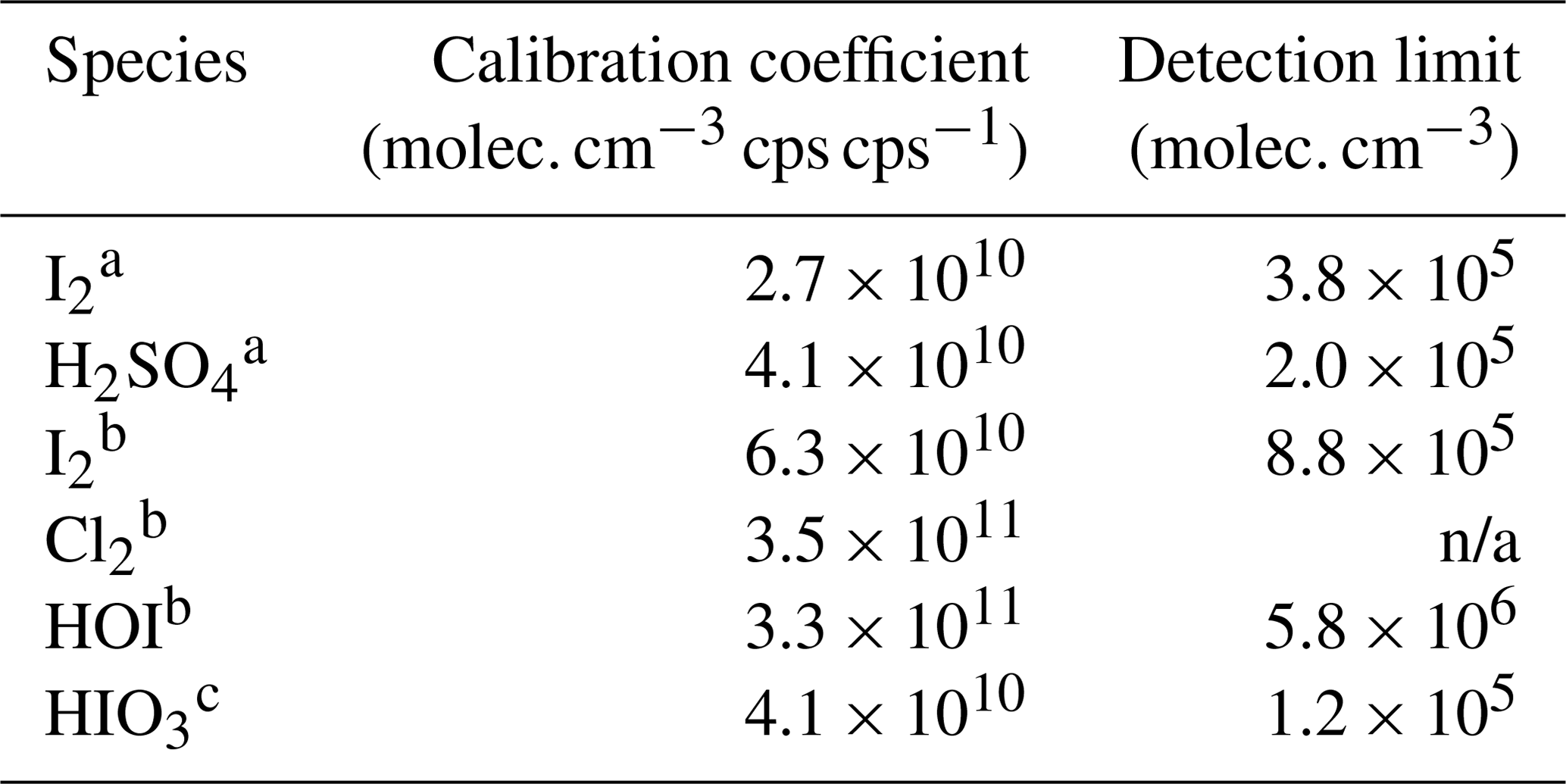
a Inter-method calibrations; b offline calibrations; c derived from dissociation enthalpies.
Further, we estimate the detection limit of the calibrated species. The detection limit is defined as the analyte concentration, corresponding to the sum of the mean signal and 3 times the standard deviations (3σ) of the background fluctuations during a 2 h background measurement. We derive the detection limit of HOI, HIO3, I2, and H2SO4 to be 5.8 × 106, 1.2 × 105, 3.8 × 105, and 2.0 × 105 molec. cm−3 (or 0.2, 0.005, 0.015, and 0.008 pptv), respectively, for a 2 min integration time.
3.4 Comparison between Br-MION-CIMS and Br-FIGAERO-CIMS
While Br-MION-CIMS and Br-FIGAERO-CIMS use the same chemical ionization scheme, their designs differ in the ion-molecule reaction chamber (IMR). MION is an atmospheric pressure (1 bar) drift tube; analyte molecules gain an electric charge in an axial laminar flow. FIGAERO is connected to a cone-shaped IMR operated at a reduced pressure (150 mbar); the sample flow is injected into the inlet via an orifice, necessarily causing turbulence and wall interactions in the IMR region. The atmospheric pressure and reduced pressure IMRs are both widely used for trace gas measurements. We thus compare iodine species measurements from Br-MION-CIMS and Br-FIGAERO-CIMS to better understand the performance and applicability of the bromide ionization scheme.
We show in Fig. 5 the same iodine oxidation event as in Fig. 2 to illustrate the time series for HIO3 ⋅ Br−, HOI ⋅ Br−, IO ⋅ Br−, and I2 ⋅ Br−, measured with Br-MION-CIMS (red circles) and Br-FIGAERO-CIMS (grey sticks), respectively. Note that the FIGAERO alternates between gas and particle measurements; here we show only the gas-phase signals. Clear and concurrent signals of HIO3, HOI, IO, and I2 are evident from both the Br-MION-CIMS and Br-FIGAERO-CIMS. Prior to the iodine oxidation event (08:11), there was no photochemical production and thus virtually no signal of oxidized iodine species in both instruments. The dark reaction of ozone with I2 did not proceed at a significant rate due to the low rate coefficient and to low levels of I2. Signals detected during this period are considered as the persistent background coming from electronic noise or other sources such as the ionizer, carrier flows, or long-term “memory” in the case of the Br-FIGAERO-CIMS. Not surprisingly, the Br-MION-CIMS has a near-zero background for all analytes. For HIO3 (Fig. 5a), the background signal in the Br-FIGAERO-CIMS is also negligible; however, IO shows a substantial persistent background (Fig. 5c) in the Br-FIGAERO-CIMS.
