the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 28 Jul 2022
| 28 Jul 2022
Temperature-dependent sensitivity of iodide chemical ionization mass spectrometers
Michael A. Robinson
J. Andrew Neuman
L. Gregory Huey
James M. Roberts
Steven S. Brown
Iodide chemical ionization mass spectrometry (CIMS) is a common analytical tool used in both laboratory and field experiments to measure a large suite of atmospherically relevant compounds. Here, we describe a systematic ion molecule reactor (IMR) temperature dependence of iodide CIMS analyte sensitivity for a wide range of analytes in laboratory experiments. Weakly bound iodide clusters, such as HCl, HONO, HCOOH, HCN, phenol, 2-nitrophenol, and acyl peroxynitrate (PAN) detected via the peroxy radical cluster, all exhibit strong IMR temperature dependence of sensitivity ranging from −3.4 % ∘C−1 to 5.9 % ∘C−1 (from 37 to 47 ∘C). Strongly bound iodide clusters, such as Br2, N2O5, ClNO2, and PAN detected via the carboxylate anion, all exhibit little to no IMR temperature dependence ranging from 0.2 % ∘C−1 to −0.9 % ∘C−1 (from 37 to 47 ∘C). The IMR temperature relationships of weakly bound clusters provide an estimate of net reaction enthalpy, and comparison with database values indicates that these clusters are in thermal equilibrium. Ground site HCOOH data collected in the summer of 2021 in Pasadena (CA) are corrected and show a reversal in the diel cycle, emphasizing the importance of this correction (35±6 % during the day, % at night). Finally, we recommend two approaches to minimize this effect in the field, namely heating or cooling the IMR; the latter technique has the added benefit of improving absolute sensitivity.
- Article
(2031 KB) - Full-text XML
-
Supplement
(989 KB) - BibTeX
- EndNote





Atmospheric trace gases participate in chemical reactions that impact air quality and climate. Quantification of these trace gases, often at parts per trillion levels, demands highly sensitive and selective measurements. For decades, chemical ionization mass spectrometry (CIMS) has been used to provide rapid and sensitive in situ measurements of a wide variety of trace gases and aerosol composition. More recently, time-of-flight mass spectrometers (ToF-MS) have been developed to measure tens or even hundreds of compounds simultaneously, making their field deployment increasingly common in the last decade (Bertram et al., 2011; Lee et al., 2014; Lopez-Hilfiker et al., 2014, 2019; Krechmer et al., 2018; Koss et al., 2018; Veres et al., 2021). CIMS instruments employ reagent ions to react selectively with analytes of interest, and a number of negative (I−, Br−, CF3O−, SF6−, CH3C(O)O−) and positive (H3O+, NO+, NH4+) reagent ions have been utilized in field and lab experiments to quantify a wide range of atmospheric trace gases (Slusher et al., 2004; Crounse et al., 2006; Huey et al., 1995; Veres et al., 2008; de Gouw and Warneke, 2007; Koss et al., 2016; Zaytsev et al., 2019). Negative ion-adduct chemistry has been used with quadrupole mass spectrometers that sample the atmosphere since 1975 (Dougherty et al., 1975). The use of iodide-adduct ion chemistry has become commonplace in atmospheric chemistry field campaigns, as this chemistry has provided sensitive measurements of molecular halogens, halogen oxides, carboxylic acids, acyl peroxynitrates (PANs), oxygenated organics, and nitrogen oxides (Slusher et al., 2004; Osthoff et al., 2008; Kercher et al., 2009; Thornton et al., 2010; Mielke et al., 2011; Riedel et al., 2012; Neuman et al., 2016).
Often iodide ions (and/or its water clusters) react with a neutral analyte molecule to form an adduct that can be identified and detected in a ToF-MS. Iodide's large mass defect, low likelihood of electron transfer, or proton abstraction often allow selective ionization chemistry that can achieve low detection limits and avoid interferences. Additionally, iodide's single natural isotope greatly simplifies complex mass spectral interpretation. Iodide-adduct formation is typically only slightly exergonic (i.e., ΔG<0), meaning that adduct formation is rapid (i.e., every collision forms a cluster ion) and relatively fragment-free (i.e., soft ionization; de Hoffmann and Stroobant, 2007). A complication of iodide CIMS, in contrast to proton transfer reactions, is that sensitivities to different analytes range several orders of magnitude (Iyer et al., 2016) which implies that product ion abundance is not solely determined by forward-reaction rates. It has been proposed that this range of sensitivities is due to the variation in binding enthalpy between the iodide anion and neutrals (Iyer et al., 2016).
The iodide anion-molecule chemistry in the presence of water is a system of reactions.
Here, I− and I−• H2O are reagent ions, A is the analyte of interest, and I• A− is a product ion. Reaction (R3) pertains to the special analyte case of peroxyacetyl radicals, which differs from most analyte cluster reaction pathways (i.e., Reactions R1 or R2). The portion of each reaction pathway depends on the analyte and reagent ([I• H2O−] or [I−]). Ambient mixing ratios of a given analyte are obtained using Eq. (1):
where k is the effective rate constant of the reaction, and t is the reaction time. This equation also holds true for other reagent ions (i.e., I−•H2O). Typically k and t are not determined separately, but the product of the two parameters, or the effective sensitivity, is determined for a given analyte (Huey, 2007).
Much has been done to characterize this ion chemistry for application to atmospheric chemistry field and laboratory studies (Huey et al., 1995; Slusher et al., 2004; Kercher et al., 2009; Lee et al., 2014; Lopez-Hilfiker et al., 2016). Analyte ionization occurs subsequent to mixing with reagent ions in a volume known as the ion molecule reactor (IMR; see Fig. 1). Instrument analyte detection sensitivity is impacted by the effective reaction rate constants and pathways of the analyte and reagent ion(s) in the IMR (Reactions R1–R3). Water vapor plays an important role in the iodide-adduct ionization efficiency, which in turn impacts instrument sensitivity (Lee et al., 2014; Slusher et al., 2004; Huey et al., 1995; Kercher et al., 2009). Instrument sensitivity for a given analyte molecule can exhibit a strong relationship to no relationship with water vapor, and this water dependence requires careful calibration of these instruments as a function of the IMR water vapor pressure (Lee et al., 2014). This instrument sensitivity dependence on water has motivated efforts to actively control water or carefully calibrate as a function of water in order to stabilize or determine sensitivity during field deployment (Lee et al., 2014; Slusher et al., 2004; Veres et al., 2021). The IMR temperature determines the importance of kinetics vs. thermodynamics for determining the outcome of ion molecule reactions, but there is limited discussion of the importance of IMR temperature in CIMS systems.
Reactions in the IMR can be in a kinetic or thermodynamic control regime, where the kinetic regime is described as a sufficiently exergonic reaction (i.e., ) with a slow reverse-reaction rate, such that equilibrium is not established in the IMR. The thermodynamic control regime is described as a moderately exergonic reaction (i.e., ΔG<0) with reverse-reaction rates ∼5 times smaller than the residence time in the IMR, allowing the ion-analyte system to reach equilibrium on this timescale. The SiF5−(HNO3) system achieves equilibrium on timescales equivalent to the residence time in most IMRs used in field-deployable CIMS instruments (∼100 ms) (Huey, 2007). A CIMS instrument using Br− reagent ions to measure the Br−(HO2) cluster exhibited thermal equilibrium behavior, with a decrease in instrument sensitivity of 20 % with an increase in the IMR temperature from 20 to 40 ∘C (Sanchez et al., 2016). Similar thermochemistry systems (i.e., similar ΔG) are likely to equilibrate on similar timescales.
Limited kinetic studies have been conducted for iodide anion-molecule reactions. Examples include the N2O5 clustering reaction (Reaction R1; cm3 s−1 at 293 K) and the carboxylate anion reaction (Reaction R3; to cm3 s−1 at 298 K; Huey et al., 1995, p. 6; Villalta and Howard, 1996). However, quantum chemical calculations predict collision rates of neutral molecules with I− for a variety of atmospherically relevant molecules to range from to cm3 s−1 (Iyer et al., 2016). If an analyte is in the thermally controlled regime, the temperature-dependent equilibrium constant of this system of reactions will affect the instrument sensitivity. A system in equilibrium is described by the linear form of the van't Hoff equation, which relates the change in the equilibrium constant (Keq) of the ion-molecule reaction to the absolute temperature (T), as follows:
where ΔrH0 is the standard reaction enthalpy, ΔrS0 is the standard reaction entropy, and R is the gas constant. The slope and intercept of the van't Hoff plot (ln Keq vs. ) give the enthalpy and entropy change, respectively (Keesee and Castleman, 1986).
