the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 07 May 2024
| 07 May 2024
Extraction, purification, and clumped isotope analysis of methane (Δ13CDH3 and Δ12CD2H2) from sources and the atmosphere
Malavika Sivan
Thomas Röckmann
Carina van der Veen
Maria Elena Popa
Measurements of the clumped isotope anomalies (Δ13CDH3 and Δ12CD2H2) of methane have shown potential for constraining methane sources and sinks. At Utrecht University, we use the Thermo Scientific Ultra high-resolution isotope-ratio mass spectrometer to measure the clumped isotopic composition of methane emitted from various sources and directly from the atmosphere.
We have developed an extraction system with three sections for extracting and purifying methane from high (> 1 %), medium (0.1 % to 1 %), and low-concentration (< 0.1 %) samples, including atmospheric air (∼ 2 ppm = 0.0002 %). Depending on the methane concentration, a quantity of sample gas is processed that delivers 3 ± 1 mL of pure methane, which is the quantity typically needed for one clumped isotope measurement. For atmospheric air with a methane mole fraction of 2 ppm, we currently process up to 1100 L of air.
The analysis is performed on pure methane, using a dual-inlet setup. The complete measurement time for all isotope signatures is about 20 h for one sample. The mean internal precision values of sample measurements are 0.3 ± 0.1 ‰ for Δ13CDH3 and 2.4 ± 0.8 ‰ for Δ12CD2H2. The long-term reproducibility, obtained from repeated measurements of a constant target gas, over almost 3 years, is around 0.15 ‰ for Δ13CDH3 and 1.2 ‰ for Δ12CD2H2. The measured clumping anomalies are calibrated via the Δ13CDH3 and Δ12CD2H2 values of the reference CH4 used for the dual-inlet measurements. These were determined through isotope equilibration experiments at temperatures between 50 and 450 °C.
We describe in detail the optimized sampling, extraction, purification, and measurement technique followed in our laboratory to measure the clumping anomalies of methane precisely and accurately. This paper highlights the extraction and one of the first global measurements of the clumping anomalies of atmospheric methane.
- Article
(2475 KB) - Full-text XML
-
Supplement
(405 KB) - BibTeX
- EndNote





Atmospheric methane, CH4, is the second most important anthropogenic greenhouse gas after CO2. The global warming potential of CH4 is 28 times greater than that of CO2 over a 100-year period. Having a shorter lifetime of ∼ 11 years (Li et al., 2022) compared to CO2 (Archer et al., 2009), CH4 responds faster to changes in its source and sink fluxes than CO2. This also means that CH4 emission reduction measures can have a relatively faster effect on atmospheric composition, reducing global warming. Global-scale measurements of CH4 mole fractions show an increasing trend since pre-industrial times. The current global mean atmospheric CH4 mole fraction as of January 2023 is 1972 ppb, while the estimated pre-industrial values were 700 to 800 ppb (Lan et al., 2022). This long-term increase is mostly attributed to anthropogenic emissions (IPCC, 2022). Precise direct atmospheric measurements have revealed significant shorter-term variations in the growth rate of atmospheric CH4, including stable levels in the early 2000s followed by an accelerating increase since 2007. Various studies have attempted to attribute this temporal change to variations in the balance between different CH4 sources and atmospheric sinks. However, these existing studies do not converge on the same conclusion. This shows that we do not fully understand the CH4 cycle yet, which means that we cannot predict its future behaviour confidently.
Major CH4 sources are often separated into these categories according to the production mechanism: biogenic (wetlands, cattle, lakes, landfills), thermogenic (natural gas, coalbed CH4, shale gas, etc.), pyrogenic (biomass burning, combustion of fossil fuels, etc.), and abiotic (volcanic and geothermal areas, gas–water–rock interactions, etc.) sources. The main CH4 sink in the troposphere is photochemical oxidation by OH and Cl radicals (Khalil et al., 1993). Part of the CH4 that reaches the stratosphere is removed by Cl and O(1D). About 10 % of the atmospheric CH4 is taken up by surface sinks (Topp and Pattey, 1997).
A method commonly used to identify different sources and sinks of CH4 is based on measurements of its bulk isotopic composition, denoted as δ13C and δD. Each source has a characteristic isotopic composition range as shown in Fig. 1a, as a result of the isotopic composition of the various substrates and the process-dependent isotopic fractionation during CH4 formation (Whiticar et al., 1986; Whiticar, 1999; Sherwood Lollar et al., 2006; Etiope and Sherwood Lollar, 2013; Conrad, 2002; Kelly et al., 2022; Menoud et al., 2020). CH4 from all these sources contribute to atmospheric CH4 with an expected isotopic composition of the source mixture around −54 ‰ for δ13C and −290 ‰ for δD (Whiticar and Schaefer, 2007) (as shown in Fig. 1a). The sink reactions preferentially remove the lighter isotopologues of CH4 from the atmosphere (Saueressig et al., 2001; Cantrell et al., 1990; Whitehill et al., 2017), resulting in an enrichment of the heavier isotopes in the residual CH4. The combined effect of emissions from the various sources and removal by the different sinks lead to an overall atmospheric CH4 bulk isotopic composition of around −48 ‰ for δ13C and −90 ‰ for δD. Many measurements have been performed to date, using analysis in the laboratory on collected samples and field-deployable instruments at various sites to study the variations in atmospheric CH4 (Menoud et al., 2020, 2021, 2022; Lu et al., 2021; Beck et al., 2012; Fernandez et al., 2022; Röckmann et al., 2016a; Sherwood et al., 2017). However, due to the overlap of some of the source signatures, it is not always possible to distinguish different sources of CH4 using the bulk isotopes (Fig. 1a).
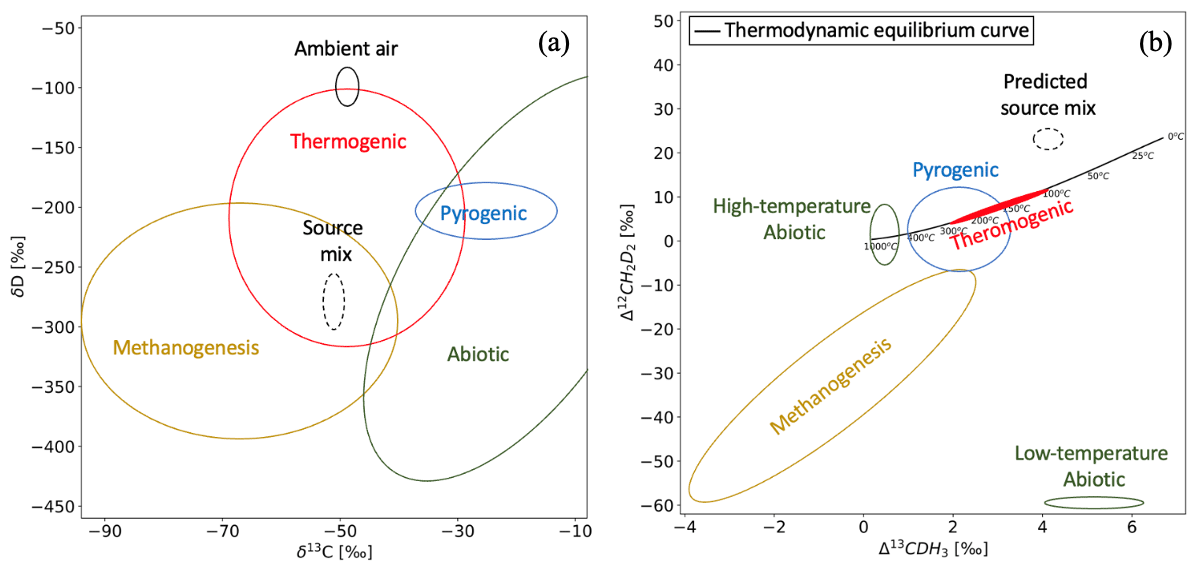
Figure 1An illustration of bulk (a) and clumped (b) isotopic composition of major CH4 sources as reported so far.
The measurement of the two most abundant clumped isotopologues (13CDH3 and 12CD2H2) of CH4 can be used as an additional tool to constrain CH4 sources (Douglas et al., 2017; Eiler, 2007; Young et al., 2017; Stolper et al., 2014). The clumping anomalies, denoted as Δ13CDH3 and Δ12CD2H2, are a measure of the deviation of the number of clumped molecules present relative to that expected from the random distribution of the light and heavy isotopes over all isotopologues of CH4. At thermodynamic equilibrium, these anomalies are temperature dependent and can thus be used to calculate the CH4 formation or equilibration temperature. In the case of thermodynamic disequilibrium, the clumped signatures can be exploited to identify various kinetic gas formation and fractionation (mixing, diffusion, etc.) processes. The clumped isotope signatures are specific to different sources and processes, independent of the bulk signatures, and thus can deliver additional information on sources and cycling of CH4 in the environment.
Measuring the clumped isotopic composition of CH4, however, poses several technical challenges. The 13CDH3 and CD2H2 molecules and H2O (which is always present in a mass spectrometer at much higher concentrations than the CH4 clumped isotopologues) have very slightly different masses, approximately 18.0409, 18.0439, and 18.0153 amu (atomic mass unit), respectively. This difference cannot be distinguished using a conventional mass spectrometer. Also, the 13CH4 and CDH3 have the same nominal mass ( 17), but these interferences can be circumvented by separating the C and H atoms, i.e. by converting the CH4 to CO2 for the δ13C measurements and to H2 for δD. For clumped isotope measurements, such an approach would eliminate the signal we are looking for; thus, the measurements need to be performed on intact CH4 molecules. In recent years, high-resolution isotope-ratio mass spectrometers have become available that can resolve these small mass differences (Eiler et al., 2013; Young et al., 2017). These new instruments can separate the ion beams around mass 18 corresponding to CH3D+, , and , facilitating the CH4 clumped isotope measurements.
Another challenge includes the measurement of low ion currents and the instrument stability required for long measurement times. The natural abundance of the clumped molecules is very low i.e. about and of the total CH4 for 13CH3D and 12CH2D2, respectively. The corresponding ion currents are proportionally low, typically around 5000 cps (counts per second) for 13CH3D+ and 100 cps for . The cumulated number of counts control the limits of the achievable precision for the rare isotopologues. Therefore, to achieve per mil-level precision, the isotopologue ratios need to be measured for a long time. This requires several millilitres (, where STP represents standard temperature and pressure) of pure CH4 for one measurement. To obtain pure CH4 for the measurements, the samples need to be purified. Isotope fractionation can occur during sample handling, extraction, and purification, potentially introducing biases and inaccuracies in the measured bulk and clumped isotopologue ratios. Careful consideration of sample preparation methods, including minimizing fractionation and optimizing purification procedures, is crucial to ensure reliable and reproducible results. Another hurdle is that there are no readily available reference gases with known clumped isotopic composition to calibrate the measurements, so these need to be prepared.
A number of studies have reported the Δ13CDH3 and Δ12CD2H2 of CH4 from various sources, e.g. natural gas seeps, rice paddies and wetlands, lake sediments, shale gas, coal mines, natural gas leakage, and laboratory incubation experiments (Wang et al., 2015; Young et al., 2017; Stolper et al., 2018; Loyd et al., 2016; Ono et al., 2021; Giunta et al., 2019). A general overview of the expected clumped isotope signatures of CH4 from different sources is illustrated in Fig. 1b. Thermogenic CH4 is usually formed in thermodynamic equilibrium and therefore lies on the thermodynamic equilibrium curve between 100 and 300 °C. Biogenic CH4 production, denoted as methanogenesis in Fig. 1b, is often characterized by disequilibrium Δ12CD2H2 values due to the kinetic isotopic fractionation associated with methanogenesis and/or combinatorial effects (Röckmann et al., 2016b; Yeung, 2016). The reported ranges of values for abiotic (produced at high and low temperatures) and pyrogenic CH4 are also shown in Fig. 1b. The predicted clumping anomaly of the atmospheric CH4 source mix resulting from the combination of all sources is about 4 ‰ for Δ13CDH3 and 20 ‰ for Δ12CD2H2, as reported by Haghnegahdar et al. (2017) (Fig. 1b).
Recent modelling studies have suggested the potential of clumped isotope measurements of atmospheric CH4, especially Δ12CD2H2, to distinguish between the main drivers of change in the CH4 burden (Chung and Arnold, 2021; Haghnegahdar et al., 2017). However, as mentioned above, the clumped isotope measurements require a few millilitres (at STP) of pure CH4. Therefore, a challenge specific to atmospheric CH4 measurements is the extraction of CH4 from very large samples of air required (thousands of litres).