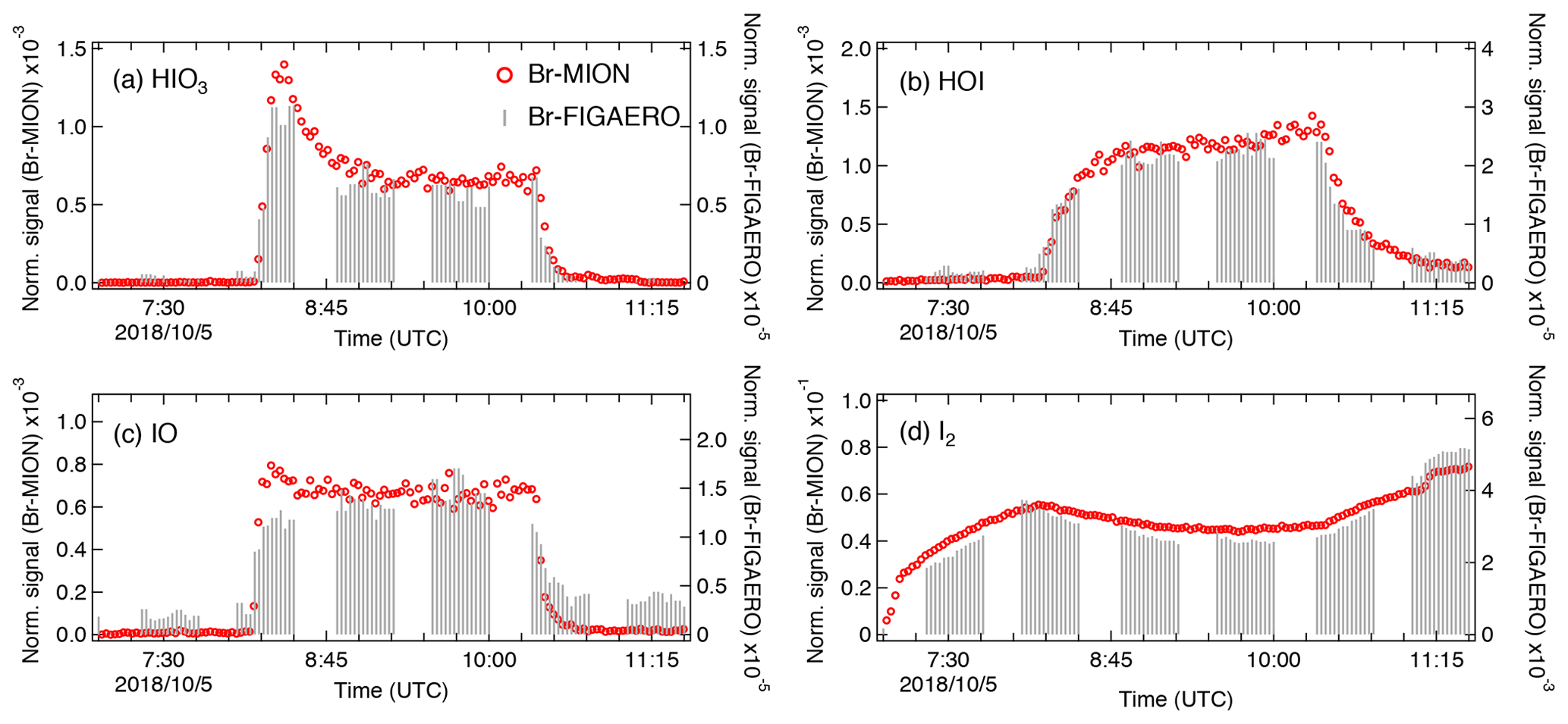
Figure 5Signal comparison of selected iodine species measured with Br-MION-CIMS and (gas-phase) Br-FIGAERO-CIMS, respectively, during the same iodine oxidation experiment shown in Fig. 2.
When we initiated the photochemistry, oxidized iodine signals rapidly built up toward an asymptote within timescales of minutes. The instrumental differences in these timescales are small for HIO3 and HOI but larger for IO. When colliding with the IMR surface, HIO3 condenses irreversibly; it thus makes sense that the Br-MION-CIMS and Br-FIGAERO-CIMS signals show the same timescale for HIO3. Semi-volatile HOI, however, can return to the gas phase from the walls depending on the surface coverage of HOI and the vapor concentration. Additionally, the heterogeneous reaction of aqueous iodide (I−) and ozone (Carpenter et al., 2013) could also contribute to the emission of HOI from the IMR wall in the FIGAERO. As the evaporation flux is typically a function of the amount of analyte on the surface, the buffering effect could degrade the instrument time response upon changes in analyte concentration. Here, however, we did not observe a significant memory effect likely because the HOI concentration was too low to fully saturate the IMR surface or because any HOI evaporation was suppressed due to an enhanced accommodation coefficient of HOI on the metal surface. We expect IO to be prone to loss on the metal surface due to its radical nature.
After the iodine oxidation event (10:21), the photochemical production of oxidized iodine species was terminated and vapor concentrations decayed exponentially due to dilution and losses to chamber walls. Memory effects could also influence the signal time constant. The dilution loss rate was around 2.1 × 10−4 s−1 (4760 s time constant) for all species in the chamber, determined by the total chamber flow rate and the chamber volume. Wall loss rates, however, vary for species with different diffusion constants. The decay rates of HIO3 are 400 s for the Br-MION-CIMS and 370 s for the Br-FIGAERO-CIMS, much faster than the dilution loss. For comparison, the time constant for H2SO4 vapor loss was 300 s. These time constants are thus consistent with wall loss (around 2.2 × 10−3 s−1). The IO decay time constant is 294 s for the Br-MION-CIMS and 435 s for the Br-FIGAERO-CIMS. The time constant for the Br-MION-CIMS indicates that the decay of IO is also driven by wall loss, so the net flux during this period was thus towards the wall rather than from the wall. Therefore, the difference in IO between instruments may well be attributed to the persistent background from the ionizer of the FIGAERO. The HOI signals have longer decay time constants in both instruments of 909 s for the Br-MION-CIMS and 714 s for the Br-FIGAERO-CIMS; this may reflect a time constant for the depletion of HOI adsorbed onto the chamber walls. The I2 signal increases after the event termination because it is no longer photolyzed while its injection continues.