Analyte sensitivity in CIMS is a product of the net rate of product ion formation, the adduct binding energy, and the instrument transmission of those adduct ions. A recent framework for understanding iodide-adduct binding energies uses a mass spectrometer ion lens voltage-scanning procedure to estimate the relative binding energies (Lopez-Hilfiker et al., 2016). This approach gives important insight into the stability of ion adducts and the extent to which they may decluster during transmission and can, thus, be used to provide inferred or relative sensitivities (Lopez-Hilfiker et al., 2016). A drawback of the voltage-scanning method for the determination of relative adduct binding energies is the potential for unintended changes in instrument ion transmission, which may result from the modulation of instrument tuning to induce the needed change in field strength. In this work, we leverage kinetic energy in the form of a temperature change as a substitute for modulation of the field strength, thereby eliminating the impact of ion transmission. The effect of temperature on instrument sensitivity has not been widely explored for the iodide-adduct CIMS instrument, where the impact has been documented for limited analytes such as the iodide carboxylate anion reaction (Reaction R3) and the Br−(HO2) cluster (Villalta and Howard, 1996; Sanchez et al., 2016).
Previously, several CIMS IMR temperature control methods have been implemented. Documented instrument deployments range from actively heating the IMR region or hardware close to the IMR (50 to 60 ∘C), such as for the Filter Inlet for Gases and Aerosols instrument (Lopez-Hilfiker et al., 2014), to insulating (Lee et al., 2018) or even actively cooling the IMR region (15 ∘C) (Neuman et al., 2002). However, thermal coupling between the IMR and ToF body, which may be difficult to temperature control in field deployments of CIMS instruments (discussed below), may introduce temperature variations in regions where ion molecule reactions occur.
We extend the impact of temperature on the abundance of product ions to a range of atmospheric trace gases and provide a framework for understanding variations in detection sensitivity with temperature for I− ion molecule reactions. Additionally we estimate thermally controlled reaction pathways in our IMR system by comparing observed reaction enthalpies to literature values. Finally, we compare our experiments to field observations and recommend temperature control strategies to improve sensitivity stability.
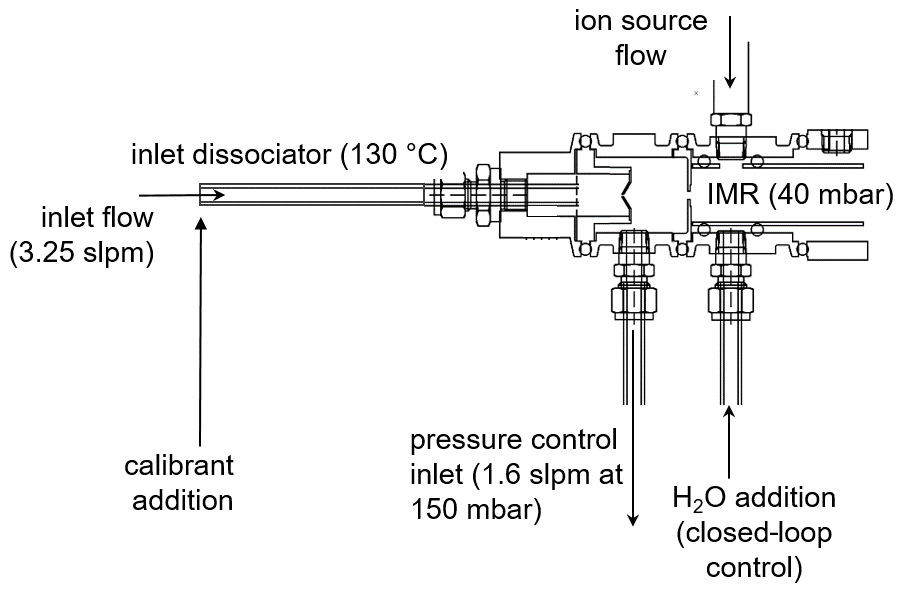
The experiments here are conducted on a modified commercially available CIMS instrument (https://www.tofwerk.com/products/api-tof/, last access: 23 July 2022) that has been used extensively for sampling from ground and airborne platforms (Veres et al., 2021). Figure 1 depicts the front end of the instrument, which differs from the commercially available instrument. The NOAA I− ToF-CIMS instrument (hereafter NOAA CIMS) utilizes a custom pressure-controlled inlet that maintains constant instrument pressures independent of ambient pressure. The NOAA CIMS notably operates at a lower IMR pressure (40 mbar, rather than the 100 mbar used in many instruments) and a lower segmented quadrupole (SSQ) pressure (1.64 mbar, rather than 2 mbar) in order to reduce the impact of secondary chemistry in the IMR and SSQ regions (Bertram et al., 2011; Lee et al., 2014). The remainder of the ToF body has not been modified.
The NOAA CIMS detects analyte molecules with I− and I−•H2O ion clusters using a ToF-MS with a mass resolving power of approximately 5000 (Δm m−1). Reagent ions are formed by flowing 1 slpm (standard liter per minute) of N2 and 9 sccm (standard cubic centimeters per minute) of 1025 ppm methyl iodide (CH3I in N2) in front of a vacuum ultraviolet krypton lamp (Restek photoionization lamp, model 108-BTEX, Restek Corporation, PA, USA). This ionization technique produces an intense and relatively interference-free source of iodide ions (Ji et al., 2020; Breitenlechner et al., 2022). These experiments (unless stated otherwise) were done with water dynamically added to the IMR (via a saturated small N2 flow, 0–100 sccm) to maintain a cluster ratio (CR; I−•H2O : I−) of 0.50±0.002, as is typical in field operation of the instrument. The added saturated N2 flow can dilute the sample in the IMR and is typically a negligible effect; however, due to the nature of these experiments (large N2 flow at high IMR temperatures), we have corrected for dilution. A detailed description of the CR closed-loop control system can be found in Sect. 1 in the Supplement.
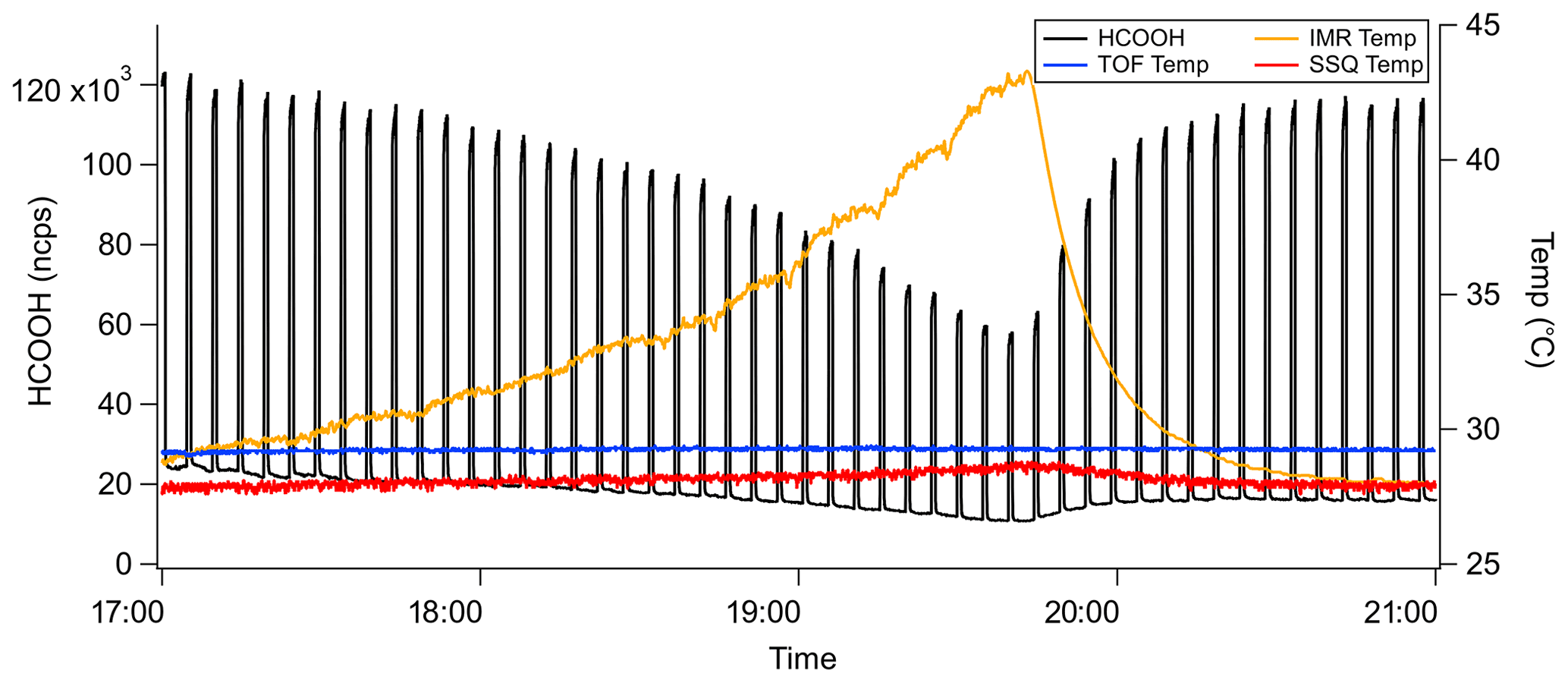
Figure 2Typical time series for an IMR temperature experiment. The product ion signal is normalized to the I−•H2O signal.