This paper presents one of the first measurements of the clumping anomalies of atmospheric methane and provides a detailed comparison to the previously reported model predictions. The paper also describes in detail the technical setups and procedures for CH4 clumped measurements at Utrecht University including (i) the extraction and purification of CH4 from high- and low-concentration samples, including the extraction from large quantities of air (∼ 1000 L); (ii) calibration of measured anomalies using gas-equilibration experiments at different temperatures; (iii) the detailed settings and procedures of the actual isotope measurements using the Thermo Scientific Ultra mass spectrometer; and (iv) the data processing and calculations involved. We also report the performance of these systems so far, in terms of precision, reproducibility, stability, etc. Thus, this paper serves as a description of our measurement technique for future reference.
2.1 Notations, definitions, and calculations
The bulk isotopic composition of CH4, denoted as δ13C and δD, is defined as follows:
where and are the isotopic ratios of and of the sample, and and are isotopic ratios of the international standards for δ13C and δD (VPDB and VSMOW)1 with their values being 0.011180 and 0.00015576, respectively (Assonov et al., 2020; Gonfiantini, 1978).
The clumped isotopic composition of CH4 is expressed as clumping anomalies Δ13CDH3 and Δ12CD2H2 relative to the clumped isotope ratio that would be obtained if the heavy isotopes, 13C and D, were distributed randomly across all isotopologues in the same sample:
where and are the isotopologue ratios of and of the sample, and and are isotope ratios of and of the sample itself. The denominators in Eqs. (2a) and (2b) give the expected random distribution of the heavier isotopes in a sample, where 4 and 6 are symmetry factors (Young et al., 2017).
2.2 Mass spectrometer specifications and measurement methods
CH4 bulk and clumped isotopic compositions were determined using the Thermo Scientific Ultra HR-IRMS (high-resolution isotope-ratio mass spectrometer, denoted Ultra hereafter). The prototype of the instrument was introduced by Eiler et al. (2013), and the characteristics of the Ultra at Utrecht University have been explained in detail by Adnew et al. (2019). The instrument is operated with the advanced Qtegra™ software package for data acquisition, instrument control, and data analysis.
The sample is introduced via one of the four variable-volume bellows into the ion source, and reference gas is provided from another bellows. After ionization in the ion source, the ion beam is accelerated, focused, and passed through a slit into the mass analyser. Three different slit widths of 250, 16, and 5 µm can be chosen in the standard setup, giving three resolution options: low resolution (LR), medium resolution (MR), and high resolution (HR), respectively. An additional “aperture” option can be turned on to achieve even higher resolution (HR+), wherein the focused ion beam is trimmed further in the y axis by an additional slit situated just before the electromagnet. However, increasing the resolution results in a decrease in intensity.
The ions are separated by energy and mass in the mass analyser, which leads to very well focussed ion beams, and they are collected with a variable detector array that supports one fixed and eight moveable detector platforms, which are equipped with nine Faraday detectors (L1, L2, L3, L4, centre, H1, H2, H3, H4) that can be read out with selectable resistors with resistances between 3×108 Ω and 1013 Ω. The three collector platforms at the high mass end (H2, H3, and H4) are additionally equipped with compact discrete dynode (CDD) ion counting detectors next to the Faraday detectors.
Characterization of the Ultra for CH4 measurements
Clumped isotope measurements of CH4 using the Ultra are performed at high resolution (5 µm entrance slit width) with aperture, i.e. HR+ setting, to get the highest possible resolution. Two Faraday collectors are read out with resistors, 1×1011 Ω for 16 and 1×1012 Ω for 17 (13CH4). To measure 17 (12CDH3) and the clumped isotopologues at 18, we use the CDD of detector H4, which has a narrow detector slit. With careful tuning, the instrument can achieve mass resolving power (5 % to 95 %) higher than 42 000, which is sufficient to separate CH4 isotopologues from each other, from contaminating isobars like H2O+, OH+, and , and the adducts formed in the source, i.e. , , and .
As the high resolution is to a large degree achieved by using a very narrow source slit, most of the ions do not pass through the slit but deposit on the slit assembly. This leads to carbon accumulation around the slit and over time obstructs the passage of ions into the mass analyser, resulting in reduced ion transmission and sensitivity. The carbon deposits can also introduce additional scattering and deflection of ions, leading to the broadening of mass peaks and decreased mass resolution. There can also be signal instabilities due to fluctuations in ion transmission. These effects together can compromise the instrument's capability to resolve closely spaced ions. Therefore, we change the source slit regularly to avoid the impact of carbon deposits. To keep track of this, the number of counts of of each measurement is monitored (Fig. S1 in the Supplement). When the counts decrease to less than 0.5 times the counts of the first measurement using a new slit, the slit is replaced. The usual lifetime of one slit is around 6 months, depending on the number of CH4 measurements done.
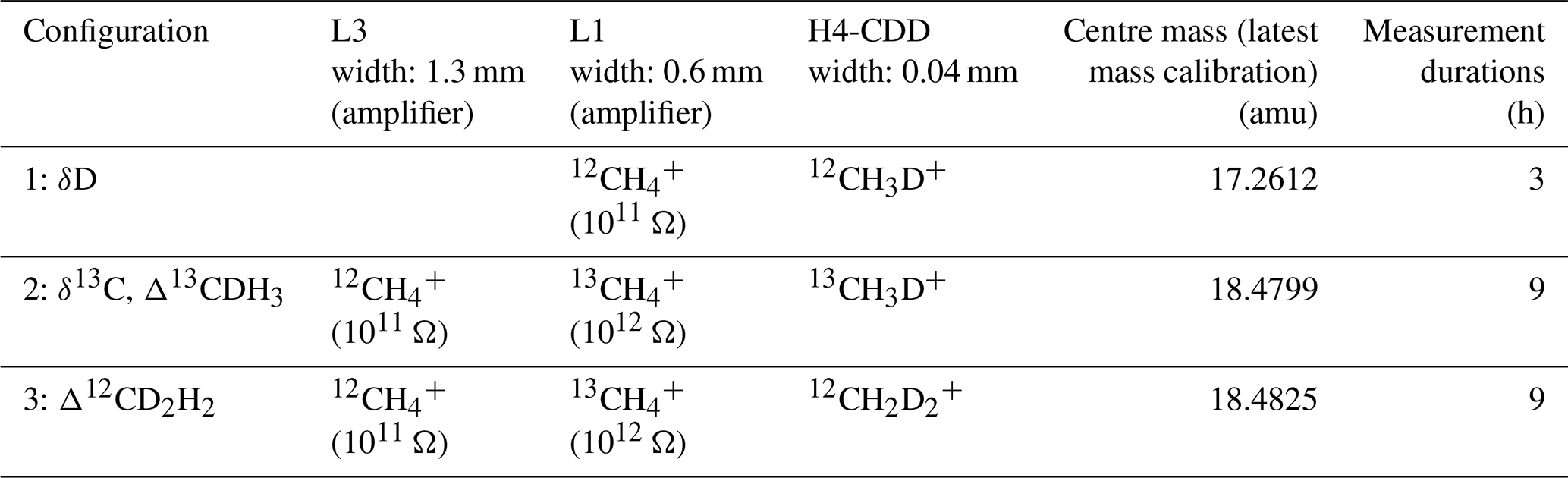
The main CH4 isotopologues, , , 12CH3D+, 13CH3D+, and , are measured in three different configurations on the Ultra. The configurations differ by the peak centre mass setting and the relative distance between the detectors, and the peak positions are finely adjusted (Fig. 3) such that the right ions are detected by each detector. The details of the three different configurations, resistors, and detectors used for the measurements on the Ultra are given in Table 1. In the first configuration, (L1) and 12CH3D+ (H4-CDD) are measured for about 3 h. The second configuration is set up to measure (L3), (L1), and 13CH3D+ (H4-CDD), and the third configuration measures (L3), (L1), and (H4-CDD). Configurations 2 and 3 are measured alternately for 18 h in seven cycles each lasting about 2.5 h. Therefore, in total, one complete measurement of all three configurations takes about 20 h. The sample and reference gases are measured alternately, each three times (meaning integrations) for a total of 201.3 s; the average of which is considered one data point. The result of one complete measurement is the average of all the data measured (outliers removed), and the internal precision is the standard error over these data points.
Table 1The details of the three different configurations, resistors, and detectors used for the measurements on the Ultra.
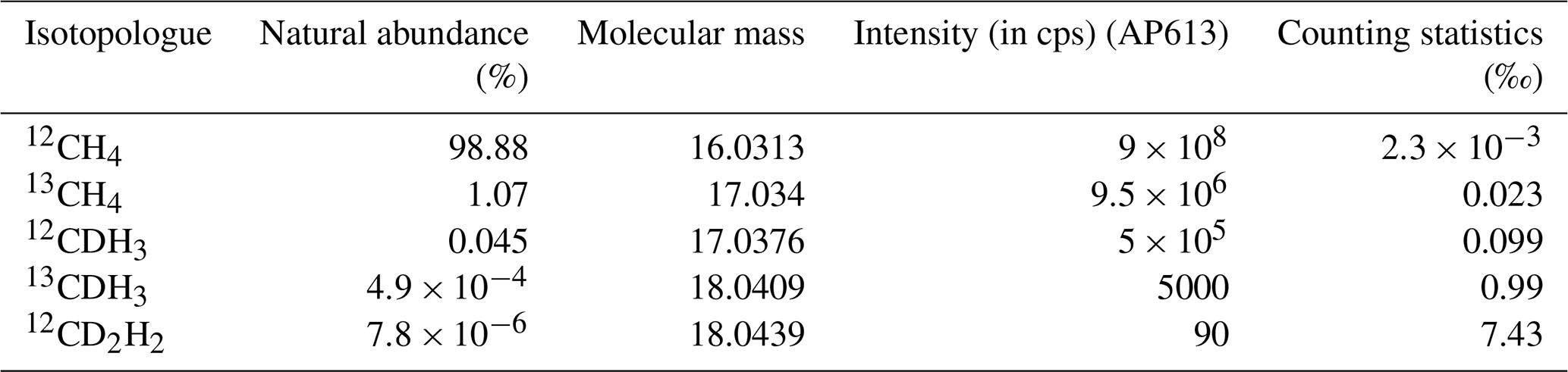
A summary of the natural abundances, molecular masses, expected intensity (in cps) (for AP613, the laboratory reference gas), and the counting statistics precision limit for all five isotopologues is given in Table 2.
Table 2A summary of the natural abundances, molecular masses, expected intensity (in cps) (for AP613, the laboratory reference gas), and the counting statistics precision limit for an integration time of 201.3 s for all five isotopologues of CH4 measured on the Ultra.
The gases are measured at a source pressure of maximum mbar. The pressure in the source is controlled by the bellows pressure, which can be set and adjusted using Qtegra. The typical pressure in the bellows required to achieve this source pressure for CH4 is around 65 to 70 mbar. We use a continuous pressure adjustment method, which means, after each integration, the bellows pressures are checked five times, and the bellows are compressed by 0.5 % each time, until the set value is attained. The tolerance of the pressure adjustment is set to 0.5 mbar, so the signal is stable within ± 0.7 %. This ensures that the instrument measures the reference and sample at the same source pressure during the entire (more than) 20 h of measurement time.
All measurements are made relative to a reference gas, which is a stainless-steel (SS) canister filled from a high purity (> 99.999 %) CH4 reference gas cylinder (AP613). The sample and the reference are measured alternately, and then the bulk and clumped isotopic composition of the samples are calculated from the isotopologue ratios as follows:
where , , , and are the values of the sample measured against the reference calculated from the measured ion intensities on the Ultra. These values are converted to the standard scales: , , , and using the formulae above. The clumping anomalies of the reference gas used for the measurements, AP613, denoted as and , were assigned using temperature-equilibration experiments which are explained in detail in the next section. The bulk isotopic composition of AP613, denoted as and , was obtained by measurements using a conventional continuous-flow IRMS system (Menoud et al., 2021).
2.3 Temperature calibration scale
To produce a CH4-clumped isotope calibration scale, we performed a series of isotope exchange experiments at various temperatures. For this, the laboratory reference gas AP613 was used, which is a commercially available pure CH4 gas cylinder with known bulk isotopic composition. CH4 from AP613 was equilibrated at temperatures ranging from 50 to 450 °C using two different catalysts: γ-Al2O3 for temperatures below 200 °C and Pt on Al2O3 for 200 to 450 °C.
