the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 22 May 2024
| 22 May 2024
Quality assurance and quality control of atmospheric organosulfates measured using hydrophilic interaction liquid chromatography (HILIC)
Ping Liu
Bo-Xuan Li
Yu-Qing Zhang
Daniel J. Bryant
Xin-Ming Wang
As a crucial constituent of fine particulate matter (PM2.5), secondary organic aerosols (SOAs) influence public health, regional air quality, and global climate patterns. This paper highlights the use of hydrophilic interaction liquid chromatography (HILIC) which effectively retains strongly polar analytes that might exhibit incomplete or no retention in reverse chromatography, resulting in superior separation efficiency.
A HILIC column was used to analyze six standards, environmental standards (1648a and 1649b), and samples collected in urban environments in Guangzhou in the Pearl River Delta region, which serve as valuable reference points for evaluating the organic composition of the atmospheric environment. The results indicate a high degree of accuracy in the analytical method. Sodium octyl-d17 sulfate serves as the internal standard, with a linear correlation coefficient of the six standards, boasting a linear correlation coefficient r ranging from 0.993–0.9991 and a slope, k, of the linear equation from 0.966–1.882. The instrument detection limits (IDLs) are established at 0.03–0.20 µg mL−1, while the method detection limits (MDLs) fall within the range of 0.30–1.75 ng m−3, demonstrating the method's exceptional sensitivity.
Since isoprene-derived organosulfates (iOSs) are highly polar due to containing a hydrophilic bond to the hydroxyl group and a hydrophobic bond to the sulfate, and as such showed strong retention using this method, this technique employs sodium ethyl sulfate and sodium octyl sulfate standards for semi-quantitative compound analysis of iOSs. The error in sample analysis (EA) ranged from 12.25 %–95.26 %, and the two standards maintained a consistent recovery rate between 116 %–131 % and 86.4 %–127 %. These findings indicate a high level of precision when semi-quantifying compounds with similar structural characteristics, affirming the analysis method's minimal relative error and underscoring its repeatability, process stability, and the reliability of its results for iOSs. To enhance the method's reliability assessment, the study analyzed polar organic components of standard particulate matter samples (1648a and 1649b), providing precise determinations of several iOSs using this method. Methyltetrol sulfate ( 215, ) is the highest concentration in the ambient samples, up to 67.3 ng m−3 in the daytime. These results serve as valuable reference points for assessing the organic composition of the atmospheric environment.
- Article
(1706 KB) - Full-text XML
- BibTeX
- EndNote





Organosulfates (OSs) represent a category of organic compounds featuring the sulfate functional group (R-OSO3H) found ubiquitously in atmospheric aerosols. OSs contribute to 5 %–30 % of the organic mass fraction within particulate matter (Shakya and Peltier, 2013, 2015; Tolocka and Turpin, 2012; Surratt et al., 2008; Lukacs et al., 2009). Their unique hydrophilic and hydrophobic characteristics influence the hygroscopicity and cloud condensation nuclei (CCN) formation potential of aerosol particles (Hansen et al., 2015), underscoring the need for a comprehensive investigation into their chemical compositions and formation mechanisms in the atmosphere. OSs are formed from the oxidation of anthropogenic precursors, such as benzene and toluene, and biogenic volatile organic compounds (VOCs), such as isoprene, monoterpenes (primarily α-pinene, β-pinene, and limonene), sesquiterpenes, aromatics, aldehydes, and others, under a variety of oxidation and sulfuric acid conditions (Surratt et al., 2008, 2010). Isoprene is the most abundant precursor of global secondary organic aerosols (SOAs) (Bates and Jacob, 2019; Hodzic et al., 2016). The epoxide pathway plays a critical role in isoprene SOA (iSOA) formation, in which isoprene epoxydiols (IEPOX) and/or hydroxymethyl-methyl-α-lactone (HMML) can react with nucleophilic sulfate, producing isoprene-derived organosulfates (iOSs) (Surratt et al., 2010; Lin et al., 2013; He et al., 2018).