Ambient air was sampled through a mass-flow-controlled (3.25 slpm) perfluoroalkoxy alkane (PFA) inlet (30 cm length, 0.64 cm internal diameter), which was heated to 130 ∘C to thermally dissociate PAN for detection via the carboxylate anion (Reaction R3) or peroxy radical cluster (Slusher et al., 2004). Ambient air is sampled through a 1000 µm diameter orifice into a pressure-controlled region maintained at 150 mbar. This pressure control guarantees that 1.2 slpm enters the IMR, independent of ambient pressure, and mixes with the ∼1 slpm flow from the ion source. The reagent ion signals during this work were typically 1×106 ion counts per second (MHz) for I−•H2O and 2 MHz for I−. The ion packet extraction frequency in the time-of-flight region of the instrument during this work was 25 000 Hz. Product cluster ions are normalized by the I−•H2O signal to account for changes in ion source output. Every 2 min, the calibrants are removed and lab air is sampled; the calibration signals are then determined as the difference between lab air alone and lab air with calibrant. Two IMR designs are investigated in this work: the standard Aerodyne Inc. IMR (ARI) and the NOAA IMR, which has been deployed on the instrument for the Atmospheric Tomography-3 aircraft campaign (AToM-3, 2017, https://espo.nasa.gov/atom/archive/browse/atom/id10, last access: 23 July 2022), the Atmospheric Tomography-4 aircraft campaign (AToM-4, 2018, https://espo.nasa.gov/atom/archive/browse/atom/id14, last access: 23 July 2022), the Fire Influence on Regional to Global Environments and Air Quality aircraft campaign (FIREX-AQ, 2019, https://www-air.larc.nasa.gov/missions/firex-aq/, last access: 23 July 2022), and at the Southwest Urban NOx and VOC experiment ground site (SUNVEx, 2021, https://csl.noaa.gov/projects/sunvex/, last access: 23 July 2022). The NOAA IMR design was deployed during SUNVEx, and the ARI IMR design was used to investigate the influence of IMR temperature in the laboratory. Both IMR designs are summarized in Table S1 in the Supplement.
2.1 IMR temperature experiments
The ion chemistry that forms unique product ions occurs primarily in the IMR. Consequently, the temperature dependence of the sensitivity was predominately examined as a function of IMR temperature. Experiments were conducted using the widely used temperature-controllable ARI IMR, which includes a cartridge heater in a stainless steel block mounted to the IMR and controlled via the instrument's computer system (Bertram et al., 2011; Krechmer et al., 2016). All temperatures are obtained from manufacturer-supplied thermistors placed on the IMR and ToF regions. Even without independently heating the IMR, the heat generated by the instrument maintains the IMR temperature at approximately 30 ∘C. The IMR temperature was increased from 30 to 45 ∘C over a period of 3 h, as shown in Fig. 2. After reaching the maximum IMR temperature, temperature control was turned off to allow the IMR to return to ambient temperature. During the entire experiment, standard additions of analyte molecules occurred every 2 min for 45 s. The normalized product ion signal and instrument temperatures during a typical IMR temperature experiment with formic acid (HCOOH) additions are shown in Fig. 2.
2.2 ToF body temperature experiments
As discussed in Sect. 3.3 below, temperature-dependent ion chemistry can occur downstream of the IMR, in which case the IMR temperature alone does not determine sensitivity. These regions contain segmented quadrupoles that guide ions, and their temperature is determined largely by the temperature of the entire ToF body, which is closely coupled with ambient temperature. Collisional dissociation occurs during voltage-scanning experiments which vary voltages between the first (SSQ) and second (big segmented quadrupole, BSQ) quadrupole ion guides, indicating that SSQ temperatures may affect sensitivity (Lopez-Hilfiker et al., 2016). While newer instruments have improved temperature control of low-pressure regions of the instrument (the IMR, SSQ, BSQ, and ToF), for many applications it may not be feasible to control the temperature of the entire ToF, as it is a very large thermal mass that is often securely mounted when installed on a mobile platform. Consequently, we have investigated the impact of a variable ToF temperature while controlling the IMR temperature, in order to represent typical field conditions. These additional experiments were conducted using a radiative heater placed next to the ToF body and an insulating blanket. Three types of experiments were performed using the ARI IMR design to examine temperature control strategies that could be implemented to stabilize sensitivity if ambient temperatures change substantially. The first is to actively cool the IMR and determine sensitivity while the ambient temperature was varied. This control strategy may afford improved sensitivity for many molecules and reduces sensitivity changes caused by large changes in ambient temperature. The IMR was cooled using a thermoelectric chiller that pumps cold water through copper tubing wrapped around the IMR. This control strategy achieved an IMR temperature of ∼18 ∘C during these experiments. The second control strategy is to heat the IMR to a temperature higher than the maximum expected ToF body temperature (in this case ∼45 ∘C). This control strategy suffers from a loss in sensitivity for some analytes, but it is more easily implemented using the commercially provided hardware compared with cooling the IMR. For both of these active control strategies, the IMR temperature was allowed to stabilize, and the radiative heater was then turned on to a set point of ∼40 ∘C, which replicates many harsh field deployment temperatures on aircraft and at ground sites. The third strategy is the absence of active temperature control. This experiment replicates conditions during prior field deployments and reveals the sensitivity changes that can occur in the absence of IMR temperature control; it was conducted on both IMR designs. The product ion signals, reagent ion signals, and temperatures during a typical experiment without IMR temperature control are shown in Fig. S2. A comparison of IMR saturated N2 flow rates during the ToF body temperature experiment between the two investigated IMR designs is provided in Fig. S3. The changes observed in added IMR saturated N2 flow is likely due to geometric differences between the two designs and the impact on mixing in the IMR.