Both catalysts were activated using the procedure explained in Eldridge et al. (2019). For each heating experiment, about 10 pellets of the catalyst were inserted into a 20 mL glass tube with a Teflon valve and evacuated to 10−3 mbar to remove adsorbed air and moisture. The tube was then filled with 140 mbar of pure O2 and heated for about 5 h at 550 °C for activation of the catalyst. After heating, the tube was evacuated overnight (for 12 to 14 h) at 550 °C and then cooled to room temperature. The pellets were not exposed to outside air once activated. After the activated pellets were cooled to room temperature, 5 to 6 mL of pure CH4 (AP613) was added to the tube and heated at the desired temperature and duration as given in Table 3.
Table 3Summary of the equilibrated gas experiments; Δ13CDH3 raw and Δ12CD2H2 raw values are relative to the reference gas, and Δ13CDH3 absolute and Δ12CD2H2 absolute are calculated using the assigned anomalies of the reference gas.

The equilibrated gases were measured on the Ultra against the reference gas, i.e. unmodified CH4 from the AP613 cylinder. The raw Δ13CDH3 and Δ12CD2H2 values are calculated using Eqs. (3c) and (3d) but assuming and to be zero. The raw values obtained in this way showed the expected dependence on temperature but with a shift due to the real clumped values of the reference being different from zero. To determine this offset, the functions from Eldridge et al. (2019) were fit to the data with an added free parameter for the offset as given in Eqs. (4a) and (4b):
The parameters a and b were then optimized, keeping the shape of the temperature dependence constant, and they were used to estimate the Δ13CDH3 and Δ12CD2H2 values of our reference gas. In practice, this was done using a Monte Carlo simulation with 1000 runs: at each run, each data point was independently applied with a random error based on the uncertainty of that measurement, assuming Gaussian distribution of the errors. The functions above were then fitted, and a set of free parameters (a and b) were obtained. The final absolute Δ13CDH3 and Δ12CD2H2 values of the reference were calculated by averaging the a and b parameters for all runs (with outliers removed), and the errors reported are the corresponding standard deviations.
2.4 CH4 extraction and purification system
The schematic of the extraction system is shown in Fig. 2.

Figure 2Schematic of high-concentration extraction system (HCES) and low-concentration extraction system (LCES) and the GC setup at IMAU. Samples are introduced to the HCES via H4 and to the LCES via L0. The pre-concentrated sample in CT2 is transferred to Trap A via a connection between L12 and H2. The acronyms used in the figures are explained in the main text (Sect. 2.4).
Precise measurements of the clumped isotopic composition of CH4 on the Ultra require about 3 ± 1 mL of pure CH4 for a single measurement. Throughout this paper, the quantity of gas is specified in millilitres (mL) (at STP, unless otherwise specified; the conversion to molar units is ).
The CH4 extraction and preconcentration procedure followed in our laboratory involves several steps depending on the sample concentration as explained below.
2.4.1 HCES
The high-concentration extraction system (HCES) is used to extract CH4 from samples with more than 1 % of CH4, i.e. extracting from up to 200 mL of sample gas. The HCES includes two empty traps (Trap C and Trap D), two traps filled with silica gel (Trap A and Trap B), and a gas chromatograph (GC) with a passive thermal conductivity detector (TCD), all connected with in. SS tubing and 316 L VIM-VAR Swagelok valves. All the parts are shown in the schematic (Fig. 2). This system is built following the one described in Young et al. (2017).
The CH4 in the sample gas is separated from the other components by GC, and then it is collected cryogenically on silica gel. The sample is introduced via valve H4 and collected in Trap A with silica gel cooled to −196 °C with liquid N2. The pressure in the system is monitored to ensure that all the sample is trapped. The sample in Trap A is introduced to the GC from Trap A using He at a flow rate of 30 mL min−1 for 5 min by warming the trap to about 70 °C using a hot water bath.
The GC has two columns used in series for the final purification of CH4: A 5 m in. o.d. SS column packed with a 5 Å molecular sieve to separate H2, Ar, O2, and N2 from hydrocarbons and a 2 m in. o.d. SS column packed with HayeSep D porous polymer to separate CH4 from the remaining higher hydrocarbons like C2H6, C3H8, etc. Wide columns of in. are used to attain separation of more than 5 mL of CH4 within 55 min.
CH4 elutes from the GC column after O2, N2, and Kr. For concentrated samples (> 5 % CH4 in air) without Kr, O2 elutes around 10 min, N2 around 22 min, and CH4 around 40 min when the GC is operated at 50 °C. After the complete elution of N2 (35 min), Trap B with silica gel is cooled with liquid N2 to collect CH4 for about 15 min. Once all the CH4 is collected, Trap B is evacuated for 10 min to remove the He carrier gas while the trap is still cooled with liquid N2. Following this, CH4 is released from Trap B by warming the trap to ∼ 70 °C (hot water bath) and collected in a sample vial filled with silica gel and cooled with liquid N2 to be transferred to the mass spectrometer.
For samples with CH4 concentrations between 1 % and 5 % CH4 in air, the sample volumes required to extract the required amount of CH4 are larger (> 100 mL). In this case, the O2 and N2 peaks are not fully resolved and not well separated from CH4. Therefore, CH4 along with traces of O2 and N2 eluted from the GC is collected in Trap A instead of the sample vial and passed through the GC a second time for further purification (same steps as above). In the second round of extraction, the O2 and N2 peaks are small and well separated from each other and from the CH4 peak. For samples with ppm levels of Kr (notably atmospheric samples), separation of pure CH4 from Kr was only achieved when the GC columns were heated at 40 °C instead of 50 °C normally used for other samples. The comparison of chromatograms before and after Kr separation was achieved is shown in Fig. 9.
After each chromatographic separation, the GC columns are baked at 200 °C for 30 min with He flow to remove CO2, the heavier hydrocarbons, and other impurities. After baking, the columns are slowly cooled to 50 °C for the next extraction. Traps A and B are heated overnight at 150 °C while pumping with a high-vacuum pump. The silica gel flask used for sample collection is evacuated until the next use.
2.4.2 LCES
Extracting CH4 from large quantities of air involves a stepwise increase of the CH4 concentration by cryogenically trapping the sample gas in successively smaller charcoal traps, until the concentration is high enough for the sample to be further processed with the HCES. The low-concentration extraction system (LCES) is made of a in. glass tube with J. Young high-vacuum PTFE valves, and the major components are an empty glass trap (GT), two Russian doll traps (RDT1 and RDT2), and two charcoal traps (CT1 and CT2) as shown in Fig. 2. A part of the LCES is from the extraction system that has been used previously for CO isotope analysis (Bergamaschi et al., 2000, 1998).
The GT and RDTs are respectively used to remove H2O and CO2 from the air. This is followed by two pre-concentration steps in CT1 and CT2, which both collect all the CH4 but only a small part of bulk air so that the CH4 concentration increases in each step. The exhaust of the low-vacuum pump which draws the air though the extraction system is connected to a G2301 greenhouse gas analyser (Picarro Inc.) to monitor CO2, CH4, and H2O concentrations during the whole extraction procedure. This ensures that a potential breakthrough is detected.
The air taken directly from outside or from a cylinder is first dried using the GT cooled to −70 °C with a dry ice and ethanol slurry. A Mg(ClO4)2 tube after the GT further dries the air sample before it is introduced to the traps for collection. RDT1 and RDT2, both cooled to −196 °C with liquid N2 and connected in series, are used to scrub CO2, N2O, H2O, traces, and other condensable gases from the air. The CO2-free air is then passed through CT1 (−196 °C), which traps CH4 quantitatively, and only part of the remaining air components (O2, N2, etc.). During this CT1 collection period, CT2 is bypassed. The flow of air is controlled using a mass flow controller (MFC 1) and is adjusted to 6 to 6.5 L min−1 to maintain a pressure lower than 230 mbar in the glass line between L1 and L6 to avoid condensation of O2 in the traps cooled with liquid N2, which is a potential danger. The glass line is partially heated using heating wires to avoid freezing of tubes and valves.
Once a quantity of about 1100 L of air has been processed, the remaining air in the glass line is pumped until P4 drops to 4 mbar. To transfer the collected air from CT1 to CT2, the liquid N2 around CT1 is replaced with a dry ice + EtOH slurry to warm the trap to −70 °C. At this temperature, the emerging N2+O2 mixture is pumped out for 3 to 4 min, while the CH4 stays in the CT1 trap. In the meantime, the bypassed CT2 is cooled to −196 °C with liquid N2. The remaining gas mix in CT1 is released by removing the dry ice slurry and heating CT1 with a hot water bath and is passed through CT2 (−196 °C). As the pressure in the line drops to 10 mbar, 0.5 L min−1 of additional pure N2 is used to transfer any remaining gas from CT1 to CT2 for 5 min via MFC 1. After this, the liquid N2 bath of CT2 is replaced with dry ice + EtOH slurry and pumped for 1 to 2 min to further concentrate the air mixture. At the end of this step, the final sample volume is less than 100 mL, and the sample can be transferred to Trap A of the HCES, cooled with liquid N2. CT2 is heated using a water bath, and, after the pressure reading on P3 drops to 0 mbar, it is flushed with pure N2 from MFC 3 (at 5 mL min−1 for 2 min) to transfer the remaining gas. Once all the sample is collected in Trap A, the high-concentration extraction procedure is followed as explained above.
For samples with medium concentrations (0.1 % to 1 % CH4) i.e. < 3 L total sample volume, the first few steps of the LCES are skipped, and the sample is directly trapped in CT2. The remaining procedure is the same as explained above.
Before each extraction, RDTs and CTs are cleaned using 0.5 L min−1 of pure N2 for 40 min while heating them with hot water baths at 70 °C to avoid contamination from the previous sample.
2.4.3 Extraction system tests with laboratory reference gas
The extraction and purification system was tested using three of our laboratory reference gases: AP613, CAL1549, and IMAU-3. Various mixtures of pure AP613 in zero air (synthetic air: O2+N2) and pure CAL1549 in zero air were used to test the extraction system, and then the extracted CH4 was measured on the Ultra. The separation of Kr from CH4 in the GC and the effect of Kr on the isotope measurements on the Ultra were tested using a 1:1 mixture of IMAU-3 and pure Kr.
To replicate the atmospheric CH4 samples, pure AP613 was mixed with zero air to a mole fraction of 2.5 ppm of methane in 1000 L. Since zero air is devoid of CO2 and H2O, GT and RDT2 were bypassed for these tests. RDT1 was still immersed in liquid N2 to ensure that even small traces of CO2 were trapped and to check that the RDTs do not influence the clumping anomalies of CH4. The rest of the procedure was followed as for normal sampling.
2.5 Quality checks for the Ultra
To establish the accuracy of the Ultra measurements, the Ultra δD and δ13C measurements were compared to conventional bulk isotope measurements. Most samples were analysed for δD and δ13C before the extraction and purification, using an independent conventional bulk isotope measurement system (Menoud et al., 2020), and the results were compared to the ones obtained from the Ultra measurements after the extraction.
Weekly “zero enrichment” measurements (same gas in both bellows) were done to check for systematic differences between the bellows (e.g. by contamination, leaks). These, together with regular measurements of the pure CAL1549 gas, were used to monitor the stability of the instrument and the reproducibility of the measurements. The internal precision of the measurements was estimated for each measurement (sample or test gas) from the 1 SE (standard error) over the whole measurement.
An inter-laboratory comparison with the Nu Panorama high-resolution mass spectrometer operated at the University of Maryland (UMD) was done for the three laboratory reference gases: AP613, CAL1549, and IMAU-3. The results of these comparisons are presented in the next section.
3.1 Ultra measurements
As described in Sect. 2.2, clumped isotope measurements on the Ultra involve measuring the different isotopologues in three configurations for a total of 20 h. Typical mass scans of the three configurations are shown in Fig. 3. The position of the peak centres (marked with red dotted lines in Fig. 3) is quite stable during the entire measurement procedure and small mass shifts are corrected every hour using the peak centre correction feature in the software.