Previous research has employed reversed-phase liquid chromatography (RPLC) for the analysis of aqueous atmospheric samples encompassing water-soluble and methanol-extractable aerosol constituents, as well as fog water (Bryant et al., 2020, 2021). This reversed-phase approach, utilizing a non-polar stationary phase and a polar mobile phase, effectively retains higher-molecular-weight OSs derived from monoterpenes (e.g., ) (Gao et al., 2006; Surratt et al., 2007b) and aromatic OSs (e.g., ) (Kundu et al., 2010; Staudt et al., 2014). However, it is less efficient for the separation of lower-molecular-weight and highly polar OSs, which elute in less than 2.5 min and co-elute with various other OSs, small organic acids, polyols, and inorganic sulfates (Stone et al., 2012). The co-elution of so many analytes leads to matrix effects, reducing the analyte's signal (Bryant et al., 2020, 2021, 2023b, a). The iOSs are hydrophilic compounds owing to their hydroxyl functional groups, and the iOSs are ionic polar compounds. Hence, an alternative approach for the iOS characterization that could accomplish simultaneous analysis of polar and water-soluble components while avoiding the drawbacks associated with current analytical methods would be highly desirable.
To address this challenge, a hydrophilic interaction liquid chromatography (HILIC) featuring an amide stationary phase has been utilized (Hettiyadura et al., 2015, 2017; Cui et al., 2018). HILIC is purposefully designed to retain molecules with ionic and polar functional groups and has demonstrated effectiveness in retaining carboxylic-acid-containing OSs like glycolic acid sulfate and lactic acid sulfate, which are among the most prevalent atmospheric OSs quantified to date (Olson et al., 2011; Hettiyadura et al., 2015, 2017; Cui et al., 2018). Since these OS compounds are easily ionized in negative mode, they can be efficiently detected in negative electrospray ionization ((−) ESI) mode (Romero and Oehme, 2005; Surratt et al., 2007a). In this experiment, a combination of HILIC chromatographic separation and tandem mass spectrometry (MS/MS) was employed to separate and detect highly polar OSs relevant to the atmosphere. A mixed standard of OSs facilitated the separation, identification, and quantification of polar, ionic, and non-volatile OSs present in the atmosphere. The HILIC separation was accomplished using an ethylene-bridged hybrid (BEH) amide column, and OSs were semi-quantified based on the calibration curve derived from alternative standards through triple-quadrupole mass spectrometry detection (TQD). This approach enabled the detection and quantification of OSs originating from isoprene within the atmosphere of the Pearl River Delta.
Recent studies have identified hundreds of OSs in the ambient environment (Iinuma et al., 2007; Surratt et al., 2008; Riva et al., 2016; Brüggemann et al., 2017, 2019; Le Breton et al., 2018; Hettiyadura et al., 2019). However, authentic standards for OSs remain scarce, with only a few commercially available or synthesized in laboratories (Staudt et al., 2014; Hettiyadura et al., 2015; Huang et al., 2018). The utilization of different surrogate standards results in considerable discrepancies in quantifying OS concentrations (Zhang et al., 2022; He et al., 2018; Surratt et al., 2008), signifying the persisting challenge of accurate quantification in OS studies. HILIC chromatography is a promising analytical technique for the separation of OSs from one another and the complex aerosol matrix. When coupled with authentic standard development and highly sensitive MS/MS detection, it offers an improved method for quantifying and speciating atmospheric OSs. Enhanced measurements of this compound class will contribute to a better understanding of SOA precursors and their formation mechanisms.
2.1 Field sampling
Sampling was undertaken during October 2018 in Guangzhou. Guangzhou is situated in the Pearl River Delta region of southern China which has large-scale land coverage of broadleaf evergreen trees, high temperatures, and strong solar radiation all year round.