2.3 Calibration sources
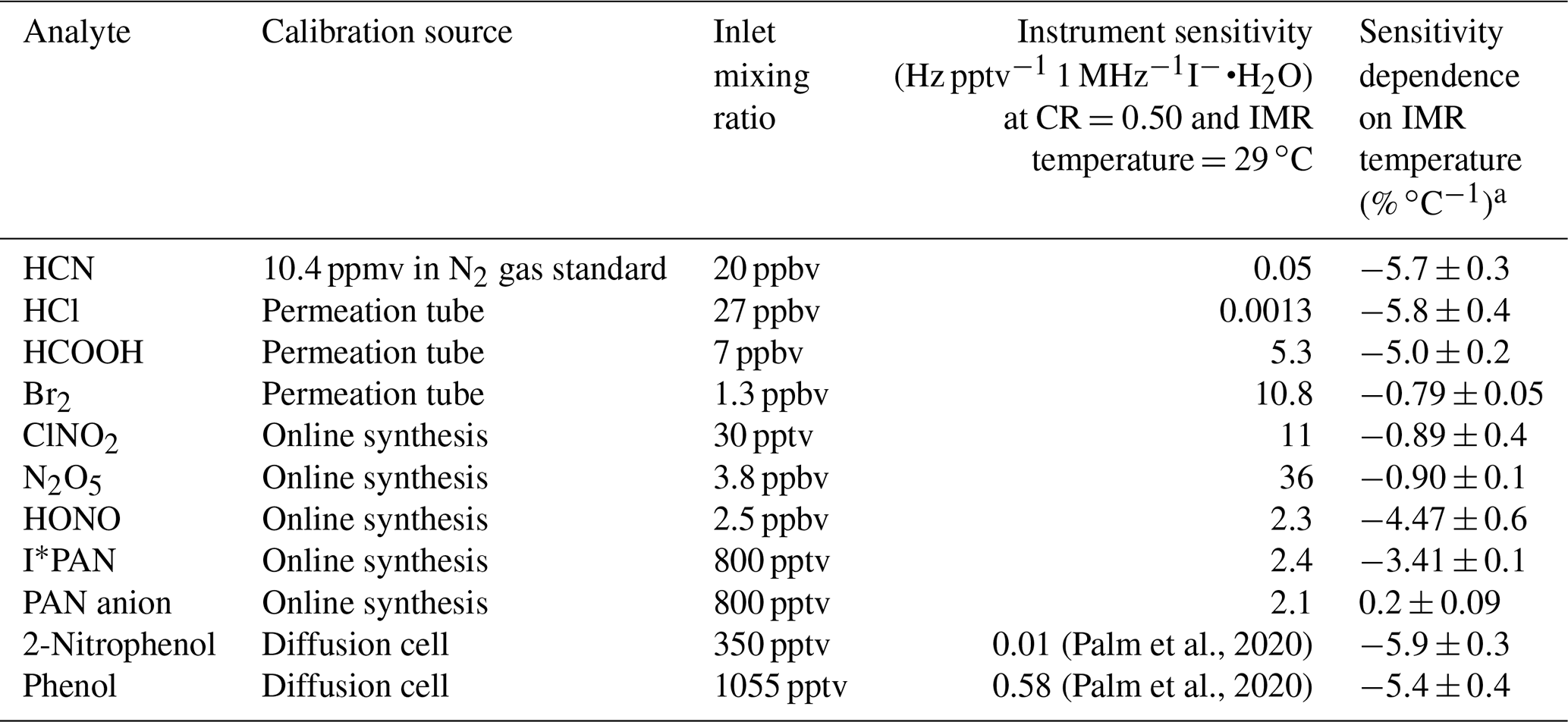
In order to understand the broader impacts of IMR temperature on detection sensitivity, a wide range of analytes (see Table 1) were added to the instrument while varying the IMR temperature. Iodide ion chemistry detection sensitivity and water dependence vary considerably among analytes (see, for example, the supplementary information of Lee et al, 2014). We have tested the IMR temperature dependence for compounds that span a wide range of sensitivities. Table 1 summarizes the calibration method, inlet mixing ratio, reagent-normalized instrument sensitivity (CR =0.5, IMR temperature =29 ∘C), and sensitivity dependence on IMR temperature. Permeation tubes (HCOOH and Br2: KIN-TEK; HCl: VICI Metronics) are held in PFA sleeves and temperature controlled (HCl and Br2 at 30 ∘C, HCOOH at 40 ∘C). A continuous 50 sccm flow of N2 is directed around the permeation tubes and through the PFA sleeves. Phenol (99 % MilliporeSigma) and 2-nitrophenol (98 % Sigma Aldrich) were added to a glass diffusion cell (Allen Scientific Glass) with a headspace flow of approximately 30 sccm of zero air.
Table 1The calibration sources, instrument sensitivity, and IMR temperature dependence of sensitivity for the analytes studied.
a Change in sensitivity from 310 to 320 K.
Most analytes detected in the NOAA CIMS lose sensitivity with increasing temperature, as shown in Fig. 3 and Table 1. Notably the sensitivity of several halogens remains nearly constant with temperature, and the PAN carboxylate anion (Reaction R3) sensitivity does not decrease with temperature. Instrument sensitivities are normalized to the sensitivity measured at 310 K, as this allows for a comparison across all field and lab operating conditions examined here. Additionally, normalization permits a comparison of the temperature dependence of analytes with absolute sensitivities (Hz pptv−1) that range several orders of magnitude between analytes (Lee et al., 2014; Iyer et al., 2016).
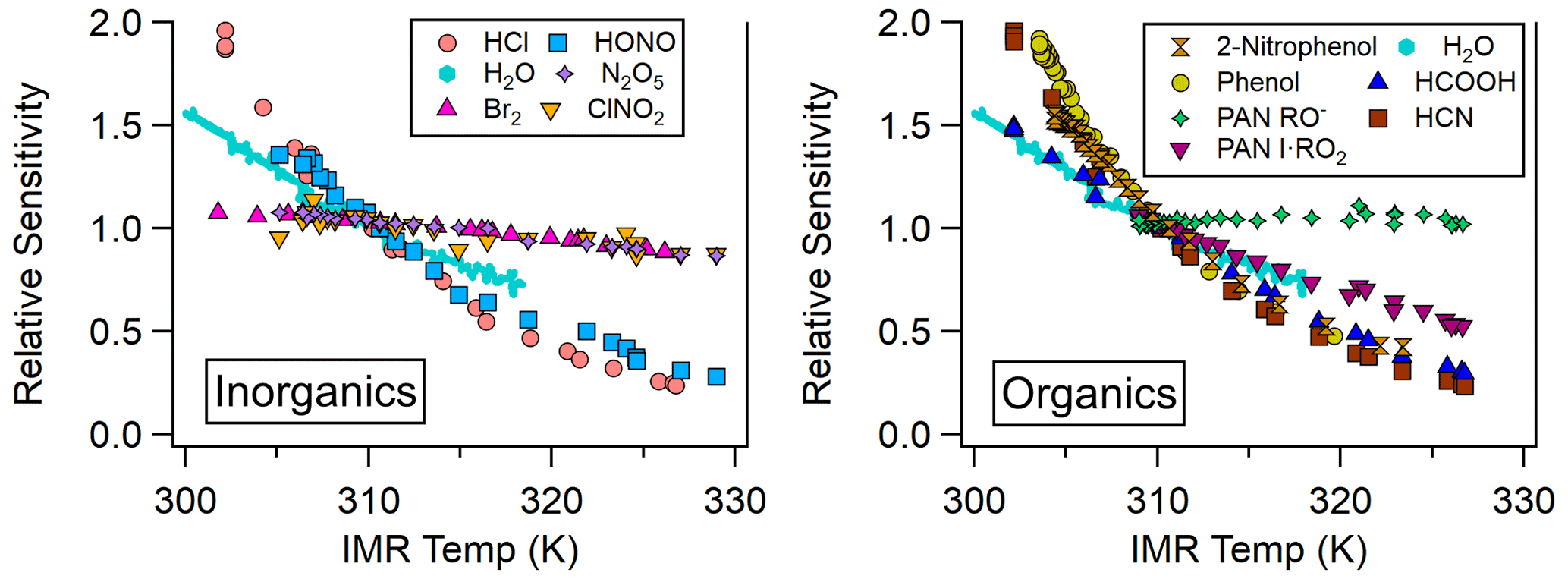
Figure 3Relative sensitivity (normalized to sensitivity at 310 K) as a function of IMR temperature for organic and inorganic atmospheric trace gases detected via iodide-adduct chemical ionization.
The analytes studied here can be grouped into two classes: strongly bound clusters with weak IMR temperature dependence (such as Br2, N2O5, and ClNO2) and weakly bound clusters with strong IMR temperature dependence (HCl, HONO, HCOOH, HCN, and PAN via peroxy radical cluster). The carboxylate anion detection of PAN behaves similarly to strongly bound clusters, but it is not clustering ion molecule chemistry and cannot be classified. The absolute sensitivities of phenol and 2-nitrophenol are currently unknown for our instrument, so we have not classified them. However, we expect them to have low sensitivity, as these molecules are detected with relatively low sensitivity in other iodide CIMS instruments (0.58 and 0.01 Hz pptv−1, respectively; Palm et al., 2020). Weakly bound inorganic analytes such as HCl and HONO exhibit reductions in sensitivity of 80 % and 67 %, respectively, for an IMR temperature increase from 31 to 50 ∘C. Weakly bound organic analytes such as phenol and HCOOH exhibit decreases in sensitivity of 75 % and 60 %, respectively, when the IMR temperature increases from 30 to 46 ∘C. Strongly bound inorganic analytes such as Br2 and N2O5 exhibit much smaller changes in sensitivity (15 %) over these IMR temperature ranges.
3.1 Impact of IMR heating on water cluster distribution
IMR heating impacts both product ion formation and the iodide water cluster distribution. We believe that iodide and its first water cluster (I−•H2O) dominate the reagent ion distribution in our IMR. The detector signal for the most abundant higher-order cluster is the second water cluster (I−•(H2O)2), which is typically 0.05 % of I−•H2O, and likely reflects the low abundance of higher-order clusters in the IMR. The IMR temperature sweep experiments were conducted to an IMR temperature of ∼55 ∘C. This is the maximum temperature in which instrument CR could be controlled to 50 %, which is the nominal operating CR during field deployment. The reduction in the iodide water cluster product ion as a function of IMR temperature is shown in Fig. 3. This experiment was conducted without IMR temperature or water control, in order to understand the influence of IMR temperature on the I−•(H2O) product ion. This thermodynamically controlled behavior of the I−•(H2O) product ion, which is also a critical reagent ion in our IMR, emphasizes the importance of temperature and CR control to maintain a constant reagent ion distribution and stable sensitivity.