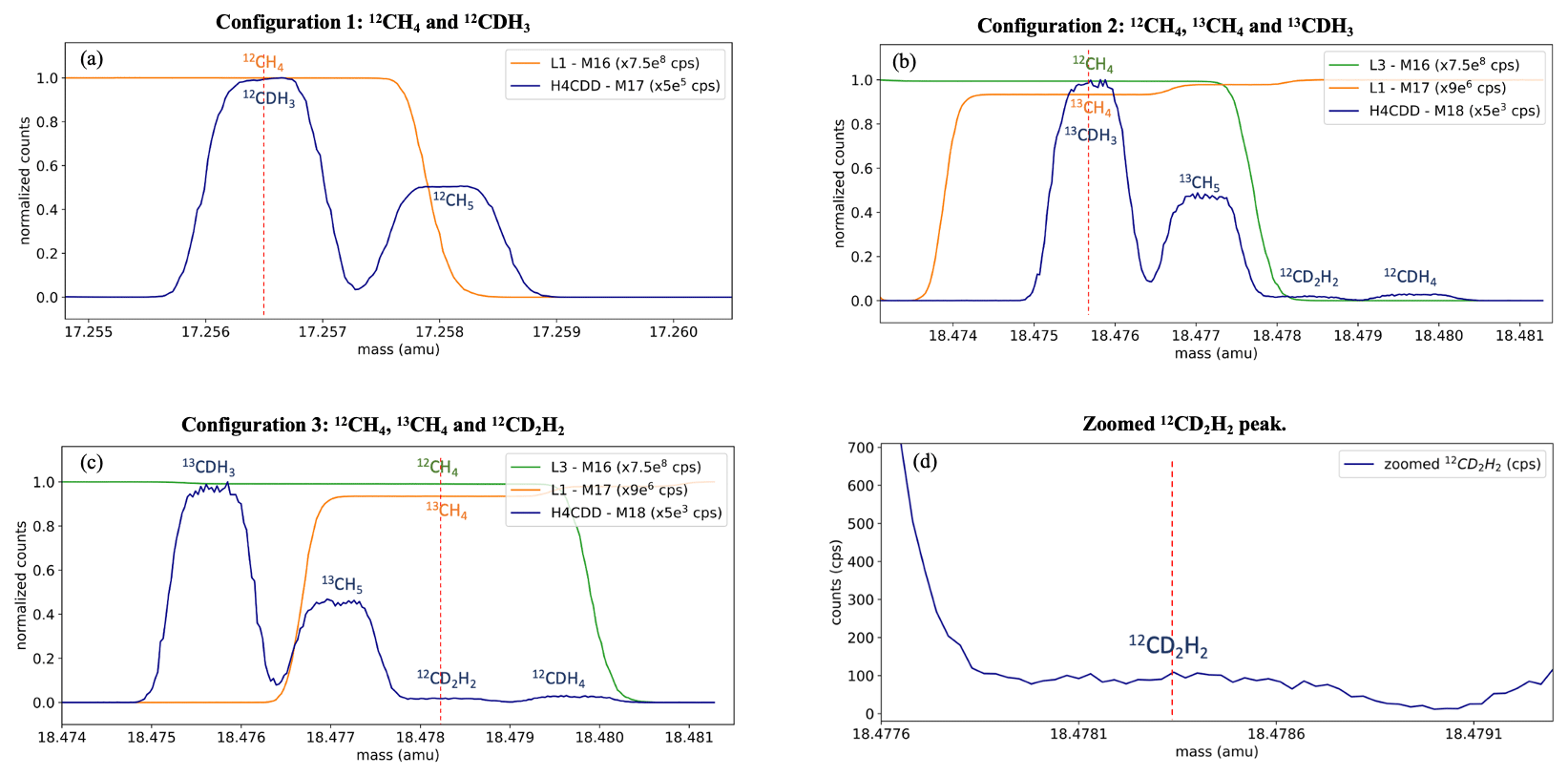
Figure 3Mass scans of three configurations to measure 12CDH3 (a), 13CH4 and 13CDH3 (b), and 13CH4 and 12CD2H2 (c, d). The x axis values correspond to the peak centre setting, i.e. mass 17 in panel (a) and mass 18 in panels (b)–(d), and the other detectors are offset to these values to show the other isotopologues on the same scale. The different detectors used, and the normalization factors are given in the legends. The dashed red line indicates the peak centre mass setting. Panel (d) shows the zoomed-in peak of 12CD2H2 and the counts measured.
3.2 Temperature equilibration experiments
The results of the heating experiments are presented in Table 3. The equilibrated gas (subsample of AP613 heated at different temperatures; Sect. 2.3) was measured against the non-equilibrated gas from AP613 (directly from the cylinder), which is the Ultra reference gas. Raw measurement values relative to the reference gas are reported as Δ13CDH3 raw and Δ12CD2H2 raw.
The measured values of heated AP613 at different temperatures were compared to the theoretical equilibrium curve, and the Δ13CDH3 and Δ12CD2H2 values of AP613 were estimated using the Monte Carlo simulations as described in Sect. 2.3. The Δ13CDH3 and Δ12CD2H2 assigned to our reference gas, AP613, are the following: Δ13CDH3=2.23 ± 0.12 ‰ and Δ12CD2H2=3.1 ± 0.9 ‰. Since this pair of values for the clumping anomalies does not lie on the thermodynamic equilibrium curve, we cannot assign a formation temperature value to AP613. The absolute values of Δ13CDH3 and Δ12CD2H2 calculated using the assigned values of AP613 are given in Table 3 and in Fig. 4.
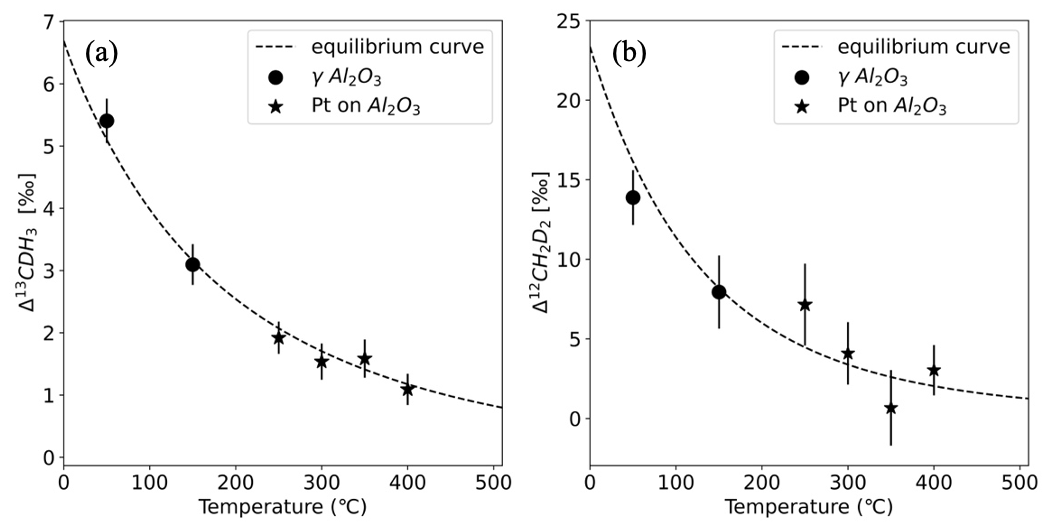
Figure 4Absolute Δ13CDH3 and Δ12CD2H2 of the equilibrated gas compared to the theoretical equilibrium curve, calculated using the assigned anomalies of the reference gas, AP613: Δ13CDH3=2.23 ± 0.12 ‰ and Δ12CD2H2=3.1 ± 0.9 ‰. The data points represent the equilibrated gas at different temperatures with the markers corresponding to the different catalysts as given in the legend. The dashed black line is the thermodynamic equilibrium curve.
3.3 Internal precision and reproducibility of the Ultra measurements
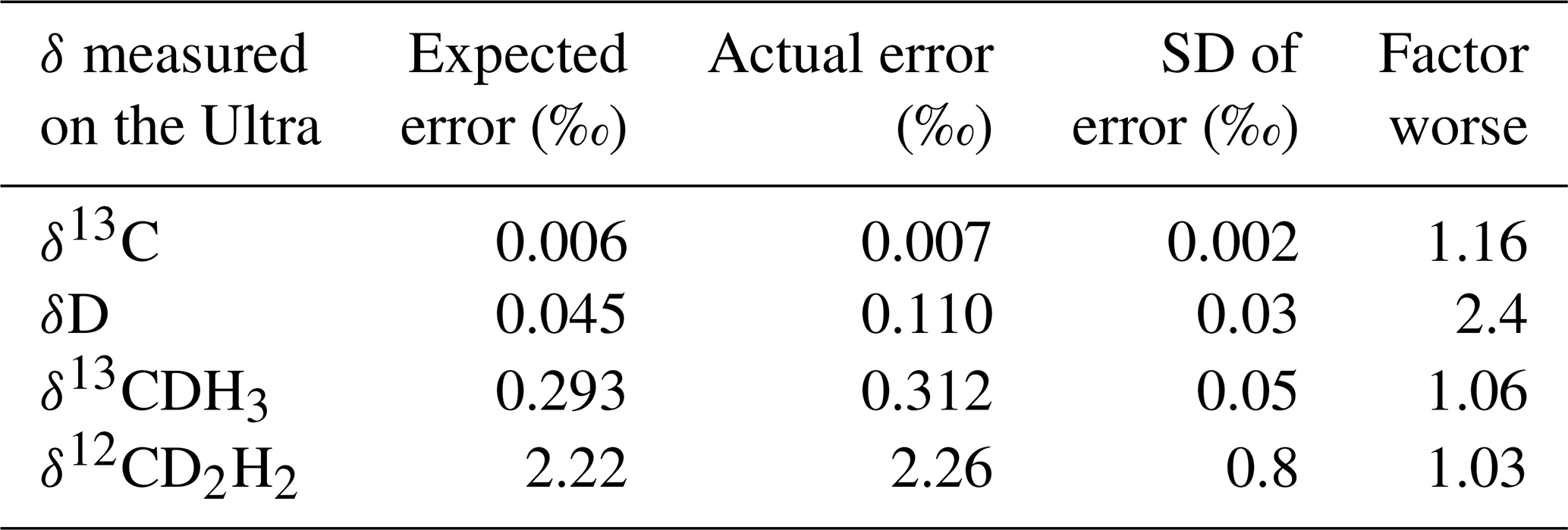
The average standard errors of the measured δ13C, δD, δ13CDH3, and δ12CD2H2 values and their comparison to the expected precision based on counting statistics of the shot noise are given in Table 4. Achieved precisions are very close to the shot noise limit for δ13C, δ13CDH3, and δ12CD2H2. Typically, δD measurements are about 2 times worse than the shot noise limit. This may be because of the following reasons: the high count rates (of the order of 105) of 12CH3D measured using the H4-CDD detector are close to the upper limit of the CDD operating range, and they are not in the optimal region. Therefore, we expect here a lower signal-to-noise ratio (meaning a higher relative error). Additionally, the peak top of 12CH3D, which is not very flat and sometimes rounded, suggest that the ion beam is slightly too wide for H4-CDD with a very narrow collector slit, which is not unexpected given the relatively high abundance. That means, very slight variations in the ion beam direction can result in relatively large variations in the quantity of ions entering the detector. However, the changes in δD between different samples are much higher than the achieved precision, which is better than the one for conventional continuous-flow IRMS (CF-IRMS) instruments.
Table 4Average standard errors of δ13C, δD, δ13CDH3, and δ12CD2H2 measurements on the Ultra and the expected errors from counting statistics of the shot noise. The “factor worse” column shows how good our measurements are compared to the shot noise limit.
The average precision (1 SE) values of calculated clumping anomalies of over 300 measurements in the last 3 years are 0.3 ± 0.1 ‰ for Δ13CDH3 and 2.4 ± 0.8 ‰ for Δ12CD2H2, depending on the CH4 sample volume and measurement duration. The precision of Δ13CDH3 and Δ12CD2H2 is calculated by propagating the error from the measured δ13C, δD, δ13CDH3, and δ12CD2H2 values, using the Eqs. (3c) and (3d).
The measurement procedure is slightly modified for samples smaller than 2 mL of CH4. In such cases, 12CD2H2 is measured relatively longer than the standard procedure, with shorter measurements of 12CDH3 to attain the maximum possible precision for Δ12CD2H2.