Applying the calibration coefficients, we convert the Br-MION-CIMS signals to absolute concentrations and subsequently correlate them with signals measured with Br-FIGAERO-CIMS. We then estimate the tentative detection limits for HOI and HIO3 in the Br-FIGAERO-CIMS to be 3.3 × 107 and 5.1 × 106 molec. cm−3 (versus 5.8 × 106 and 1.2 × 105 molec. cm−3 in the Br-MION-CIMS), respectively, at 3σ of the background signal for a 2 min integration time during a 2 h period; they are in general 1 order of magnitude higher than those in the Br-MION-CIMS. This is in line with the higher background signals observed in the Br-FIGAERO-CIMS. Note that for both HOI and HIO3 the uncertainties introduced from the correlational analysis are negligible compared to the limited accuracy of the calibration sources (55 % for HOI and % for HIO3). We are unable to estimate the I2 detection limit in the Br-FIGAERO-CIMS due to a lack of I2 background measurement; but Br-FIGAERO-CIMS can and did detect I2 at the low parts per trillion by volume level with good fidelity.
We confirm in this study that bromide chemical ionization is a suitable technique for the time-resolved, highly sensitive, and simultaneous measurements of iodine species and sulfuric acid. The Br-MION-CIMS shows constant sensitivity throughout the relative humidity range of 40 % to 80 % at −10 ∘C as long as the sum of the two reagent ions (Br− + H2O ⋅ Br−) is used for signal normalization. This demonstrates the applicability of this technique to field measurements in the ambient marine environment.
We quantify iodine species and sulfuric acid via offline calibrations (i.e., permeation tube and calibrator) and inter-method calibrations (i.e., CE-DOAS and pre-calibrated nitrate-CIMS). Further, we calculate the binding enthalpies between the calibrated species and reagent ions, which qualitatively agree with the corresponding calibration coefficients. This indicates that the quantum chemical calculations can be employed along with the calibration experiments to determine the sensitivities for unquantifiable species; more work is required to further establish the correlation between calibration coefficients and binding enthalpies.
Further, using inter-method and offline calibrations, we estimate the detection limits of HOI, HIO3, I2, and H2SO4 in Br-MION-CIMS to be 5.8 × 106, 1.2 × 105, 3.8 × 105, and 2.0 × 105 molec. cm−3, respectively, for a 2 min integration time during a 2 h period. To our knowledge, the simultaneous measurements of various iodine species and sulfuric acid with low detection limits are unprecedented for online techniques. Detection limits for HOI and HIO3 in the Br-FIGAERO-CIMS are 3.3 × 107 and 5.1 × 106 molec. cm−3, which are in general 1 order of magnitude higher than those in the Br-MION-CIMS. The signal comparison between the two instruments also shows that the Br-CIMS can be coupled to both the atmospheric pressure and the reduced pressure interfaces for iodine species and sulfuric acid measurements in the marine environment.
Data available on request from the authors.
The supplement related to this article is available online at: https://doi.org/10.5194/amt-14-4187-2021-supplement.
MW, XCH, YJT, and HF wrote the manuscript. XCH, YJT, MW, and MiS designed the experiments. XCH, YJT, and JS carried out the Br-MION-CIMS measurements, MW, DC, and VH carried out the Br-FIGAERO-CIMS measurements, and HF carried out the CE-DOAS measurements. YJT, XCH, HF, DC, JS, and MaS performed the calibrations. SI, XCH, and TK carried out the quantum chemical calculations. MW performed the comparison analysis of the Br-FIGAERO-CIMS and Br-MION-CIMS. NMD, TK, MR, RV, and MiS commented on the manuscript. All other co-authors participated in either the development or preparations of the CLOUD facility and the instruments and/or collecting and analyzing the data.
The authors declare that they have no conflict of interest.
We thank the European Organization for Nuclear Research (CERN) for supporting CLOUD with important technical and financial resources and for providing a particle beam from the CERN Proton Synchrotron. We also thank Juhani Virkanen and Heini Ali-Kovero for providing assistance in the laboratory analytical experiments.
This research has been funded by the US National Science Foundation (grant nos. AGS-1531284, AGS-1801574 and AGS-1801280), the Academy of Finland (grant nos. 296628, 328290, Centre of Excellence 1118615), and the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (GASPARCON; grant no. 714621). The FIGAERO-CIMS was supported by an MRI grant for the US NSF (grant no. AGS-1531284), as well as the Wallace Research Foundation.
Open-access funding was provided by the Helsinki University Library.
This paper was edited by Hendrik Fuchs and reviewed by two anonymous referees.
Bitter, M., Ball, S. M., Povey, I. M., and Jones, R. L.: A broadband cavity ringdown spectrometer for in-situ measurements of atmospheric trace gases, Atmos. Chem. Phys., 5, 2547–2560, https://doi.org/10.5194/acp-5-2547-2005, 2005.
Carpenter, L. J., MacDonald, S. M., Shaw, M. D., Kumar, R., Saunders, R. W., Parthipan, R., Wilson, J., and Plane, J. M. C.: Atmospheric iodine levels influenced by sea surface emissions of inorganic iodine, Nat. Geosci., 6, 108–111, https://doi.org/10.1038/ngeo1687, 2013.
Chai, J.-D. and Head-Gordon, M.: Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections, Phys. Chem. Chem. Phys., 10, 6615–6620, https://doi.org/10.1039/b810189b, 2008.
Chance, R. J., Shaw, M., Telgmann, L., Baxter, M., and Carpenter, L. J.: A comparison of spectrophotometric and denuder based approaches for the determination of gaseous molecular iodine, Atmos. Meas. Tech., 3, 177–185, https://doi.org/10.5194/amt-3-177-2010, 2010.
Dias, A., Ehrhart, S., Vogel, A., Williamson, C., Almeida, J., Kirkby, J., Mathot, S., Mumford, S., and Onnela, A.: Temperature uniformity in the CERN CLOUD chamber, Atmos. Meas. Tech., 10, 5075–5088, https://doi.org/10.5194/amt-10-5075-2017, 2017.
Dillon, T. J., Tucceri, M. E., and Crowley, J. N.: Laser induced fluorescence studies of iodine oxide chemistry: Part II. The reactions of IO with CH3O2, CF3O2 and O3, Phys. Chem. Chem. Phys., 8, 5185–5198, https://doi.org/10.1039/b611116e, 2006.