Figure 4The van't Hoff relationship for a selection of atmospheric trace gases using iodide-adduct ion chemistry in the NOAA CIMS (normalized at 310 K).
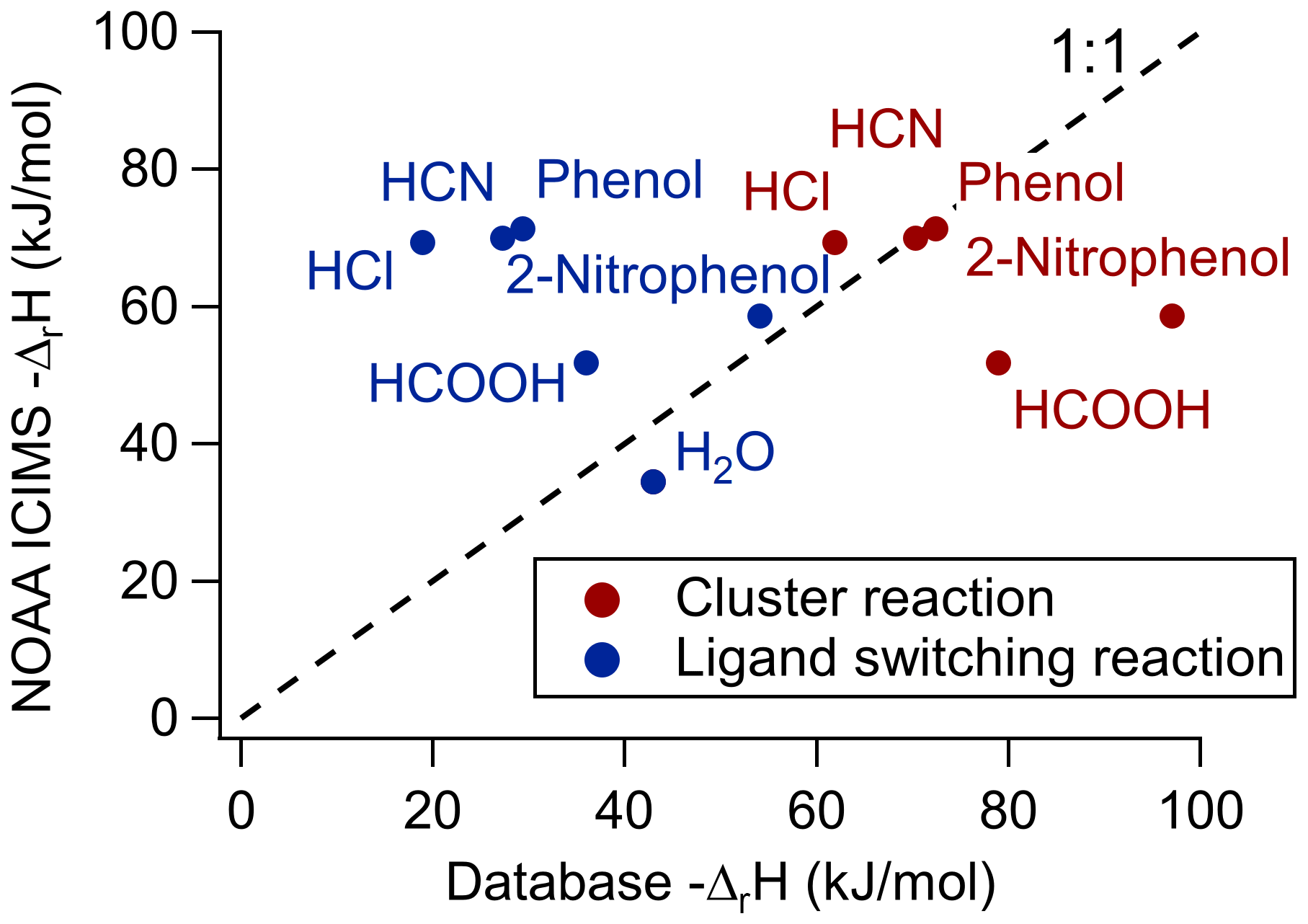
Figure 5NOAA-CIMS-measured reaction enthalpies vs. the NIST WebBook database reaction enthalpies for either the direct-clustering pathway or ligand-switching pathway. A one-to-one line is included to indicate where the measured reaction enthalpy agrees with the database.
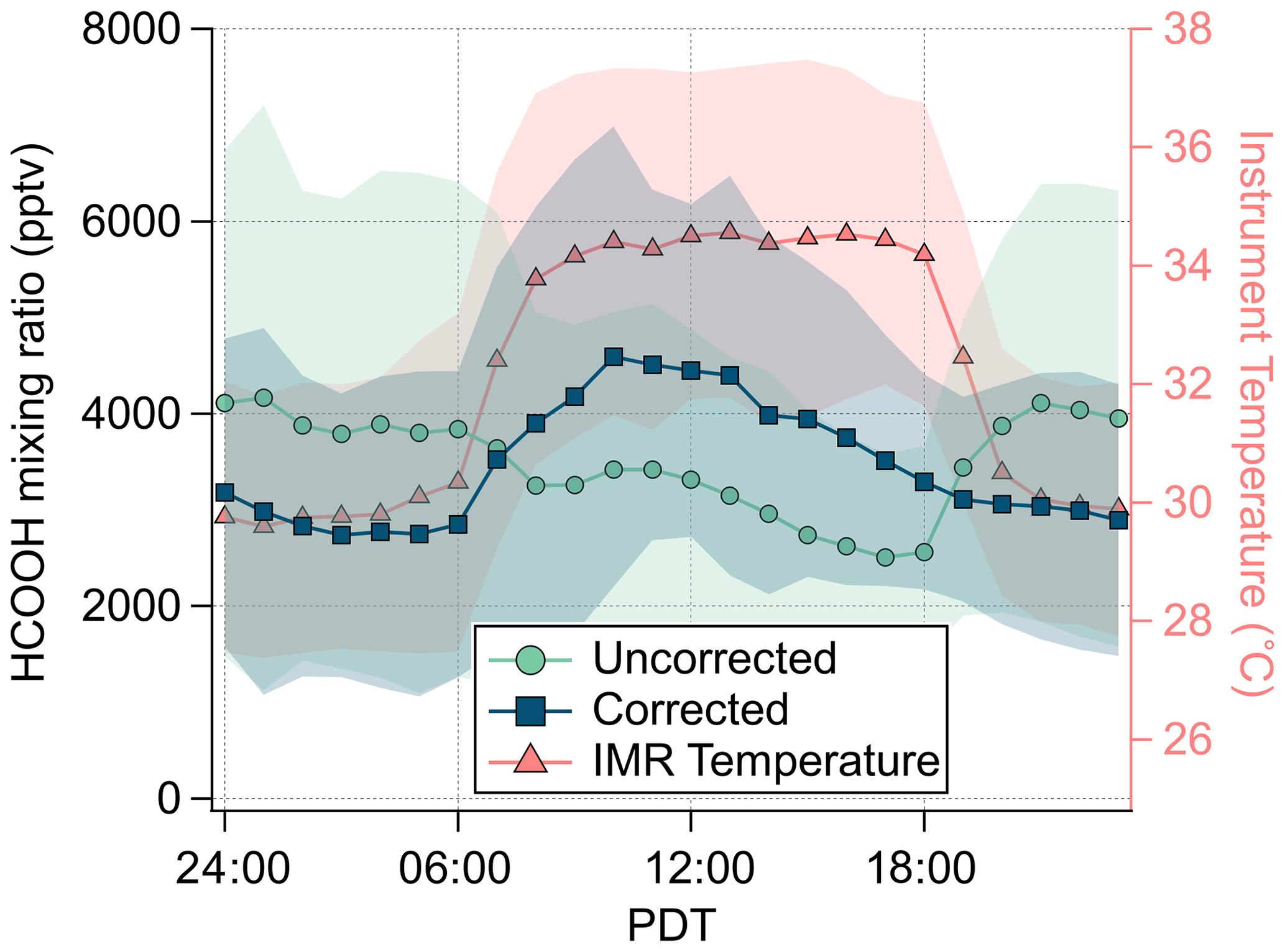
Figure 6Campaign average diurnal mixing ratio of HCOOH (uncorrected and corrected for IMR temperature) and IMR temperature. Shading represents 1 standard deviation.