The results of the zero enrichment measurements using AP613 are shown in Fig. 5. The mean of these measurements done over 3 years is 2.3 ± 0.1 ‰ for Δ13CDH3 and 3.2 ± 0.3 ‰ for Δ12CD2H2, and all the data points fall symmetrically around the values of AP613 calibrated based on the heating experiments (2.2 ± 0.1 ‰ and 3.1 ± 0.9 ‰ for Δ13CDH3 and Δ12CD2H2, respectively). The standard deviation of these measurements, 0.4 ‰ for Δ13CDH3 and 2.1 ‰ for Δ12CD2H2, is close to the typical measurement error. Together, these measurements show that there are no other large sources of errors in the sample measurements (e.g. leaks in the inlet and/or room temperature variations) and that both bellows used for the measurements behave similarly.
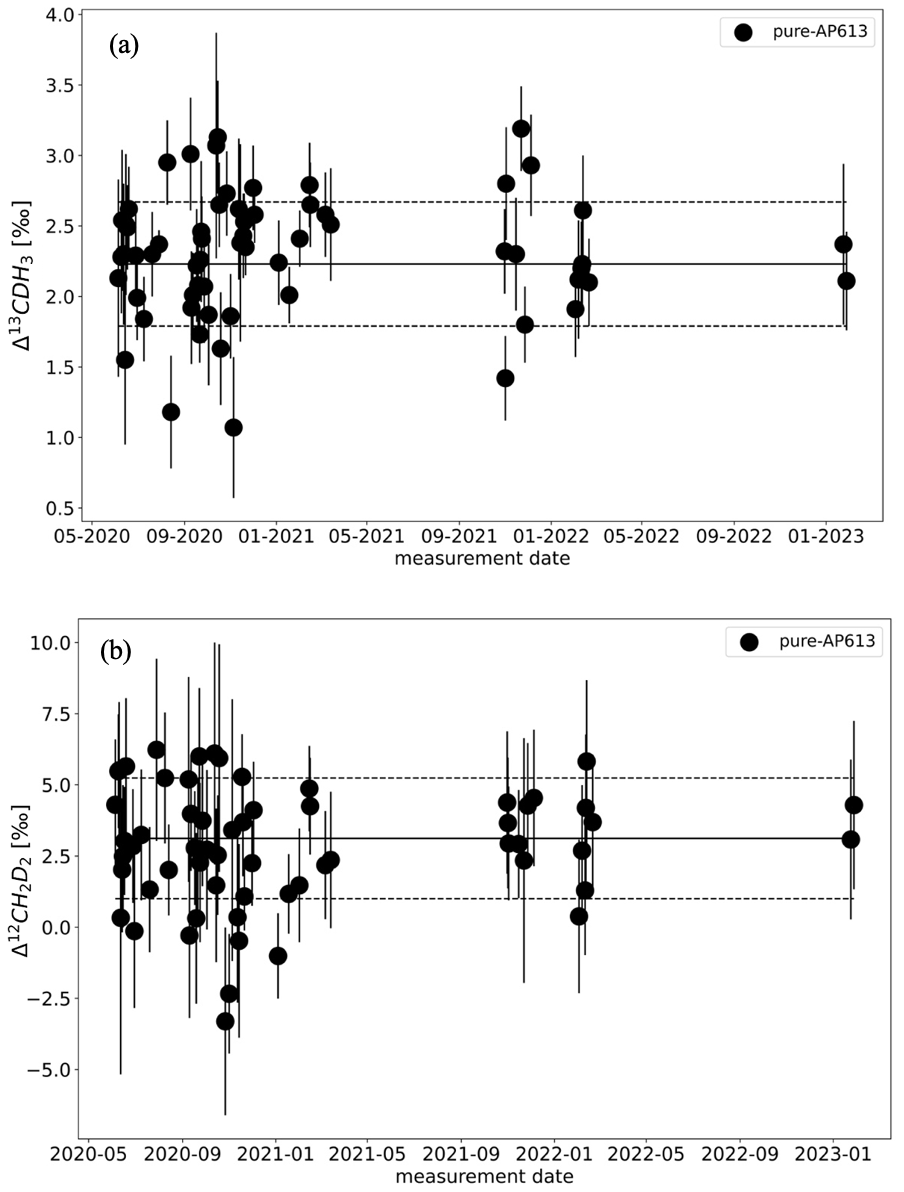
Figure 5Results of the zero enrichment measurements, each dot representing the calculated clumping anomalies Δ13CDH3 (a) and Δ12CD2H2 (b) of gas AP613. The solid black line represents the values of AP613 assigned from the temperature calibration experiments, and the dashed black lines indicate the 1σ SD of these measurements over 3 years.
The reproducibility of the measurements on the Ultra was quantified by repeated measurements of pure CAL1549 as shown in Fig. 6. Long-term reproducibility, estimated as 1σ standard deviation of the measurements of pure CAL1549 over almost 3 years, is around 0.15 ‰ for Δ13CDH3 and 1.2 ‰ for Δ12CD2H2. This external reproducibility is consistent with the individual measurement uncertainty, which is on average 0.3 ‰ for Δ13CDH3 and 2.3 ‰ for Δ12CD2H2 for these measurements.

Figure 6Results of the measurements of pure CAL1549 for Δ13CDH3 (a) and Δ12CD2H2 (b). The solid black line represents the average value of these measurements, and the dashed black line is the standard deviation (1σ) of the eight measurements shown.
3.4 Inter-laboratory calibration
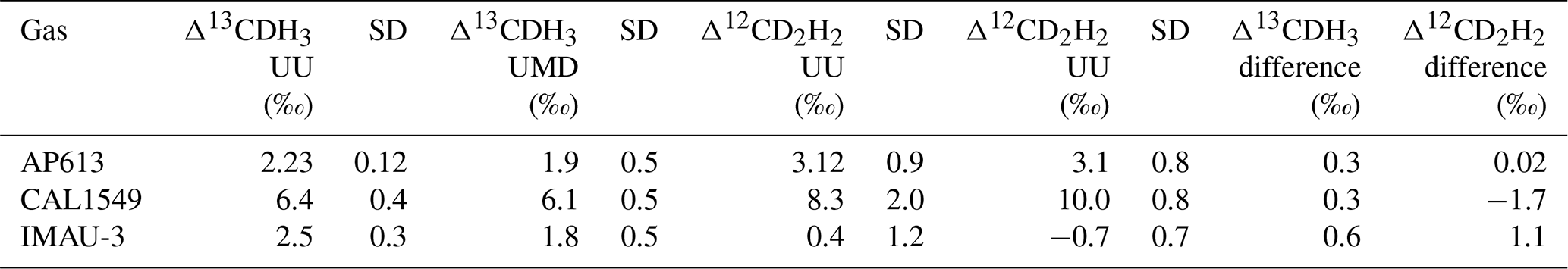
Three of our gases (AP613, CAL1549, and IMAU-3) were measured on both the Thermo Scientific Ultra at Utrecht University (UU) and the Nu Panorama at the University of Maryland (UMD). The results of these measurements are given in Table 5.
Table 5Comparison of Δ13CDH3 and Δ12CD2H2 measurements of the three reference gases (AP613, CAL1549, and IMAU-3) on the Ultra at UU and the Panorama at UMD.
The values assigned to AP613 using our heating experiments (Sect. 3.2) agree well with the measured value of the non-heated pure AP613 on the Panorama as shown in Fig. 7. The other two gases are also within the measurement uncertainty (1σ).
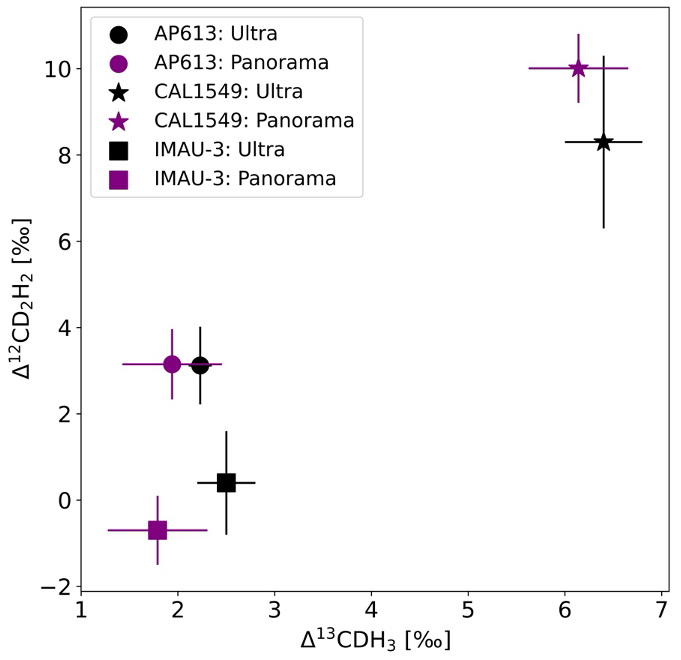
Figure 7The clumping anomalies of AP613, CAL1549, and IMAU-3 measured on the UU-Ultra (black) and the UMD-Panorama (purple). The symbols dot, star, and square represent the gases AP613, CAL1549, and IMAU-3, respectively.
3.5 Extraction test with known gas
As mentioned earlier, mixtures of pure CH4 from AP613 or CAL1549 with zero air were used to test and characterize the extraction system. The CH4 extracted from these mixtures was measured against the AP613 reference gas on the Ultra. The results of the measurements are presented in Fig. 8 as the difference between the expected and the measured values. We expect this difference to be zero within the measurement uncertainty if the extraction procedure does not introduce any isotopic fractionation. Pure CH4 from CAL1549 was also passed through the extraction system (hereby denoted as pure CAL1549 extracted) using the normal extraction procedure to check for any contamination or fractionation associated with gas introduction and collection via the extraction system.

Figure 8Test results of the extraction system with different mixtures of laboratory reference gases as stated in the legend. Each coloured dot and star represent the difference between the measured and expected Δ13CDH3 (a) and Δ12CD2H2 (b) values, respectively, of extracted-AP613 and extracted CAL1549 as given in the legend. The dashed black line is the standard deviation (1σ) of the difference for Δ13CDH3 and Δ12CD2H2, respectively.
The standard deviation values of the difference between the expected and the measured values of these extraction tests are 0.4 ‰ for Δ13CDH3 and 2.8 ‰ for Δ12CD2H2. Most of these extracted reference gas measurements are within this unexpected uncertainty (1σ). When the difference was more than about 2σ, additional tests were performed or parts of the system were replaced or cleaned for longer until the measurements were good enough. Typically, large offsets from the expected values are caused by incomplete trapping and releasing of gas from the silica gel used in Traps A and B of HCES. This is solved by conditioning the silica gel for longer (than the standard procedure; Sect. 2.4.1) at 150 °C.
The effect of Kr on the measurements was investigated using a 1:1 mixture of IMAU-3 and pure Kr. This mixture was directly measured on the Ultra and compared with the values of pure IMAU-3. The δ13C, δD, Δ13CDH3, and Δ12CD2H2 values of the mixture measured on the Ultra are −34.6 ‰, −242.0 ‰, 7.45 ± 0.37 ‰, and 65.7 ± 2.3 ‰, respectively, whereas those of pure IMAU-3 are −36.6 ‰, −200.0 ‰, 2.5 ± 0.3 ‰, and 0.4 ± 1.2 ‰, respectively. This shows that Kr introduces a strong bias in the measurements of both the bulk and clumped isotopic composition of CH4. Therefore, it is very important to remove Kr from the sample before measuring the CH4 isotopic composition on the Ultra.
3.6 Chromatograms
Accurate and precise measurements of Δ13CDH3 and Δ12CD2H2 on the Ultra require 3 ± 1 mL of pure CH4. CH4 from sample mixtures pre-concentrated in the extraction system is separated from the bulk sample using the GC, as explained in detail above. Chromatograms for samples with different CH4 concentrations are illustrated in Fig. 9. When the total sample volume is above 100 mL, O2 and N2 are not completely separated from CH4; therefore, a second round of GC purification is needed (Fig. 9b and c). For atmospheric CH4 samples, separation of Kr from CH4 is attained only when the GC columns are kept at 40 °C (Fig. 9e) instead of the usual 50 °C (Fig. 9d) used for other CH4 samples.

Figure 9GC chromatograms of different sample mixtures as shown in the legends. (a) Chromatogram of 20 % CH4 +80 % zero air: 25 mL sample volume (5 mL CH4). (b, c) Chromatograms of the first and second rounds of 1 % CH4 + 99 % zero air: 250 mL sample volume (2.5 mL CH4). (d) Chromatogram of a pre-concentrated atmospheric air: 70 mL sample volume (2 mL CH4), when GC columns were heated at 50 °C and Kr is not separated from CH4. (e) Chromatogram of pre-concentrated atmospheric air when GC columns are heated at 40 °C and Kr and CH4 are well separated.