Duplissy, J., Merikanto, J., Franchin, A., Tsagkogeorgas, G., Kangasluoma, J., Wimmer, D., Vuollekoski, H., Schobesberger, S., Lehtipalo, K., Flagan, R. C., Brus, D., Donahue, N. M., Vehkamaki, H., Almeida, J., Amorim, A., Barmet, P., Bianchi, F., Breitenlechner, M., Dunne, E. M., Guida, R., Henschel, H., Junninen, H., Kirkby, J., Kurten, A., Kupc, A., Maattanen, A., Makhmutov, V., Mathot, S., Nieminen, T., Onnela, A., Praplan, A. P., Riccobono, F., Rondo, L., Steiner, G., Tome, A., Walther, H., Baltensperger, U., Carslaw, K. S., Dommen, J., Hansel, A., Petaja, T., Sipila, M., Stratmann, F., Vrtala, A., Wagner, P. E., Worsnop, D. R., Curtius, J., and Kulmala, M.: Effect of ions on sulfuric acid-water binary particle formation: 2. Experimental data and comparison with QC-normalized classical nucleation theory, J. Geophys. Res., 121, 1752–1775, https://doi.org/10.1002/2015JD023539, 2016.
Feller, D.: The role of databases in support of computational chemistry calculations, J. Comput. Chem., 17, 1571–1586, https://doi.org/10.1002/(SICI)1096-987X(199610)17:13<1571::AID-JCC9>3.0.CO;2-P, 1996.
Finley, B. D. and Saltzman, E. S.: Observations of Cl2, Br2, and I2 in coastal marine air, J. Geophys. Res.-Atmos., 113, D21301, https://doi.org/10.1029/2008JD010269, 2008.
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., Scalmani, G., Barone, V., Mennucci, B., Petersson, G. A., Nakatsuji, H., Caricato, M., Li, X., Hratchian, H. P., Izmaylov, A. F., Bloino, J., Zheng, G., Sonnenberg, J. L., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Montgomery Jr., J. A., Peralta, J. E., Ogliaro, F., Bearpark, M., Heyd, J. J., Brothers, E., Kudin, K. N., Staroverov, V. N., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A., Burant, J. C., Iyengar, S. S., Tomasi, J., Cossi, M., Rega, N., Millam, J. M., Klene, M., Knox, J. E., Cross, J. B., Bakken, V., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R. E., Yazyev, O., Austin, A. J., Cammi, R., Pomelli, C., Ochterski, J. W., Martin, R. L., Morokuma, K., Zakrzewski, V. G., Voth, G. A., Salvador, P., Dannenberg, J. J., Dapprich, S., Daniels, A. D., Farkas, Ö., Foresman, J. B., Ortiz, J. V, Cioslowski, J., and Fox, D. J.: Gaussian09 Revision D.01, Gaussian Inc. Wallingford CT, Gaussian 09 Revis. C.01, available at: http://www.gaussian.com (last access: 1 December 2020), 2010.
Gallagher, M. S., King, D. B., Whung, P. Y., and Saltzman, E. S.: Performance of the HPLC/fluorescence SO2 detector during the GASIE instrument intercomparison experiment, J. Geophys. Res.-Atmos., 102, 16247–16254, https://doi.org/10.1029/97jd00700, 1997.
Gómez Martín, J. C., Blahins, J., Gross, U., Ingham, T., Goddard, A., Mahajan, A. S., Ubelis, A., and Saiz-Lopez, A.: In situ detection of atomic and molecular iodine using Resonance and Off-Resonance Fluorescence by Lamp Excitation: ROFLEX, Atmos. Meas. Tech., 4, 29–45, https://doi.org/10.5194/amt-4-29-2011, 2011.
Gómez Martín, J. C., Gálvez, O., Baeza-Romero, M. T., Ingham, T., Plane, J. M. C., and Blitz, M. A.: On the mechanism of iodine oxide particle formation, Phys. Chem. Chem. Phys., 15, 15612–15622, https://doi.org/10.1039/c3cp51217g, 2013.
He, X.: From the measurement of halogenated species to iodine particle formation, M.Sc. thesis, Institute for Atmospheric and Earth System Research, University of Helsinki, Finland, 71 pp., 2017.
He, X.-C., Iyer, S., Sipilä, M., Ylisirniö, A., Peltola, M., Kontkanen, J., Baalbaki, R., Simon, M., Kürten, A., Tham, Y. J., Pesonen, J., Ahonen, L. R., Amanatidis, S., Amorim, A., Baccarini, A., Beck, L., Bianchi, F., Brilke, S., Chen, D., Chiu, R., Curtius, J., Dada, L., Dias, A., Dommen, J., Donahue, N. M., Duplissy, J., El Haddad, I., Finkenzeller, H., Fischer, L., Heinritzi, M., Hofbauer, V., Kangasluoma, J., Kim, C., Koenig, T. K., Kubečka, J., Kvashnin, A., Lamkaddam, H., Lee, C. P., Leiminger, M., Li, Z., Makhmutov, V., Xiao, M., Marten, R., Nie, W., Onnela, A., Partoll, E., Petäjä, T., Salo, V.-T., Schuchmann, S., Steiner, G., Stolzenburg, D., Stozhkov, Y., Tauber, C., Tomé, A., Väisänen, O., Vazquez-Pufleau, M., Volkamer, R., Wagner, A. C., Wang, M., Wang, Y., Wimmer, D., Winkler, P. M., Worsnop, D. R., Wu, Y., Yan, C., Ye, Q., Lehtinen, K., Nieminen, T., Manninen, H. E., Rissanen, M., Schobesberger, S., Lehtipalo, K., Baltensperger, U., Hansel, A., Kerminen, V.-M., Flagan, R. C., Kirkby, J., Kurtén, T., and Kulmala, M.: Determination of the collision rate coefficient between charged iodic acid clusters and iodic acid using the appearance time method, Aerosol Sci. Technol., 55, 231–242, https://doi.org/10.1080/02786826.2020.1839013, 2021a.