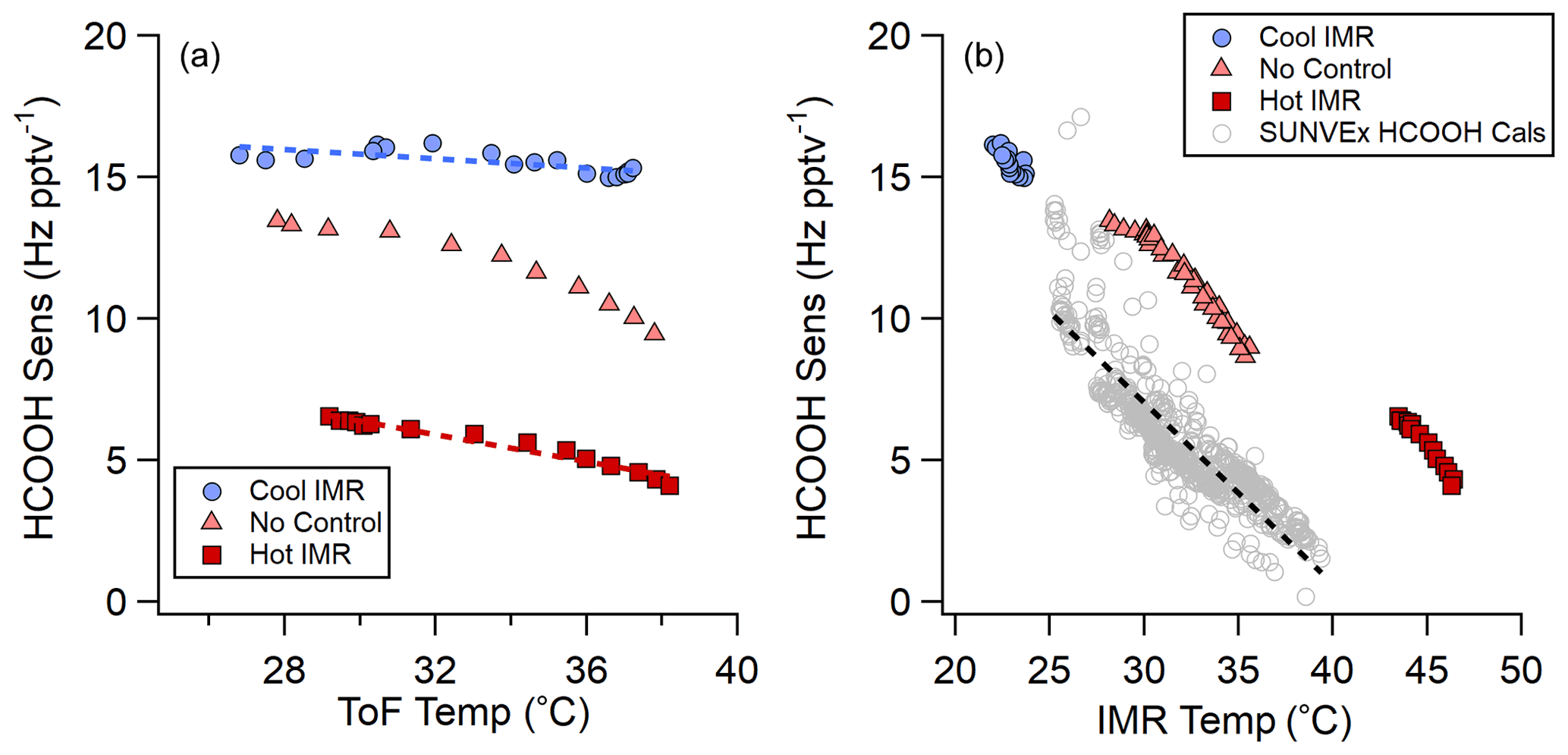
Figure 7Comparison of absolute HCOOH sensitivity between laboratory IMR temperature experiments (ARI IMR design) and SUNVEx field standard additions (NOAA IMR design).
3.2 IMR temperature dependence gives insight into iodide ion chemistry
Analyte systems that equilibrate in the IMR can be investigated further with the van't Hoff relationship (Keesee and Castleman, 1986), as shown in Fig. 4. The positive slopes indicate that the net reactions of these analytes are exothermic (i.e., ΔrH<0). Analyte systems in equilibrium give a measurement of the net reaction enthalpy in the IMR, according to the slope of the van't Hoff relationship (Eq. 2) (see Fig. S4 for HCN example). These measured reaction enthalpies can be represented by a combination of the two predominant reaction pathways' (Reactions R1 and R2) reaction enthalpies. These condition-specific reaction enthalpies are compared to reported values (NIST Chemistry WebBook, SRD 69, 2021), as shown in Fig. 5. In order to compare measured reaction enthalpies to literature values, the ligand-switching (Reaction R2) reaction enthalpies must be calculated. This is done by subtracting the single-water-cluster reaction enthalpy from the analyte direct-cluster reaction enthalpy (blue in Fig. 5), and an example calculation is shown below for the HCOOH ligand-switching reaction:
The direct-cluster and ligand-switching reaction enthalpies are shown in red and blue, respectively, in Fig. 5. The area where the one-to-one line falls between these values is an indication of the apparent branching ratio between each reaction pathway in the IMR under these conditions. For the case of HCOOH, the net reaction enthalpy is kJ mol−1. Consequently, this thermochemical system lies between the ligand-switching reaction enthalpy of kJ mol−1 and the direct-cluster reaction enthalpy of kJ mol−1. This result, which is only valid for conditions in the IMR during this experiment, implies that HCOOH reacts both through the ligand-switching pathway and the direct-cluster pathway. A similar conclusion can be drawn for 2-nitrophenol, as the measured net reaction enthalpy of kJ mol−1 falls between the reported ligand-switching reaction enthalpy of kJ mol−1 and direct-clustering reaction enthalpy of kJ mol−1. These analytes differ from HCN, HCl, and phenol which (within experimental uncertainty) all fall close to the one-to-one line for the direct-clustering pathway (see Fig. 5). For example, the literature reported value for the HCl clustering reaction is kJ mol−1, which compares well within the experimentally determined value of kJ mol−1. The involvement of the ligand-switching chemistry may explain the relatively high sensitivity for HCOOH compared with extremely low sensitivity direct-clustering analytes, such as HCN, HCl, and phenol.
3.3 Field deployment and IMR temperature control
The impact on HCOOH sensitivity due to IMR temperature changes during the SUNVEx field campaign (NOAA IMR) are shown in Fig. 6. During the day (09:00 to 18:00 PDT, Pacific Daylight Time), elevated instrument temperatures caused a decrease in HCOOH sensitivity, requiring a campaign average correction of 35±6 % (average ±1 standard deviation). At night (20:00 to 06:00 PDT), instrument temperatures were lower than lab-determined sensitivities, thereby increasing HCOOH sensitivity and requiring a campaign average correction of %. The diel dependence of HCOOH is reversed if this temperature correction is not applied, making it critical to accurately interpret these data.
Sufficient energy can decluster even the most strongly bound clusters, as shown by this work and SSQ voltage-scanning experiments (Lopez-Hilfiker et al., 2016). Therefore, in order to maintain stable sensitivities in the instrument, understanding the locations where declustering occurs is important for implementing efficient IMR temperature control strategies in the field. During the SUNVEx field deployment of the NOAA CIMS in 2021, the instrument was installed at a ground site where the IMR temperatures ranged from 25 to 40 ∘C (298 to 313 K) (see Fig. S5). The temperature changes were caused both by the PAN inlet dissociator that was only on during the day and ambient temperature fluctuations. This temperature range is not atypical for field locations (see Fig. S6 for a research aircraft example). Hourly HCOOH standard additions indicated a significant change in instrument sensitivity as a function of IMR temperature (see Fig. 7b). The variations in sensitivity with temperature in lab experiments and field observations are similar; however, absolute sensitivities differ between the two IMR designs (SUNVEx – NOAA IMR, shown in gray in Fig. 7b; lab experiments – ARI IMR, red, salmon, and blue points in Fig. 7b). Small differences in these slopes indicate that IMR temperature laboratory experiments do not fully mimic the behavior of the instrument when deployed in a harsh environment, suggesting that ion-analyte declustering occurs not only in the IMR but further into the instrument, likely in the SSQ or BSQ.
Several ToF body experiments were conducted on the ARI IMR to determine effective methods for stabilizing sensitivity when environmental temperatures change (Fig. 7). Instrument temperatures (described as ToF temperature) varied from 28 to 38 ∘C over the course of these experiments, as shown in Fig. 7a. The heated IMR control strategy attempted to maintain the IMR at 45 ∘C while the ToF temperature varied. The cooled IMR control strategy attempted to maintain the IMR temperature at 18 ∘C while the ToF temperature varied. Both IMR temperature control strategies improved HCOOH sensitivity stability compared with an uncontrolled instrument. Although, HCOOH sensitivity is reduced with the heated IMR control (red squares), this control strategy still reduces sensitivity drift (∼50 % reduction) compared with an uncontrolled IMR (salmon triangles in Fig. 7). Cooling the IMR (blue circles in Fig. 7) both improves absolute sensitivity and reduces sensitivity drift further (∼75 % reduction in sensitivity drift) compared with uncontrolled IMR temperature operation.