3.7 Propagation of error from clumping anomaly to the formation temperature
The clumping anomalies, Δ13CDH3 and Δ12CD2H2, can be used to calculate the formation temperature of CH4 when it is formed in thermodynamic equilibrium. The average precision of the Ultra measurements is 0.3 ‰ for Δ13CDH3 and 2.4 ‰ for Δ12CD2H2. When propagated into the calculated temperatures (Eqs. 4a and 4b), the measurement error has a non-linear effect across the temperature range of 0 to 1000 °C. This is because of the polynomial function that defines the relation between the clumping anomalies and temperatures as given in Eqs. (4a) and (4b). Figure 10 shows that the formation temperatures can be predicted with relatively low uncertainty at lower temperatures. For example, at 50 °C the formation temperature can be estimated as °C from Δ13CDH3 and °C from Δ12CD2H2. At 400 °C, for the same measurement precision, the temperature estimated from Δ13CDH3 is °C and from Δ12CD2H2 it is °C. Although the absolute clumped isotope effects are larger for Δ12CD2H2 than for Δ13CDH3, formation temperatures calculated from Δ13CDH3 give a more precise temperature estimate because of the better measurement precision for Δ13CDH3.
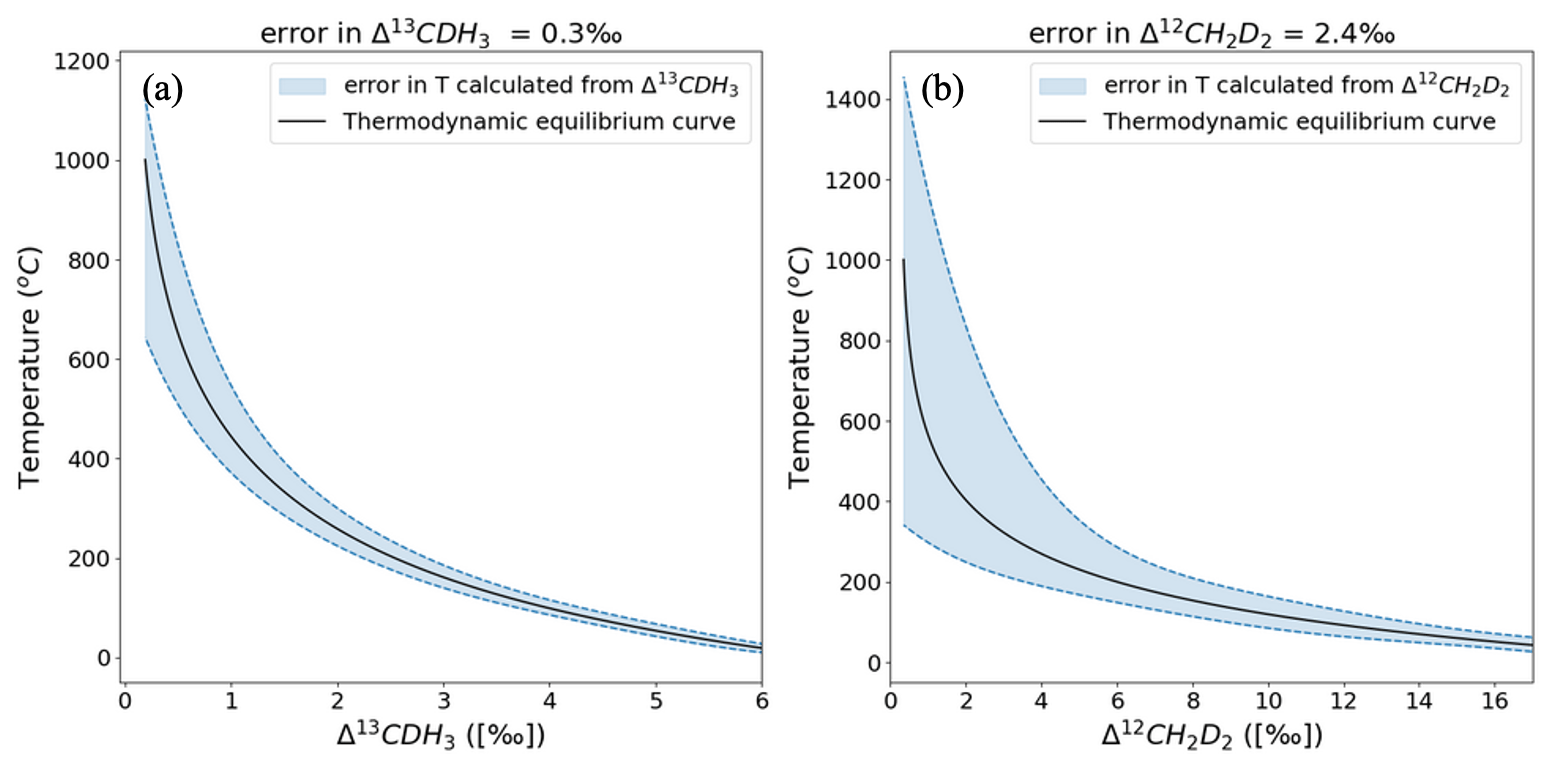
Figure 10Error in the formation temperatures calculated from Δ13CDH3 (a) and Δ12CD2H2 (b). The black solid line represents the thermodynamic equilibrium curve, and the blue dashed lines give the upper and lower limits of the errors of temperatures propagated from the errors in the measured clumping anomaly.
3.8 Overview of different samples measured
3.8.1 Samples with different source signatures
CH4 samples collected from different origins and laboratory experiments were extracted and measured with the setup explained in Sect. 2.4. An overview of the bulk and clumped isotopic composition of some of these samples from different sources of CH4 is presented in Fig. 11 (Table S1 in the Supplement). The precision of individual measurements is in the range of 0.2 ‰ to 0.5 ‰ for Δ13CDH3 and 1.4 ‰ to 4 ‰ for Δ12CD2H2, depending on the sample volume.
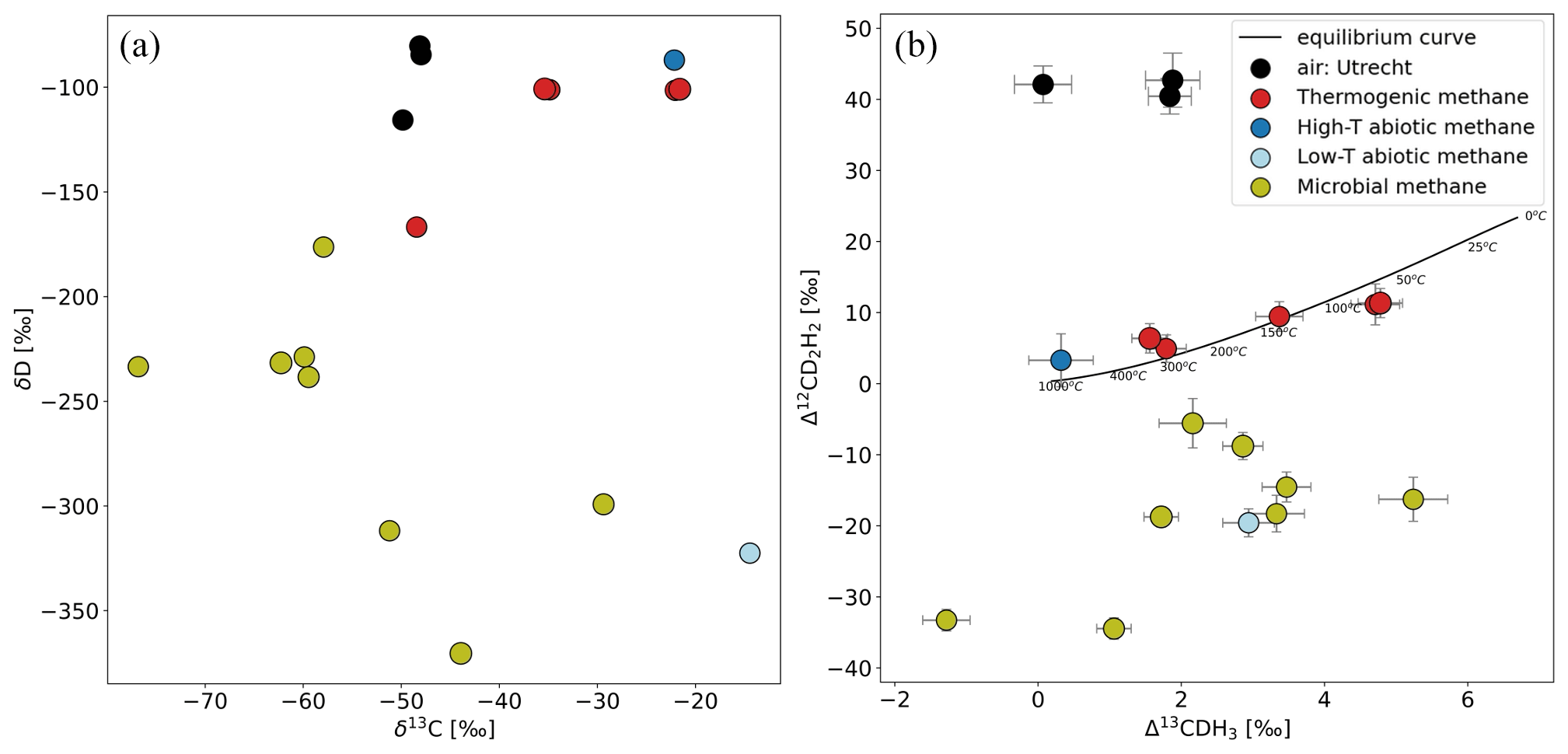
Figure 11Comparison of δ13C and δD (a) and Δ13CDH3 and Δ12CD2H2 (b) of samples from different source types and atmospheric air measured outside IMAU. The overview of the samples shown in this figure is given in Table S1. The solid black line represents the thermodynamic equilibrium curve with corresponding temperature values.
Most of the samples of thermogenic origin lie on or close to the thermodynamic equilibrium line; therefore, the formation temperature of CH4 can be calculated for them. All the samples with a microbial origin (e.g. incubation experiments with methanogens, CH4 from lake water and sediments) have depleted Δ12CD2H2 values. The low-temperature abiotic CH4 also has negative Δ12CD2H2. This is in line with previous studies that also show that the production of CH4 by methanogens and in rocks abiotically at lower temperatures is affected by kinetic fractionation and/or combinatorial effect that leads to negative Δ12CD2H2. So far, about 80 samples have been measured on the Ultra from very different origins with clumping anomalies ranging from −1 ‰ to 6 ‰ for Δ13CDH3 and from −40 ‰ to 45 ‰ for Δ12CD2H2.
3.8.2 Ambient air measurement
Using the low-concentration extraction system (LCES), we extracted and measured several samples of atmospheric air sampled in Utrecht, and the results of the first measurements are given in Table 6.
Table 6Results of δ13C, δD, Δ13CDH3 and Δ12CD2H2 of atmospheric CH4 (airs A, B, and C) sampled in Utrecht and the comparison of the measured values to the model predictions in Haghnegahdar et al. (2017) and Chung and Arnold (2021).

The solid black dots in Fig. 11b show the results of the first measurements of the clumping anomaly of atmospheric CH4 in Utrecht (0 ‰ to 2 ‰ for Δ13CDH3 and 40 ‰ to 43 ‰ for Δ12CD2H2). The air samples in Table 6 were sampled under three different atmospheric conditions: (i) clean air from the north (air A), (ii) clean air from the south (air B), and (iii) air with high CH4 content due to local/regional pollution (air C). The values of the clumped isotopic composition of all three air samples are characterized by a very high anomaly for Δ12CD2H2 and a low anomaly for Δ13CDH3. The first measurements of atmospheric methane reported by Haghnegahdar et al. (2023) of air sampled from various atmospheric scenarios in and around Maryland, USA, are compatible (0 ‰ to 3 ‰ for Δ13CDH3 and 42 ‰ to 55 ‰ for Δ12CD2H2) with our measured values.
Firstly, comparing these values to the ones of CH4 emitted from various sources, it is evident that atmospheric CH4 has a distinct clumped signature, particularly in Δ12CD2H2. The large positive anomaly for Δ12CD2H2 of atmospheric CH4 can be explained by a strong clumped isotope fractionation due to the sink reactions of CH4 in the atmosphere (Haghnegahdar et al., 2017). The distinct differences between various source types and the offset of atmospheric CH4 also suggest that more measurements of the clumping anomaly of air, especially Δ12CD2H2, can provide more information about the different sources and sink reactions that determine atmospheric CH4 levels.