He, X.-C., Tham, Y. J., Dada, L., Wang, M., Finkenzeller, H., Stolzenburg, D., Iyer, S., Simon, M., Kürten, A., Shen, J., Rörup, B., Rissanen, M., Schobesberger, S., Baalbaki, R., Wang, D. S., Koenig, T. K., Jokinen, T., Sarnela, N., Beck, L. J., Almeida, J., Amanatidis, S., Amorim, A., Ataei, F., Baccarini, A., Bertozzi, B., Bianchi, F., Brilke, S., Caudillo, L., Chen, D., Chiu, R., Chu, B., Dias, A., Ding, A., Dommen, J., Duplissy, J., El Haddad, I., Gonzalez Carracedo, L., Granzin, M., Hansel, A., Heinritzi, M., Hofbauer, V., Junninen, H., Kangasluoma, J., Kemppainen, D., Kim, C., Kong, W., Krechmer, J. E., Kvashin, A., Laitinen, T., Lamkaddam, H., Lee, C. P., Lehtipalo, K., Leiminger, M., Li, Z., Makhmutov, V., Manninen, H. E., Marie, G., Marten, R., Mathot, S., Mauldin, R. L., Mentler, B., Möhler, O., Müller, T., Nie, W., Onnela, A., Petäjä, T., Pfeifer, J., Philippov, M., Ranjithkumar, A., Saiz-Lopez, A., Salma, I., Scholz, W., Schuchmann, S., Schulze, B., Steiner, G., Stozhkov, Y., Tauber, C., Tomé, A., Thakur, R. C., Väisänen, O., Vazquez-Pufleau, M., Wagner, A. C., Wang, Y., Weber, S. K., Winkler, P. M., Wu, Y., Xiao, M., Yan, C., Ye, Q., Ylisirniö, A., Zauner-Wieczorek, M., Zha, Q., Zhou, P., Flagan, R. C., Curtius, J., Baltensperger, U., Kulmala, M., Kerminen, V.-M., Kurtén, T., Donahue, N. M., Volkamer, R., Kirkby, J., Worsnop, D. R., and Sipilä, M.: Role of iodine oxoacids in atmospheric aerosol nucleation, Science, 371, 589–595, 2021b.
Iyer, S., Lopez-Hilfiker, F., Lee, B. H., Thornton, J. A., and Kurtén, T.: Modeling the Detection of Organic and Inorganic Compounds Using Iodide-Based Chemical Ionization, J. Phys. Chem. A, 120, 576–587, https://doi.org/10.1021/acs.jpca.5b09837, 2016.
Junninen, H., Ehn, M., Petäjä, T., Luosujärvi, L., Kotiaho, T., Kostiainen, R., Rohner, U., Gonin, M., Fuhrer, K., Kulmala, M., and Worsnop, D. R.: A high-resolution mass spectrometer to measure atmospheric ion composition, Atmos. Meas. Tech., 3, 1039–1053, https://doi.org/10.5194/amt-3-1039-2010, 2010.
Kazantseva, N. N., Ernepesova, A., Khodjamamedov, A., Geldyev, O. A., and Krumgalz, B. S.: Spectrophotometric analysis of iodide oxidation by chlorine in highly mineralized solutions, Anal. Chim. Acta, 456, 105–119, https://doi.org/10.1016/S0003-2670(01)01625-7, 2002.
Kendall, R. A., Dunning, T. H., and Harrison, R. J.: Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions, J. Chem. Phys., 96, 6796, https://doi.org/10.1063/1.462569, 1992.
Kercher, J. P., Riedel, T. P., and Thornton, J. A.: Chlorine activation by N2O5: simultaneous, in situ detection of ClNO2 and N2O5 by chemical ionization mass spectrometry, Atmos. Meas. Tech., 2, 193–204, https://doi.org/10.5194/amt-2-193-2009, 2009.
Kirkby, J., Curtius, J., Almeida, J., Dunne, E., Duplissy, J., Ehrhart, S., Franchin, A., Gagné, S., Ickes, L., Kürten, A., Kupc, A., Metzger, A., Riccobono, F., Rondo, L., Schobesberger, S., Tsagkogeorgas, G., Wimmer, D., Amorim, A., Bianchi, F., Breitenlechner, M., David, A., Dommen, J., Downard, A., Ehn, M., Flagan, R. C., Haider, S., Hansel, A., Hauser, D., Jud, W., Junninen, H., Kreissl, F., Kvashin, A., Laaksonen, A., Lehtipalo, K., Lima, J., Lovejoy, E. R., Makhmutov, V., Mathot, S., Mikkilä, J., Minginette, P., Mogo, S., Nieminen, T., Onnela, A., Pereira, P., Petäjä, T., Schnitzhofer, R., Seinfeld, J. H., Sipilä, M., Stozhkov, Y., Stratmann, F., Tomé, A., Vanhanen, J., Viisanen, Y., Vrtala, A., Wagner, P. E., Walther, H., Weingartner, E., Wex, H., Winkler, P. M., Carslaw, K. S., Worsnop, D. R., Baltensperger, U., and Kulmala, M.: Role of sulphuric acid, ammonia and galactic cosmic rays in atmospheric aerosol nucleation, Nature, 476, 429–433, https://doi.org/10.1038/nature10343, 2011.
Kirkby, J., Duplissy, J., Sengupta, K., Frege, C., Gordon, H., Williamson, C., Heinritzi, M., Simon, M., Yan, C., Almeida, J., Trostl, J., Nieminen, T., Ortega, I. K., Wagner, R., Adamov, A., Amorim, A., Bernhammer, A. K., Bianchi, F., Breitenlechner, M., Brilke, S., Chen, X. M., Craven, J., Dias, A., Ehrhart, S., Flagan, R. C., Franchin, A., Fuchs, C., Guida, R., Hakala, J., Hoyle, C. R., Jokinen, T., Junninen, H., Kangasluoma, J., Kim, J., Krapf, M., Kurten, A., Laaksonen, A., Lehtipalo, K., Makhmutov, V., Mathot, S., Molteni, U., Onnela, A., Perakyla, O., Piel, F., Petaja, T., Praplan, A. P., Pringle, K., Rap, A., Richards, N. A. D. D., Riipinen, I., Rissanen, M. P., Rondo, L., Sarnela, N., Schobesberger, S., Scott, C. E., Seinfeld, J. H., Sipila, M., Steiner, G., Stozhkov, Y., Stratmann, F., Tome, A., Virtanen, A., Vogel, A. L., Wagner, A. C., Wagner, P. E., Weingartner, E., Wimmer, D., Winkler, P. M., Ye, P. L., Zhang, X., Hansel, A., Dommen, J., Donahue, N. M., Worsnop, D. R., Baltensperger, U., Kulmala, M., Carslaw, K. S., Curtius, J., Tomé, A., Virtanen, A., Vogel, A. L., Wagner, A. C., Wagner, P. E., Weingartner, E., Wimmer, D., Winkler, P. M., Ye, P. L., Zhang, X., Hansel, A., Dommen, J., Donahue, N. M., Worsnop, D. R., Baltensperger, U., Kulmala, M., Carslaw, K. S., and Curtius, J.: Ion-induced nucleation of pure biogenic particles, Nature, 533, 521–526, https://doi.org/10.1038/nature17953, 2016.