Iodide ion chemistry, commonly used in ToF-CIMS instruments that sample the atmosphere, imparts a temperature dependence to analyte sensitivity. The sensitivity variation with temperature for some analytes is large (5 % ∘C−1 from 36 to 40 ∘C). Importantly, sensitivity can be largely controlled by temperature stabilizing the IMR. Further, the dependence of sensitivity on temperature gives insights into the rates of ion molecule reactions that control the product ion abundance as well as providing information on absolute sensitivity. Measuring the temperature dependence of the sensitivity allows for the determination of iodide analyte reaction enthalpies. These enthalpies reveal the reactions that control the abundance of product ions in the IMR. Weakly bound clusters are strongly temperature dependent and can be detected more sensitively at lower IMR temperatures. This work was prompted by the regular addition of a weakly bound analyte (HCOOH), which has a sensitivity temperature dependence similar to other weakly bound analytes. We recommend that a similar regular calibration approach is taken for all CIMS field deployments. These considerations are likely not unique to the iodide ion adduct system but are more generally applicable to other CIMS ion chemistries. We recommend careful examination of IMR temperature dependencies of instrument sensitivities, especially for weakly bound clusters (many organics) where the effect may be large. Temperature control of the IMR region can help reduce the impact, but further declustering (either due to temperature or collisional fragmentation) of the product ion may occur in the instrument ion optics. It is worth noting that higher-pressure IMR systems may be more susceptible to thermal effects due to the increased time to reach equilibrium. Future work on expanding the range of analyte classes (for example highly functionalized organics) and ion chemistries will be of interest to improve our skill at predicting CIMS absolute sensitivity.
Data from the SUNVEx campaign are available to the general public at https://csl.noaa.gov/groups/csl7/measurements/2021sunvex/GroundLA/DataDownload/ (last access: 23 July 2022) as of 1 September 2022.
The supplement related to this article is available online at: https://doi.org/10.5194/amt-15-4295-2022-supplement.
MAR, JAN, and PRV designed the experiments, and MAR and JAN carried them out. MAR, JAN, and PRV executed the field project. LGH, JMR, and SSB all provided critical support and comments on CIMS calibration experiments. MAR and JAN prepared the manuscript with contributions from all co-authors.
The contact author has declared that none of the authors has any competing interests.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
We thank Paul Wennberg and the California Institute of Technology for hosting the SUNVEx experiment.
The NOAA Chemical Sciences Laboratory acknowledges support for this work from the California Air Resources Board (grant no. 20RD002). This work was supported in part by the NOAA cooperative agreement with CIRES (grant no. NA17OAR4320101)
This paper was edited by Hendrik Fuchs and reviewed by two anonymous referees.
Bertram, T. H., Kimmel, J. R., Crisp, T. A., Ryder, O. S., Yatavelli, R. L. N., Thornton, J. A., Cubison, M. J., Gonin, M., and Worsnop, D. R.: A field-deployable, chemical ionization time-of-flight mass spectrometer, Atmos. Meas. Tech., 4, 1471–1479, https://doi.org/10.5194/amt-4-1471-2011, 2011.
Breitenlechner, M., Novak, G. A., Neuman, J. A., Rollins, A. W., and Veres, P. R.: A versatile vacuum ultraviolet ion source for reduced pressure bipolar chemical ionization mass spectrometry, Atmos. Meas. Tech., 15, 1159–1169, https://doi.org/10.5194/amt-15-1159-2022, 2022.
Crounse, J. D., McKinney, K. A., Kwan, A. J., and Wennberg, P. O.: Measurement of Gas-Phase Hydroperoxides by Chemical Ionization Mass Spectrometry, Anal. Chem., 78, 6726–6732, https://doi.org/10.1021/ac0604235, 2006.
de Gouw, J. and Warneke, C.: Measurements of volatile organic compounds in the earth's atmosphere using proton-transfer-reaction mass spectrometry, Mass Spectrom. Rev., 26, 223–257, https://doi.org/10.1002/mas.20119, 2007.
de Hoffmann, E. and Stroobant, V.: Ion Sources: Gas-Phase Ion-Molecule Reactions, in: Mass Spectrometry: Principles and Applications, 3rd edn., John Wiley & Sons, 72–76, ISBN 9780470033104, 2007.
Dougherty, R. C., Roberts, J. D., and Biros, F. J.: Positive and negative chemical ionization mass spectra of some aromatic chlorinated pesticides, Anal. Chem., 47, 54–59, https://doi.org/10.1021/ac60351a054, 1975.
Huey, G. L., Hanson, D. R., and Howard, C. J.: Reactions of SF and I− with Atmospheric Trace Gases, J. Phys. Chem., 99, 5001–5008, https://doi.org/10.1021/j100014a021, 1995.
Huey, L. G.: Measurement of trace atmospheric species by chemical ionization mass spectrometry: Speciation of reactive nitrogen and future directions, Mass Spectrom. Rev., 26, 166–184, https://doi.org/10.1002/mas.20118, 2007.
Iyer, S., Lopez-Hilfiker, F., Lee, B. H., Thornton, J. A., and Kurtén, T.: Modeling the Detection of Organic and Inorganic Compounds Using Iodide-Based Chemical Ionization, J. Phys. Chem. A, 120, 576–587, https://doi.org/10.1021/acs.jpca.5b09837, 2016.
Ji, Y., Huey, L. G., Tanner, D. J., Lee, Y. R., Veres, P. R., Neuman, J. A., Wang, Y., and Wang, X.: A vacuum ultraviolet ion source (VUV-IS) for iodide–chemical ionization mass spectrometry: a substitute for radioactive ion sources, Atmos. Meas. Tech., 13, 3683–3696, https://doi.org/10.5194/amt-13-3683-2020, 2020.
Keesee, R. G. and Castleman, Jr., A. W.: Thermochemical Data on Gas-Phase Ion-Molecule Association and Clustering Reactions, J. Phys. Chem. Ref. Data, 15, 1011–1071, 1986.
Kercher, J. P., Riedel, T. P., and Thornton, J. A.: Chlorine activation by N2O5: simultaneous, in situ detection of ClNO2 and N2O5 by chemical ionization mass spectrometry, Atmos. Meas. Tech., 2, 193–204, https://doi.org/10.5194/amt-2-193-2009, 2009.
Koss, A. R., Warneke, C., Yuan, B., Coggon, M. M., Veres, P. R., and de Gouw, J. A.: Evaluation of NO+ reagent ion chemistry for online measurements of atmospheric volatile organic compounds, Atmos. Meas. Tech., 9, 2909–2925, https://doi.org/10.5194/amt-9-2909-2016, 2016.
Koss, A. R., Sekimoto, K., Gilman, J. B., Selimovic, V., Coggon, M. M., Zarzana, K. J., Yuan, B., Lerner, B. M., Brown, S. S., Jimenez, J. L., Krechmer, J., Roberts, J. M., Warneke, C., Yokelson, R. J., and de Gouw, J.: Non-methane organic gas emissions from biomass burning: identification, quantification, and emission factors from PTR-ToF during the FIREX 2016 laboratory experiment, Atmos. Chem. Phys., 18, 3299–3319, https://doi.org/10.5194/acp-18-3299-2018, 2018.
Krechmer, J., Lopez-Hilfiker, F., Koss, A., Hutterli, M., Stoermer, C., Deming, B., Kimmel, J., Warneke, C., Holzinger, R., Jayne, J., Worsnop, D., Fuhrer, K., Gonin, M., and de Gouw, J.: Evaluation of a New Reagent-Ion Source and Focusing Ion–Molecule Reactor for Use in Proton-Transfer-Reaction Mass Spectrometry, Anal. Chem., 90, 12011–12018, https://doi.org/10.1021/acs.analchem.8b02641, 2018.
Lee, B. H., Lopez-Hilfiker, F. D., Mohr, C., Kurtén, T., Worsnop, D. R., and Thornton, J. A.: An Iodide-Adduct High-Resolution Time-of-Flight Chemical-Ionization Mass Spectrometer: Application to Atmospheric Inorganic and Organic Compounds, Environ. Sci. Technol., 48, 6309–6317, https://doi.org/10.1021/es500362a, 2014.