Secondly, the bulk isotopic composition (Table 6) shows as expected lower values for the polluted air C compared to the clean air A and air B, indicating regional contributions from biogenic sources as is typical for the Netherlands (Röckmann et al., 2016b; Menoud et al., 2021). However, in the case of the clumped isotopes, the air from the north is quite different in Δ13CDH3, while the values for the polluted and clean air from the south are not very different, unlike the bulk isotopes. At this point we, cannot draw strong conclusions, as we only have one measurement per condition and no information on the potential variability. More measurements of Δ13CDH3 and Δ12CD2H2 of air are needed to understand if short-term local/regional atmospheric changes affect the clumping anomaly of air.
Lastly, although the measured Δ12CD2H2 of atmospheric CH4 has very high values compared to the emissions from sources, our measurement results are still far lower than recent model predictions (Chung and Arnold, 2021; Haghnegahdar et al., 2017) (Table 6). The difference can be either due to the inaccuracy in (i) source signatures of all the different sources that contribute to atmospheric CH4 mole fraction or (ii) the theoretical values of the kinetic isotopic fractionation factor (i.e. KIE, kinetic isotopic effect) of the sink reactions of CH4 with OH and Cl and the soil sink reactions.
We used a box model to see how the clumping anomaly of air reacts to these two parameters. The model uses clumping anomalies of the source mixture and the KIEs of OH and Cl sinks as input and gives the expected anomalies of air as output. We work with three scenarios as discussed in detail below and illustrated in Fig. 12.

Figure 12Δ13CDH3 versus Δ12CD2H2 space showing the different scenarios discussed. The solid black line represents the thermodynamic equilibrium curve. The pink dot is the value of air predicted from the source mix shown as the unfilled black circle. The solid black dot is the value of air measured on the Ultra. The three arrows show the three scenarios mentioned in the text. The dashed black circle is the new source mix calculated using scenario 3.
Scenario 1. This involves replicating the values in the study of Haghnegahdar et al. (2017). If we assume that the predicted clumping anomaly of the mixture of sources in the atmosphere (Δ13CDH3=4 ‰, Δ12CD2H2=20 ‰) is accurate, then our model also gives higher values of Δ12CD2H2 and Δ13CDH3 of air as in that study, with the same KIE used (OH: 1.92 for 12CD2H2, 1.33 for 13CDH3; Cl: 2.2 for 12CD2H2, 1.46 for 13CDH3). This was done to verify that our simple model works well for this study.
Scenario 2. This involves calculating the KIEs required to arrive at the measured values of air with the same source mix as used in Haghnegahdar et al. (2017). To get the measured values from the predicted source mix, the KIEs must be lowered to 1.79 for 12CD2H2 and 1.325 for 13CDH3 for reaction with OH and 1.9 for 12CD2H2 and 1.45 for 13CDH3 for reaction with Cl. This relatively small change causes a difference of about 60 ‰ in Δ12CD2H2 between scenarios 1 and 2. Therefore, the clumping anomalies are very sensitive to the KIEs of the sink reactions.
Scenario 3. This involves calculating the clumping anomaly of the source mixture that is consistent with the KIEs used in Haghnegahdar et al. (2017) and the atmospheric air measurements presented here. In this case, the clumped isotope anomaly of the source mixture must be heavily depleted, especially in Δ12CD2H2 (Δ13CDH3=0 ‰, ‰), to get the measured values using the KIEs in scenario 1. This is much lower than the predicted value and would imply a strong underestimation of CH4 sources with depleted clumping anomalies such as biogenic sources.
Given the rather high number of clumped isotope measurements of CH4 sources that have been published to date, it seems unrealistic that the clumping anomaly of the source mix is so depleted in Δ12CD2H2 as calculated in scenario 3, which would imply that the KIE was previously indeed overestimated. These simple isotope mass balance calculations show that we need very precise estimations of the sink KIEs and more accurate measurements of the sources to completely understand the atmospheric CH4 budget using clumping anomalies.
We have presented a new versatile analytical setup for extraction, sample preparation, and measurement of the clumped isotope composition of CH4 on the Thermo Scientific Ultra instrument, including samples at atmospheric concentration. The extraction and GC purification techniques do not cause significant isotopic fractionation and preserve the signatures of the CH4 source. Currently, the system has been tested and works well for sample volumes of up to 1100 L. The typical precisions of samples measured on the Ultra are 0.3 ± 0.1 ‰ for Δ13CDH3 and 2.4 ± 0.8 ‰ for Δ12CD2H2. The long-term reproducibility, obtained from repeated measurements of a pure methane laboratory standard over almost 3 years, is around 0.15 ‰ for Δ13CDH3 and 1.2 ‰ for Δ12CD2H2. The standard deviation values of the difference between the expected and the measured values of all the extraction tests performed are 0.4 ‰ for Δ13CDH3 and 2.8 ‰ for Δ12CD2H2. The total measurement time is around 20 h. The system and the measurement procedure can be adjusted to optimize the sample volume required and long measurement times. The first measurements of samples from various sources yield results in general agreement with published values. We have measured about 80 samples on the Ultra from very different origins and a wide range of clumping anomalies: −1 ‰ to 6 ‰ for Δ13CDH3 and −40 ‰ to 45 ‰ for Δ12CD2H2. Our measurements of atmospheric CH4 show enriched Δ12CD2H2 values, but they are not as high as recently predicted by clumped isotope models. It is unlikely that the discrepancy can be explained only by an underestimation of sources with negative Δ12CD2H2, but we show that a small adjustment in the KIEs of the sinks could reconcile atmospheric and source clumped isotope compositions. The precision of atmospheric CH4 measurements can still be improved by extracting CH4 from much larger samples (2000 L).
Data supporting this study are openly available at https://doi.org/10.5281/zenodo.8269713 (Sivan, 2023.)
The supplement related to this article is available online at: https://doi.org/10.5194/amt-17-2687-2024-supplement.
All authors contributed to the design of the study. MS undertook the laboratory work with help from CvdV and MEP. MS wrote the article with input from all co-authors.
At least one of the (co-)authors is a member of the editorial board of Atmospheric Measurement Techniques. The peer-review process was guided by an independent editor, and the authors also have no other competing interests to declare.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
We would like to acknowledge the contributions of Dipayan Paul and Sönke Szidat for their important pioneering steps in testing the extraction line and setting up the Ultra measurement procedure. We would like to thank Edward Young for his help with building the high-concentration extraction line. We would also like to thank Jiayang Sun and James Farquhar from the University of Maryland for the measurements of our reference gases on the Nu Panorama instrument. We also thank our collaborators for contributing to the sample set used in this paper. Maria Elena Popa, Malavika Sivan, and the Thermo Scientific Ultra instrument are supported by the Netherlands Earth Science System Centre (NESSC), with funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie programme (grant agreement no. 847504).
This research has been supported by the Netherlands Earth System Science Centre (grant no. 847504).
This paper was edited by Christian Brümmer and reviewed by two anonymous referees.
Adnew, G. A., Hofmann, M. E. G., Paul, D., Laskar, A., Surma, J., Albrecht, N., Pack, A., Schwieters, J., Koren, G., Peters, W., and Röckmann, T.: Determination of the triple oxygen and carbon isotopic composition of CO2 from atomic ion fragments formed in the ion source of the 253 Ultra high-resolution isotope ratio mass spectrometer, Rapid Commun. Mass Sp., 33, 1363–1380, https://doi.org/10.1002/rcm.8478, 2019.
Archer, D., Eby, M., Brovkin, V., Ridgwell, A., Cao, L., Mikolajewicz, U., Caldeira, K., Matsumoto, K., Munhoven, G., Montenegro, A., and Tokos, K.: Atmospheric Lifetime of Fossil Fuel Carbon Dioxide, Annu. Rev. Earth Pl. Sc., 37, 117–134, https://doi.org/10.1146/annurev.earth.031208.100206, 2009.
Assonov, S., Groening, M., Fajgelj, A., Hélie, J. F., and Hillaire-Marcel, C.: Preparation and characterisation of IAEA-603, a new primary reference material aimed at the VPDB scale realisation for δ(13) C and δ(18) O determination, Rapid Commun. Mass Sp., 34, e8867, https://doi.org/10.1002/rcm.8867, 2020.
Beck, V., Chen, H., Gerbig, C., Bergamaschi, P., Bruhwiler, L., Houweling, S., Röckmann, T., Kolle, O., Steinbach, J., Koch, T., Sapart, C. J., van der Veen, C., Frankenberg, C., Andreae, M. O., Artaxo, P., Longo, K. M., and Wofsy, S. C.: Methane airborne measurements and comparison to global models during BARCA, J. Geophys. Res.-Atmos., 117, D15310, https://doi.org/10.1029/2011JD017345, 2012.
Bergamaschi, P., Brenninkmeijer, C. A. M., Hahn, M., Röckmann, T., Scharffe, D. H., Crutzen, P. J., Elansky, N. F., Belikov, I. B., Trivett, N. B. A., and Worthy, D. E. J.: Isotope analysis based source identification for atmospheric CH4 and CO sampled across Russia using the Trans-Siberian railroad, J. Geophys. Res.-Atmos., 103, 8227–8235, https://doi.org/10.1029/97JD03738, 1998.
Bergamaschi, P., Bräunlich, M., Marik, T., and Brenninkmeijer, C. A. M.: Measurements of the carbon and hydrogen isotopes of atmospheric methane at Izaña, Tenerife: Seasonal cycles and synoptic-scale variations, J. Geophys. Res.-Atmos., 105, 14531–14546, https://doi.org/10.1029/1999JD901176, 2000.
Cantrell, C. A., Shetter, R. E., McDaniel, A. H., Calvert, J. G., Davidson, J. A., Lowe, D. C., Tyler, S. C., Cicerone, R. J., and Greenberg, J. P.: Carbon kinetic isotope effect in the oxidation of methane by the hydroxyl radical, J. Geophys. Res.-Atmos., 95, 22455–22462, https://doi.org/10.1029/JD095iD13p22455, 1990.
Chung, E. and Arnold, T.: Potential of Clumped Isotopes in Constraining the Global Atmospheric Methane Budget, Global Biogeochem. Cy., 35, e2020GB006883, https://doi.org/10.1029/2020GB006883, 2021.
Conrad, R.: Control of microbial methane production in wetland rice fields, Nutr. Cycl. Agroecosys., 64, 59–69, https://doi.org/10.1023/A:1021178713988, 2002.
Douglas, P. M. J., Stolper, D. A., Eiler, J. M., Sessions, A. L., Lawson, M., Shuai, Y., Bishop, A., Podlaha, O. G., Ferreira, A. A., Santos Neto, E. V., Niemann, M., Steen, A. S., Huang, L., Chimiak, L., Valentine, D. L., Fiebig, J., Luhmann, A. J., Seyfried, W. E., Etiope, G., Schoell, M., Inskeep, W. P., Moran, J. J., and Kitchen, N.: Methane clumped isotopes: Progress and potential for a new isotopic tracer, Org. Geochem., 113, 262–282, https://doi.org/10.1016/j.orggeochem.2017.07.016, 2017.
Eiler, J. M.: “Clumped-isotope” geochemistry – The study of naturally-occurring, multiply-substituted isotopologues, Earth Planet. Sc. Lett., 262, 309–327, https://doi.org/10.1016/j.epsl.2007.08.020, 2007.
Eiler, J. M., Clog, M., Magyar, P., Piasecki, A., Sessions, A., Stolper, D., Deerberg, M., Schlueter, H.-J., and Schwieters, J.: A high-resolution gas-source isotope ratio mass spectrometer, Int. J. Mass Spectrom., 335, 45–56, https://doi.org/10.1016/j.ijms.2012.10.014, 2013.
Eldridge, D. L., Korol, R., Lloyd, M. K., Turner, A. C., Webb, M. A., Miller III, T. F., and Stolper, D. A.: Comparison of Experimental vs. Theoretical Abundances of 13CH3D and 12CH2D2 for Isotopically Equilibrated Systems from 1 to 500 °C, ACS Earth and Space Chemistry, 3, 2747–2764, https://doi.org/10.1021/acsearthspacechem.9b00244, 2019.
Etiope, G. and Sherwood Lollar, B.: Abiotic Methane on Earth, Rev. Geophys., 51, 276–299, https://doi.org/10.1002/rog.20011, 2013.