Koenig, T. K., Baidar, S., Campuzano-Jost, P., Cuevas, C. A., Dix, B., Fernandez, R. P., Guo, H., Hall, S. R., Kinnison, D., Nault, B. A., Ullmann, K., Jimenez, J. L., Saiz-Lopez, A., and Volkamer, R.: Quantitative detection of iodine in the stratosphere, P. Natl. Acad. Sci. USA, 117, 1860–1866, https://doi.org/10.1073/pnas.1916828117, 2020.
Kürten, A., Rondo, L., Ehrhart, S., and Curtius, J.: Calibration of a Chemical Ionization Mass Spectrometer for the Measurement of Gaseous Sulfuric Acid, J. Phys. Chem. A, 116, 6375–6386, https://doi.org/10.1021/Jp212123n, 2012.
Lawler, M. J., Sander, R., Carpenter, L. J., Lee, J. D., von Glasow, R., Sommariva, R., and Saltzman, E. S.: HOCl and Cl2 observations in marine air, Atmos. Chem. Phys., 11, 7617–7628, https://doi.org/10.5194/acp-11-7617-2011, 2011.
Lee, B. H., Lopez-Hilfiker, F. D., Mohr, C., Kurten, T., Worsnop, D. R., and Thornton, J. A.: An iodide-adduct high-resolution time-of-flight chemical-ionization mass spectrometer: application to atmospheric inorganic and organic compounds, Env. Sci Technol, 48, 6309–6317, https://doi.org/10.1021/es500362a, 2014.
Lee, B. H., Lopez-Hilfiker, F. D., Veres, P. R., McDuffie, E. E., Fibiger, D. L., Sparks, T. L., Ebben, C. J., Green, J. R., Schroder, J. C., Campuzano-Jost, P., Iyer, S., D'Ambro, E. L., Schobesberger, S., Brown, S. S., Wooldridge, P. J., Cohen, R. C., Fiddler, M. N., Bililign, S., Jimenez, J. L., Kurtén, T., Weinheimer, A. J., Jaegle, L., and Thornton, J. A.: Flight Deployment of a High-Resolution Time-of-Flight Chemical Ionization Mass Spectrometer: Observations of Reactive Halogen and Nitrogen Oxide Species, J. Geophys. Res.-Atmos., 123, 7670–7686, https://doi.org/10.1029/2017JD028082, 2018.
Leigh, R. J., Ball, S. M., Whitehead, J., Leblanc, C., Shillings, A. J. L., Mahajan, A. S., Oetjen, H., Lee, J. D., Jones, C. E., Dorsey, J. R., Gallagher, M., Jones, R. L., Plane, J. M. C., Potin, P., and McFiggans, G.: Measurements and modelling of molecular iodine emissions, transport and photodestruction in the coastal region around Roscoff, Atmos. Chem. Phys., 10, 11823–11838, https://doi.org/10.5194/acp-10-11823-2010, 2010.
Lopez-Hilfiker, F. D., Mohr, C., Ehn, M., Rubach, F., Kleist, E., Wildt, J., Mentel, Th. F., Lutz, A., Hallquist, M., Worsnop, D., and Thornton, J. A.: A novel method for online analysis of gas and particle composition: description and evaluation of a Filter Inlet for Gases and AEROsols (FIGAERO), Atmos. Meas. Tech., 7, 983–1001, https://doi.org/10.5194/amt-7-983-2014, 2014.
Lopez-Hilfiker, F. D., Iyer, S., Mohr, C., Lee, B. H., D'Ambro, E. L., Kurtén, T., and Thornton, J. A.: Constraining the sensitivity of iodide adduct chemical ionization mass spectrometry to multifunctional organic molecules using the collision limit and thermodynamic stability of iodide ion adducts, Atmos. Meas. Tech., 9, 1505–1512, https://doi.org/10.5194/amt-9-1505-2016, 2016.
Marcy, T. P., Fahey, D. W., Gao, R. S., Popp, P. J., Richard, E. C., Thompson, T. L., Rosenlof, K. H., Ray, E. A., Salawitch, R. J., Atherton, C. S., Bergmann, D. J., Ridley, B. A., Weinheimer, A. J., Loewenstein, M., Weinstock, E. M., and Mahoney, M. J.: Quantifying Stratospheric Ozone in the Upper Troposphere with in Situ Measurements of HCl, Science, 304, 261–265, https://doi.org/10.1126/science.1093418, 2004.
McFiggans, G., Coe, H., Burgess, R., Allan, J., Cubison, M., Alfarra, M. R., Saunders, R., Saiz-Lopez, A., Plane, J. M. C., Wevill, D., Carpenter, L., Rickard, A. R., and Monks, P. S.: Direct evidence for coastal iodine particles from Laminaria macroalgae – linkage to emissions of molecular iodine, Atmos. Chem. Phys., 4, 701–713, https://doi.org/10.5194/acp-4-701-2004, 2004.
Meinen, J., Thieser, J., Platt, U., and Leisner, T.: Technical Note: Using a high finesse optical resonator to provide a long light path for differential optical absorption spectroscopy: CE-DOAS, Atmos. Chem. Phys., 10, 3901–3914, https://doi.org/10.5194/acp-10-3901-2010, 2010.
Neese, F.: The ORCA program system, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2, 73–78, https://doi.org/10.1002/wcms.81, 2012.
O'Dowd, C. D., Jimenez, J. L., Bahreini, R., Flagan, R. C., Seinfeld, J. H., Hämerl, K., Pirjola, L., Kulmala, M., and Hoffmann, T.: Marine aerosol formation from biogenic iodine emissions, Nature, 417, 632–636, https://doi.org/10.1038/nature00775, 2002.