Field sampling was conducted using a PM2.5 sampler (Tisch Environmental Inc., Ohio, USA) equipped with quartz filters (Whatman, 17.6 cm × 23.4 cm) at a flow rate of 1.13 m3 min−1. Additionally, field blanks were collected at a monthly interval. Blank filters were covered with aluminum foil and baked at 500 °C for 24 h to remove organic material, and pre- and post-sampling flow rates were measured with a calibrated rotameter. All filters were handled using clean techniques, which included storage of filters in plastic Petri dishes lined with pre-cleaned aluminum foil and manipulation with pre-cleaned stainless-steel forceps. Post-sampling, filters were stored frozen in the dark. One field blank was collected for every five samples and stored in a container with silica gel. After sampling, the filter samples were stored at −20 °C.
2.2 PM sample extraction and preparation
Following the procedure outlined by Hettiyadura et al. (2015), an 82 mm diameter circular section was excised from the quartz membrane using a cutter. This section was subsequently cut into small pieces with forceps that had been cleaned with acetonitrile (ACN). The samples were then carefully placed into a clean 100 mL beaker. To this, 300 µL of a solution with ACN and ultrapure water (95:5, by volume) containing sodium octyl-d17 sulfate at a concentration of 5.3 µg mL−1 was introduced as an internal standard. Subsequently, 15 mL of ACN of chromatographic purity and ultrapure water (95:5, by volume) was added in three separate increments, with the beaker covered with aluminum foil to prevent the organic solvent from evaporating, and was extracted by ultra-sonication extraction in an ice-water bath for 20 min. The resulting solution was then filtered through a polypropylene membrane syringe filter (0.45 µm; 25 pp, Sigma-Aldrich), and the process was repeated three times to consolidate the solution. The solution was then concentrated to an approximate volume of 5 mL using a rotary evaporator and transferred to 1.5 mL vials, and the solvent was blown to dryness using a micro-scale nitrogen evaporation system at 35 °C under a high-purity nitrogen stream. Extracts were then re-constituted with ACN and ultrapure water (95:5, by volume) to a final volume of 300 µL. The solution was thoroughly mixed and then stored in a freezer at −20 °C for subsequent analysis.
2.3 Instrumentation and reagents
OS sample analysis was performed using ultra-performance liquid chromatography electrospray triple-quadrupole tandem mass spectrometry (UPLC/ESI-TQD-MS/MS; Agilent 6400, USA) with a BEH amide column (2.1 mm × 100 mm, 1.7 µm; ACQUITY UPLC, Waters) in full-scan mode. The column temperature was held at 35 °C, and the mobile-phase flow rate was 0.5 mL min−1. The injection volume of samples and standards was 5 µL. Mobile phase A (organic phase) was ACN and water (95:5, by volume) buffered with an ammonium acetate buffer (10 mm, pH 9), and mobile phase B (aqueous phase) was 100 % water buffered with an ammonium acetate buffer (10 mm, pH 9). The software MassHunter (version B.02) was used to acquire and process all data.
The purchased standards were sodium methyl sulfate (98 %, Sigma-Aldrich), sodium ethyl sulfate (> 98 %, Sigma-Aldrich), sodium octyl sulfate (99 %, Alfa Aesar), sodium dodecyl sulfate (99.0 %, Sigma-Aldrich), sodium hexadecyl sulfate (99 %, Alfa Aesar), sodium octadecyl sulfate (99 %, Alfa Aesar), sodium octyl-d17 sulfate (99.1 %, CDN), chromatographic pure acetonitrile, (ACN, 99.9 %, CNW), ammonium acetate (99.0 %, CNW), and ammonia (20 %–22 %, CNW).