Fernandez, J. M., Maazallahi, H., France, J. L., Menoud, M., Corbu, M., Ardelean, M., Calcan, A., Townsend-Small, A., van der Veen, C., Fisher, R. E., Lowry, D., Nisbet, E. G., and Röckmann, T.: Street-level methane emissions of Bucharest, Romania and the dominance of urban wastewater, Atmospheric Environment: X, 13, 100153, https://doi.org/10.1016/j.aeaoa.2022.100153, 2022.
Giunta, T., Young, E. D., Warr, O., Kohl, I., Ash, J. L., Martini, A., Mundle, S. O. C., Rumble, D., Pérez-Rodríguez, I., Wasley, M., LaRowe, D. E., Gilbert, A., and Sherwood Lollar, B.: Methane sources and sinks in continental sedimentary systems: New insights from paired clumped isotopologues 13CH3D and 12CH2D2, Geochim. Cosmochim. Ac., 245, 327–351, https://doi.org/10.1016/j.gca.2018.10.030, 2019.
Gonfiantini, R.: Standards for stable isotope measurements in natural compounds, Nature, 271, 534–536, https://doi.org/10.1038/271534a0, 1978.
Haghnegahdar, M. A., Schauble, E. A., and Young, E. D.: A model for 12CH2D2 and 13CH3D as complementary tracers for the budget of atmospheric CH4, Global Biogeochem. Cy., 31, 1387–1407, https://doi.org/10.1002/2017GB005655, 2017.
Haghnegahdar, M. A., Sun, J., Hultquist, N., Hamovit, N. D., Kitchen, N., Eiler, J., Ono, S., Yarwood, S. A., Kaufman, A. J., Dickerson, R. R., Bouyon, A., Magen, C., and Farquhar, J.: Tracing sources of atmospheric methane using clumped isotopes, P. Natl. Acad. Sci. USA, 120, e2305574120, https://doi.org/10.1073/pnas.2305574120, 2023.
IPCC: Climate Change 2022: Impacts, Adaptation, and Vulnerability. Contribution of Working Group II to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change, edited by: Pörtner, H.-O., Roberts, D. C., Tignor, M., Poloczanska, E. S., Mintenbeck, K., Alegría, A., Craig, M., Langsdorf, S., Löschke, S., Möller, V., Okem, A., and Rama, B., Cambridge University Press, Cambridge University Press, Cambridge, UK and New York, NY, USA, 3056 pp., https://doi.org/10.1017/9781009325844, 2022.
Kelly, B. F. J., Lu, X., Harris, S. J., Neininger, B. G., Hacker, J. M., Schwietzke, S., Fisher, R. E., France, J. L., Nisbet, E. G., Lowry, D., van der Veen, C., Menoud, M., and Röckmann, T.: Atmospheric methane isotopes identify inventory knowledge gaps in the Surat Basin, Australia, coal seam gas and agricultural regions, Atmos. Chem. Phys., 22, 15527–15558, https://doi.org/10.5194/acp-22-15527-2022, 2022.
Khalil, M. A. K., Shearer, M. J., and Rasmussen, R. A.: Methane Sinks Distribution, Atmospheric Methane: Sources, Sinks, and Role in Global Change, Springer Berlin Heidelberg, 168–179, ISBN: 978-3-642-84605-2, 1993.
Lan, X., Thoning, K. W., and Dlugokencky, E. J.: Trends in globally-averaged CH4, N2O, and SF6, NOAA Global Monitoring Laboratory measurements, Version 2024-04, https://doi.org/10.15138/P8XG-AA10, 2022.
Li, Q., Fernandez, R. P., Hossaini, R., Iglesias-Suarez, F., Cuevas, C. A., Apel, E. C., Kinnison, D. E., Lamarque, J.-F., and Saiz-Lopez, A.: Reactive halogens increase the global methane lifetime and radiative forcing in the 21st century, Nat. Commun., 13, 2768, https://doi.org/10.1038/s41467-022-30456-8, 2022.
Loyd, S. J., Sample, J., Tripati, R. E., Defliese, W. F., Brooks, K., Hovland, M., Torres, M., Marlow, J., Hancock, L. G., Martin, R., Lyons, T., and Tripati, A. E.: Methane seep carbonates yield clumped isotope signatures out of equilibrium with formation temperatures, Nat. Commun., 7, 12274, https://doi.org/10.1038/ncomms12274, 2016.
Lu, X., Harris, S. J., Fisher, R. E., France, J. L., Nisbet, E. G., Lowry, D., Röckmann, T., van der Veen, C., Menoud, M., Schwietzke, S., and Kelly, B. F. J.: Isotopic signatures of major methane sources in the coal seam gas fields and adjacent agricultural districts, Queensland, Australia, Atmos. Chem. Phys., 21, 10527–10555, https://doi.org/10.5194/acp-21-10527-2021, 2021.
Menoud, M., van der Veen, C., Scheeren, B., Chen, H., Szénási, B., Morales, R. P., Pison, I., Bousquet, P., Brunner, D., and Röckmann, T.: Characterisation of methane sources in Lutjewad, The Netherlands, using quasi-continuous isotopic composition measurements, Tellus B, 72, 1–20, https://doi.org/10.1080/16000889.2020.1823733, 2020.
Menoud, M., van der Veen, C., Necki, J., Bartyzel, J., Szénási, B., Stanisavljević, M., Pison, I., Bousquet, P., and Röckmann, T.: Methane (CH4) sources in Krakow, Poland: insights from isotope analysis, Atmos. Chem. Phys., 21, 13167–13185, https://doi.org/10.5194/acp-21-13167-2021, 2021.
Menoud, M., van der Veen, C., Lowry, D., Fernandez, J. M., Bakkaloglu, S., France, J. L., Fisher, R. E., Maazallahi, H., Stanisavljević, M., Nȩcki, J., Vinkovic, K., Łakomiec, P., Rinne, J., Korbeń, P., Schmidt, M., Defratyka, S., Yver-Kwok, C., Andersen, T., Chen, H., and Röckmann, T.: New contributions of measurements in Europe to the global inventory of the stable isotopic composition of methane, Earth Syst. Sci. Data, 14, 4365–4386, https://doi.org/10.5194/essd-14-4365-2022, 2022.
Ono, S., Rhim, J. H., Gruen, D. S., Taubner, H., Kölling, M., and Wegener, G.: Clumped isotopologue fractionation by microbial cultures performing the anaerobic oxidation of methane, Geochim. Cosmochim. Ac., 293, 70–85, https://doi.org/10.1016/j.gca.2020.10.015, 2021.
Röckmann, T., Eyer, S., van der Veen, C., Popa, M. E., Tuzson, B., Monteil, G., Houweling, S., Harris, E., Brunner, D., Fischer, H., Zazzeri, G., Lowry, D., Nisbet, E. G., Brand, W. A., Necki, J. M., Emmenegger, L., and Mohn, J.: In situ observations of the isotopic composition of methane at the Cabauw tall tower site, Atmos. Chem. Phys., 16, 10469–10487, https://doi.org/10.5194/acp-16-10469-2016, 2016a.
Röckmann, T., Popa, M. E., Krol, M. C., and Hofmann, M. E. G.: Statistical clumped isotope signatures, Sci. Rep.-UK, 6, 31947, https://doi.org/10.1038/srep31947, 2016b.
Saueressig, G., Crowley, J. N., Bergamaschi, P., Brühl, C., Brenninkmeijer, C. A. M., and Fischer, H.: Carbon 13 and D kinetic isotope effects in the reactions of CH4 with O(1D) and OH: New laboratory measurements and their implications for the isotopic composition of stratospheric methane, J. Geophys. Res.-Atmos., 106, 23127–23138, https://doi.org/10.1029/2000JD000120, 2001.
Sherwood, O. A., Schwietzke, S., Arling, V. A., and Etiope, G.: Global Inventory of Gas Geochemistry Data from Fossil Fuel, Microbial and Burning Sources, version 2017, Earth Syst. Sci. Data, 9, 639–656, https://doi.org/10.5194/essd-9-639-2017, 2017.
Sherwood Lollar, B., Lacrampe-Couloume, G., Slater, G. F., Ward, J., Moser, D. P., Gihring, T. M., Lin, L. H., and Onstott, T. C.: Unravelling abiogenic and biogenic sources of methane in the Earth's deep subsurface, Chem. Geol., 226, 328–339, https://doi.org/10.1016/j.chemgeo.2005.09.027, 2006.
Sivan, M.: Extraction, purification, and clumped isotope analysis of methane (Δ13CDH3 and Δ12CD2H2) from different sources and the atmosphere, Version v1, Zenodo [data set], https://doi.org/10.5281/zenodo.8269713, 2023.
Stolper, D. A., Sessions, A. L., Ferreira, A. A., Santos Neto, E. V., Schimmelmann, A., Shusta, S. S., Valentine, D. L., and Eiler, J. M.: Combined 13C–D and D–D clumping in methane: Methods and preliminary results, Geochim. Cosmochim. Ac., 126, 169–191, https://doi.org/10.1016/j.gca.2013.10.045, 2014.
Stolper, D. A., Lawson, M., Formolo, M. J., Davis, C. L., Douglas, P. M. J., and Eiler, J. M.: The utility of methane clumped isotopes to constrain the origins of methane in natural gas accumulations, Geological Society, London, Special Publications, 468, 23–52, https://doi.org/10.1144/SP468.3, 2018.
Topp, E. and Pattey, E.: Soils as sources and sinks for atmospheric methane, Can. J. Soil Sci., 77, 167–177, https://doi.org/10.4141/s96-107, 1997.
Wang, D. T., Gruen, D. S., Lollar, B. S., Hinrichs, K. U., Stewart, L. C., Holden, J. F., Hristov, A. N., Pohlman, J. W., Morrill, P. L., Könneke, M., Delwiche, K. B., Reeves, E. P., Sutcliffe, C. N., Ritter, D. J., Seewald, J. S., McIntosh, J. C., Hemond, H. F., Kubo, M. D., Cardace, D., Hoehler, T. M., and Ono, S.: Methane cycling. Nonequilibrium clumped isotope signals in microbial methane, Science, 348, 428–431, https://doi.org/10.1126/science.aaa4326, 2015.
Whitehill, A. R., Joelsson, L. M. T., Schmidt, J. A., Wang, D. T., Johnson, M. S., and Ono, S.: Clumped isotope effects during OH and Cl oxidation of methane, Geochim. Cosmochim. Ac., 196, 307–325, https://doi.org/10.1016/j.gca.2016.09.012, 2017.
Whiticar, M. and Schaefer, H.: Constraining past global tropospheric methane budgets with carbon and hydrogen isotope ratios in ice, Philos. T. Roy. Soc. A, 365, 1793–1828, https://doi.org/10.1098/rsta.2007.2048, 2007.
Whiticar, M. J.: Carbon and hydrogen isotope systematics of bacterial formation and oxidation of methane, Chem. Geol., 161, 291–314, https://doi.org/10.1016/S0009-2541(99)00092-3, 1999.
Whiticar, M. J., Faber, E., and Schoell, M.: Biogenic methane formation in marine and freshwater environments: CO2 reduction vs. acetate fermentation – Isotope evidence, Geochim. Cosmochim. Ac., 50, 693–709, https://doi.org/10.1016/0016-7037(86)90346-7, 1986.
Yeung, L. Y.: Combinatorial effects on clumped isotopes and their significance in biogeochemistry, Geochim. Cosmochim. Ac., 172, 22–38, https://doi.org/10.1016/j.gca.2015.09.020, 2016.
Young, E. D., Kohl, I. E., Sherwood Lollar, B., Etiope, G., Rumble, D., Li, S., Haghnegahdar, M. A., Schauble, E. A., McCain, K. A., Foustoukos, D. I., Sutclife, C., Warr, O., Ballentine, C. J., Onstott, T. C., Hosgormez, H., Neubeck, A., Marques, J. M., Pérez-Rodríguez, I., Rowe, A. R., LaRowe, D. E., Magnabosco, C., Yeung, L. Y., Ash, J. L., and Bryndzia, L. T.: The relative abundances of resolved 12CH2D2 and 13CH3D and mechanisms controlling isotopic bond ordering in abiotic and biotic methane gases, Geochim. Cosmochim. Ac., 203, 235–264, https://doi.org/10.1016/j.gca.2016.12.041, 2017.
Vienna Peedee Belemnite; Vienna Standard Mean Ocean Water