Passananti, M., Zapadinsky, E., Zanca, T., Kangasluoma, J., Myllys, N., Rissanen, M. P., Kurtén, T., Ehn, M., Attoui, M., and Vehkamäki, H.: How well can we predict cluster fragmentation inside a mass spectrometer?, Chem. Commun., 55, 5946–5949, https://doi.org/10.1039/c9cc02896j, 2019.
Pfeifer, J., Simon, M., Heinritzi, M., Piel, F., Weitz, L., Wang, D., Granzin, M., Müller, T., Bräkling, S., Kirkby, J., Curtius, J., and Kürten, A.: Measurement of ammonia, amines and iodine compounds using protonated water cluster chemical ionization mass spectrometry, Atmos. Meas. Tech., 13, 2501–2522, https://doi.org/10.5194/amt-13-2501-2020, 2020.
Prados-Roman, C., Cuevas, C. A., Fernandez, R. P., Kinnison, D. E., Lamarque, J.-F., and Saiz-Lopez, A.: A negative feedback between anthropogenic ozone pollution and enhanced ocean emissions of iodine, Atmos. Chem. Phys., 15, 2215–2224, https://doi.org/10.5194/acp-15-2215-2015, 2015.
Raso, A. R. W., Custard, K. D., May, N. W., Tanner, D., Newburn, M. K., Walker, L., Moore, R. J., Huey, L. G., Alexander, L., Shepson, P. B., and Pratt, K. A.: Active molecular iodine photochemistry in the Arctic, P. Natl. Acad. Sci. USA, 114, 10053–10058, https://doi.org/10.1073/pnas.1702803114, 2017.
Riplinger, C. and Neese, F.: An efficient and near linear scaling pair natural orbital based local coupled cluster method, J. Chem. Phys., 138, 034106, https://doi.org/10.1063/1.4773581, 2013.
Rissanen, M. P., Mikkilä, J., Iyer, S., and Hakala, J.: Multi-scheme chemical ionization inlet (MION) for fast switching of reagent ion chemistry in atmospheric pressure chemical ionization mass spectrometry (CIMS) applications, Atmos. Meas. Tech., 12, 6635–6646, https://doi.org/10.5194/amt-12-6635-2019, 2019.
Rothman, L. S., Gordon, I. E., Barber, R. J., Dothe, H., Gamache, R. R., Goldman, A., Perevalov, V. I., Tashkun, S. A., and Tennyson, J.: HITEMP, the high-temperature molecular spectroscopic database, J. Quant. Spectrosc. Radiat. Transf., 111, 2139–2150, https://doi.org/10.1016/j.jqsrt.2010.05.001, 2010.
Saiz-Lopez, A., Plane, J. M. C., Baker, A. R., Carpenter, L. J., Von Glasow, R., Gómez Martín, J. C., McFiggans, G., and Saunders, R. W.: Atmospheric chemistry of iodine, Chem. Rev., 112, 1773–1804, https://doi.org/10.1021/cr200029u, 2012.
Sanchez, J., Tanner, D. J., Chen, D., Huey, L. G., and Ng, N. L.: A new technique for the direct detection of HO2 radicals using bromide chemical ionization mass spectrometry (Br-CIMS): initial characterization, Atmos. Meas. Tech., 9, 3851–3861, https://doi.org/10.5194/amt-9-3851-2016, 2016.
Schnitzhofer, R., Metzger, A., Breitenlechner, M., Jud, W., Heinritzi, M., De Menezes, L.-P., Duplissy, J., Guida, R., Haider, S., Kirkby, J., Mathot, S., Minginette, P., Onnela, A., Walther, H., Wasem, A., Hansel, A., and the CLOUD Team: Characterisation of organic contaminants in the CLOUD chamber at CERN, Atmos. Meas. Tech., 7, 2159–2168, https://doi.org/10.5194/amt-7-2159-2014, 2014.
Schwehr, K. A., Santschi, P. H., and Elmore, D.: The dissolved organic iodine species of the isotopic ratio of 129I 127I: A novel tool for tracing terrestrial organic carbon in the estuarine surface waters of Galveston Bay, Texas, Limnol. Oceanogr. Methods, 3, 326–337, https://doi.org/10.4319/lom.2005.3.326, 2005.
Sherwen, T., Evans, M. J., Carpenter, L. J., Andrews, S. J., Lidster, R. T., Dix, B., Koenig, T. K., Sinreich, R., Ortega, I., Volkamer, R., Saiz-Lopez, A., Prados-Roman, C., Mahajan, A. S., and Ordóñez, C.: Iodine's impact on tropospheric oxidants: a global model study in GEOS-Chem, Atmos. Chem. Phys., 16, 1161–1186, https://doi.org/10.5194/acp-16-1161-2016, 2016.
Simpson, W. R., Brown, S. S., Saiz-Lopez, A., Thornton, J. A., and Von Glasow, R.: Tropospheric Halogen Chemistry: Sources, Cycling, and Impacts, Chem. Rev., 115, 4035–4062, https://doi.org/10.1021/cr5006638, 2015.
Sipilä, M., Sarnela, N., Jokinen, T., Henschel, H., Junninen, H., Kontkanen, J., Richters, S., Kangasluoma, J., Franchin, A., Peräkylä, O., Rissanen, M. P., Ehn, M., Vehkamäki, H., Kurten, T., Berndt, T., Petäjä, T., Worsnop, D., Ceburnis, D., Kerminen, V. M., Kulmala, M., and O'Dowd, C.: Molecular-scale evidence of aerosol particle formation via sequential addition of HIO3, Nature, 537, 532–534, https://doi.org/10.1038/nature19314, 2016.
Spietz, P., Gómez Martín, J., and Burrows, J. P.: Effects of column density on I2 spectroscopy and a determination of I2 absorption cross section at 500 nm, Atmos. Chem. Phys., 6, 2177–2191, https://doi.org/10.5194/acp-6-2177-2006, 2006.
Spolaor, A., Gabrieli, J., Martma, T., Kohler, J., Björkman, M. B., Isaksson, E., Varin, C., Vallelonga, P., Plane, J. M. C., and Barbante, C.: Sea ice dynamics influence halogen deposition to Svalbard, The Cryosphere, 7, 1645–1658, https://doi.org/10.5194/tc-7-1645-2013, 2013.