Lee, B. H., Lopez-Hilfiker, F. D., Veres, P. R., McDuffie, E. E., Fibiger, D. L., Sparks, T. L., Ebben, C. J., Green, J. R., Schroder, J. C., Campuzano-Jost, P., Iyer, S., D'Ambro, E. L., Schobesberger, S., Brown, S. S., Wooldridge, P. J., Cohen, R. C., Fiddler, M. N., Bililign, S., Jimenez, J. L., Kurtén, T., Weinheimer, A. J., Jaegle, L., and Thornton, J. A.: Flight deployment of a high-resolution time-of-flight chemical ionization mass spectrometer: Observations of reactive halogen and nitrogen oxide species, J. Geophys. Res.-Atmos., 123, 7670–7686, https://doi.org/10.1029/2017JD028082, 2018.
Lopez-Hilfiker, F. D., Mohr, C., Ehn, M., Rubach, F., Kleist, E., Wildt, J., Mentel, Th. F., Lutz, A., Hallquist, M., Worsnop, D., and Thornton, J. A.: A novel method for online analysis of gas and particle composition: description and evaluation of a Filter Inlet for Gases and AEROsols (FIGAERO), Atmos. Meas. Tech., 7, 983–1001, https://doi.org/10.5194/amt-7-983-2014, 2014.
Lopez-Hilfiker, F. D., Iyer, S., Mohr, C., Lee, B. H., D'Ambro, E. L., Kurtén, T., and Thornton, J. A.: Constraining the sensitivity of iodide adduct chemical ionization mass spectrometry to multifunctional organic molecules using the collision limit and thermodynamic stability of iodide ion adducts, Atmos. Meas. Tech., 9, 1505–1512, https://doi.org/10.5194/amt-9-1505-2016, 2016.
Lopez-Hilfiker, F. D., Pospisilova, V., Huang, W., Kalberer, M., Mohr, C., Stefenelli, G., Thornton, J. A., Baltensperger, U., Prevot, A. S. H., and Slowik, J. G.: An extractive electrospray ionization time-of-flight mass spectrometer (EESI-TOF) for online measurement of atmospheric aerosol particles, Atmos. Meas. Tech., 12, 4867–4886, https://doi.org/10.5194/amt-12-4867-2019, 2019.
Mielke, L. H., Furgeson, A., and Osthoff, H. D.: Observation of ClNO2 in a Mid-Continental Urban Environment, Environ. Sci. Technol., 45, 8889–8896, https://doi.org/10.1021/es201955u, 2011.
Neuman, J. A., Huey, L. G., Dissly, R. W., Fehsenfeld, F. C., Flocke, F., Holecek, J. C., Holloway, J. S., Hubler, G., Jakoubek, R., Nicks Jr., D. K., Parrish, D. D., Ryerson, T. B., Sueper, D. T., and Weinheimer, A. J.: Fast-response airborne in situ measurements of HNO3 during the Texas 2000 Air Quality Study, J. Geophys. Res.-Atmos., 107, 4436, https://doi.org/10.1029/2001JD001437, 2002.
Neuman, J. A., Trainer, M., Brown, S. S., Min, K.-E., Nowak, J. B., Parrish, D. D., Peischl, J., Pollack, I. B., Roberts, J. M., Ryerson, T. B., and Veres, P. R.: HONO emission and production determined from airborne measurements over the Southeast U.S., J. Geophys. Res.-Atmos., 121, 9237–9250, https://doi.org/10.1002/2016JD025197, 2016.
NIST Chemistry WebBook, SRD 69: https://webbook.nist.gov/chemistry/ion/, last access: 15 December 2021.
Osthoff, H. D., Roberts, J. M., Ravishankara, A. R., Williams, E. J., Lerner, B. M., Sommariva, R., Bates, T. S., Coffman, D., Quinn, P. K., Dibb, J. E., Stark, H., Burkholder, J. B., Talukdar, R. K., Meagher, J., Fehsenfeld, F. C., and Brown, S. S.: High levels of nitryl chloride in the polluted subtropical marine boundary layer, Nat. Geosci., 1, 324–328, https://doi.org/10.1038/ngeo177, 2008.
Palm, B. B., Peng, Q., Fredrickson, C. D., Lee, B. H., Garofalo, L. A., Pothier, M. A., Kreidenweis, S. M., Farmer, D. K., Pokhrel, R. P., Shen, Y., Murphy, S. M., Permar, W., Hu, L., Campos, T. L., Hall, S. R., Ullmann, K., Zhang, X., Flocke, F., Fischer, E. V., and Thornton, J. A.: Quantification of organic aerosol and brown carbon evolution in fresh wildfire plumes, P. Natl. Acad. Sci. USA, 202012218, 29469–29477, https://doi.org/10.1073/pnas.2012218117, 2020.
Riedel, T. P., Bertram, T. H., Crisp, T. A., Williams, E. J., Lerner, B. M., Vlasenko, A., Li, S.-M., Gilman, J., de Gouw, J., Bon, D. M., Wagner, N. L., Brown, S. S., and Thornton, J. A.: Nitryl Chloride and Molecular Chlorine in the Coastal Marine Boundary Layer, Environ. Sci. Technol., 46, 10463–10470, https://doi.org/10.1021/es204632r, 2012.
Sanchez, J., Tanner, D. J., Chen, D., Huey, L. G., and Ng, N. L.: A new technique for the direct detection of HO2 radicals using bromide chemical ionization mass spectrometry (Br-CIMS): initial characterization, Atmos. Meas. Tech., 9, 3851–3861, https://doi.org/10.5194/amt-9-3851-2016, 2016.
Slusher, D. L., Huey, L. G., Tanner, D. J., Flocke, F. M., and Roberts, J. M.: A thermal dissociation–chemical ionization mass spectrometry (TD-CIMS) technique for the simultaneous measurement of peroxyacyl nitrates and dinitrogen pentoxide, J. Geophys. Res., 109, D19315, https://doi.org/10.1029/2004JD004670, 2004.
Thornton, J. A., Kercher, J. P., Riedel, T. P., Wagner, N. L., Cozic, J., Holloway, J. S., Dubé, W. P., Wolfe, G. M., Quinn, P. K., Middlebrook, A. M., Alexander, B., and Brown, S. S.: A large atomic chlorine source inferred from mid-continental reactive nitrogen chemistry, Nature, 464, 271–274, https://doi.org/10.1038/nature08905, 2010.
Veres, P., Roberts, J. M., Warneke, C., Welsh-Bon, D., Zahniser, M., Herndon, S., Fall, R., and de Gouw, J.: Development of negative-ion proton-transfer chemical-ionization mass spectrometry (NI-PT-CIMS) for the measurement of gas-phase organic acids in the atmosphere, Int. J. Mass Spectrom., 274, 48–55, https://doi.org/10.1016/j.ijms.2008.04.032, 2008.
Veres, P. R., Neuman, J. A., Bertram, T. H., Assaf, E., Wolfe, G. M., Williamson, C. J., Weinzierl, B., Tilmes, S., Thompson, C. R., Thames, A. B., Schroder, J. C., Saiz-Lopez, A., Rollins, A. W., Roberts, J. M., Price, D., Peischl, J., Nault, B. A., Møller, K. H., Miller, D. O., Meinardi, S., Li, Q., Lamarque, J.-F., Kupc, A., Kjaergaard, H. G., Kinnison, D., Jimenez, J. L., Jernigan, C. M., Hornbrook, R. S., Hills, A., Dollner, M., Day, D. A., Cuevas, C. A., Campuzano-Jost, P., Burkholder, J. B., Bui, T. P., Brune, W. H., Brown, S. S., Brock, C. A., Bourgeois, I., Blake, D. R., Apel, E. C., and Ryerson, T. B.: Correction for Veres et al., Global airborne sampling reveals a previously unobserved dimethyl sulfide oxidation mechanism in the marine atmosphere, P. Natl. Acad. Sci. USA, 118, e2113268118, https://doi.org/10.1073/pnas.2113268118, 2021.
Villalta, P. W. and Howard, C. J.: Direct Kinetics Study of the CH3C(O)O2+ NO Reaction Using Chemical Ionization Mass Spectrometry, J. Phys. Chem., 100, 13624–13628, https://doi.org/10.1021/jp9614153, 1996.
Zaytsev, A., Breitenlechner, M., Koss, A. R., Lim, C. Y., Rowe, J. C., Kroll, J. H., and Keutsch, F. N.: Using collision-induced dissociation to constrain sensitivity of ammonia chemical ionization mass spectrometry (NH CIMS) to oxygenated volatile organic compounds, Atmos. Meas. Tech., 12, 1861–1870, https://doi.org/10.5194/amt-12-1861-2019, 2019.