Stolzenburg, D., Simon, M., Ranjithkumar, A., Kürten, A., Lehtipalo, K., Gordon, H., Ehrhart, S., Finkenzeller, H., Pichelstorfer, L., Nieminen, T., He, X.-C., Brilke, S., Xiao, M., Amorim, A., Baalbaki, R., Baccarini, A., Beck, L., Bräkling, S., Caudillo Murillo, L., Chen, D., Chu, B., Dada, L., Dias, A., Dommen, J., Duplissy, J., El Haddad, I., Fischer, L., Gonzalez Carracedo, L., Heinritzi, M., Kim, C., Koenig, T. K., Kong, W., Lamkaddam, H., Lee, C. P., Leiminger, M., Li, Z., Makhmutov, V., Manninen, H. E., Marie, G., Marten, R., Müller, T., Nie, W., Partoll, E., Petäjä, T., Pfeifer, J., Philippov, M., Rissanen, M. P., Rörup, B., Schobesberger, S., Schuchmann, S., Shen, J., Sipilä, M., Steiner, G., Stozhkov, Y., Tauber, C., Tham, Y. J., Tomé, A., Vazquez-Pufleau, M., Wagner, A. C., Wang, M., Wang, Y., Weber, S. K., Wimmer, D., Wlasits, P. J., Wu, Y., Ye, Q., Zauner-Wieczorek, M., Baltensperger, U., Carslaw, K. S., Curtius, J., Donahue, N. M., Flagan, R. C., Hansel, A., Kulmala, M., Lelieveld, J., Volkamer, R., Kirkby, J., and Winkler, P. M.: Enhanced growth rate of atmospheric particles from sulfuric acid, Atmos. Chem. Phys., 20, 7359–7372, https://doi.org/10.5194/acp-20-7359-2020, 2020.
Thalman, R. and Volkamer, R.: Temperature dependent absorption cross-sections of O2–O2 collision pairs between 340 and 630 nm and at atmospherically relevant pressure, Phys. Chem. Chem. Phys., 15, 15371–15381, https://doi.org/10.1039/c3cp50968k, 2013.
Tham, Y. J., Wang, Z., Li, Q., Yun, H., Wang, W., Wang, X., Xue, L., Lu, K., Ma, N., Bohn, B., Li, X., Kecorius, S., Größ, J., Shao, M., Wiedensohler, A., Zhang, Y., and Wang, T.: Significant concentrations of nitryl chloride sustained in the morning: investigations of the causes and impacts on ozone production in a polluted region of northern China, Atmos. Chem. Phys., 16, 14959–14977, https://doi.org/10.5194/acp-16-14959-2016, 2016.
Tham, Y. J., He, X.-C., Li, Q., Cuevas, C. A., Shen, J., Kalliokoski, J., Yan, C., Iyer, S., Lehmusjärvi, T., Jang, S., Thakur, R. C., Beck, L., Kemppainen, D., Olin, M., Sarnela, N., Mikkilä, J., Hakala, J., Marbouti, M., Yao, L., Li, H., Huang, W., Wang, Y., Wimmer, D., Zha, Q., Virkanen, J., Spain, T. G., Doherty, S., Jokinen, T., Bianchi, F., Petäjä, T., Worsnop, D. R., Mauldin, R. L., Ovadnevaite, J., Ceburnis, D., Maier, N. M., Kulmala, M., O'Dowd, C., Dal Maso, M., Saiz-Lopez, A., and Sipilä, M.: Direct field evidence of autocatalytic iodine release from atmospheric aerosol, P. Natl. Acad. Sci. USA, 118, e2009951118, https://doi.org/10.1073/pnas.2009951118, 2021.
Vandaele, A. C., Hermans, C., Simon, P. C., Carleer, M., Colin, R., Fally, S., Mérienne, M. F., Jenouvrier, A., and Coquart, B.: Measurements of the NO2 absorption cross-section from 42 000 cm−1 to 10 000 cm−1 (238–1000 nm) at 220 K and 294 K, J. Quant. Spectrosc. Radiat. Transf., 59, 171–184, https://doi.org/10.1016/S0022-4073(97)00168-4, 1998.
Vaughan, S., Gherman, T., Ruth, A. A., and Orphal, J.: Incoherent broad-band cavity-enhanced absorption spectroscopy of the marine boundary layer species I2, IO and OIO, Phys. Chem. Chem. Phys., 10, 4471–4477, https://doi.org/10.1039/b802618a, 2008.
Wang, M., Kong, W., Marten, R., He, X. C., Chen, D., Pfeifer, J., Heitto, A., Kontkanen, J., Dada, L., Kürten, A., Yli-Juuti, T., Manninen, H. E., Amanatidis, S., Amorim, A., Baalbaki, R., Baccarini, A., Bell, D. M., Bertozzi, B., Bräkling, S., Brilke, S., Murillo, L. C., Chiu, R., Chu, B., De Menezes, L. P., Duplissy, J., Finkenzeller, H., Carracedo, L. G., Granzin, M., Guida, R., Hansel, A., Hofbauer, V., Krechmer, J., Lehtipalo, K., Lamkaddam, H., Lampimäki, M., Lee, C. P., Makhmutov, V., Marie, G., Mathot, S., Mauldin, R. L., Mentler, B., Müller, T., Onnela, A., Partoll, E., Petäjä, T., Philippov, M., Pospisilova, V., Ranjithkumar, A., Rissanen, M., Rörup, B., Scholz, W., Shen, J., Simon, M., Sipilä, M., Steiner, G., Stolzenburg, D., Tham, Y. J., Tomé, A., Wagner, A. C., Wang, D. S., Wang, Y., Weber, S. K., Winkler, P. M., Wlasits, P. J., Wu, Y., Xiao, M., Ye, Q., Zauner-Wieczorek, M., Zhou, X., Volkamer, R., Riipinen, I., Dommen, J., Curtius, J., Baltensperger, U., Kulmala, M., Worsnop, D. R., Kirkby, J., Seinfeld, J. H., El-Haddad, I., Flagan, R. C., and Donahue, N. M.: Rapid growth of new atmospheric particles by nitric acid and ammonia condensation, Nature, 581, 184–189, https://doi.org/10.1038/s41586-020-2270-4, 2020.
Washenfelder, R. A., Langford, A. O., Fuchs, H., and Brown, S. S.: Measurement of glyoxal using an incoherent broadband cavity enhanced absorption spectrometer, Atmos. Chem. Phys., 8, 7779–7793, https://doi.org/10.5194/acp-8-7779-2008, 2008.