2.4 Separation and detection of OSs
2.4.1 Separation
The separation was optimized using a gradient elution method. Mobile phase A remained at 100 % from 0 to 2 min, after which it decreased to 85 % from 2 to 4 min and remained constant at 85 % until 11 min. To re-equilibrate the column before the next injection, mobile phase A was reinstated to 100 % between 11 and 11.5 min, and this composition was maintained until 20 min. The cleaning needle solvent employed a mixture of acetonitrile and ultrapure water (in a volume ratio of 80:20).
2.4.2 Detection
In the negative-ion mode, the identification of OSs was achieved via TQD-MS, specifically utilizing an ACQUITY system as the mass spectrometer (Waters, USA). The detector operated in full-scan mode, with the first quadrupole selecting deprotonated molecules, the second quadrupole identifying fragments, and the third quadrupole analyzing product ions.
2.4.3 Optimization of experimental conditions
The choice of the fragmentation voltage directly impacts the instrument's ability to target specific compounds, while the collision energy plays a crucial role in determining the extent of fragmentation and the response of secondary fragment ions. To illustrate, when analyzing the most common compounds in the sample, and without connecting the chromatographic separation column, a 5 µL aliquot of the environmental sample was injected every 0.7 min. In this production-scanning mode, the target ions generated after ionization in the ion source were detected. The first fragmentation voltage was set to 80 V, and, with each subsequent scan, the voltage was incrementally increased by 5 V until it reached 180 V. The analysis revealed that the optimal response was achieved at 135 V. Consequently, 135 V was selected as the optimal fragmentation voltage for quantitative analysis of the actual samples.
Table 1Optimal fragmentation voltage and collision energy of different standards.
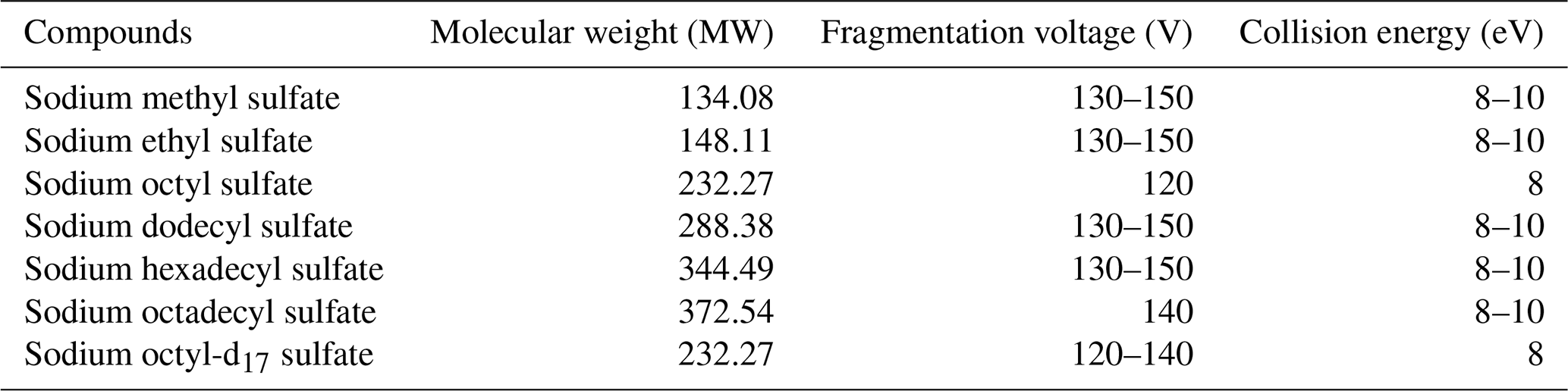
For compounds with intricate chemical structures, further analysis was carried out using MS/MS. Similarly, an energy level of 8 eV was employed in the collision cell during the OS daughter ion scanning. Table 1 displays the optimal fragmentation voltage and collision energy for different standards.
The determination of other optimal conditions for the ESI source followed a similar methodology, as presented in Table 2, including a capillary voltage of 2700 V, source temperature of 150 °C, sheath gas temperature of 400 °C, source gas (N2) flow rate at 1.7 L min−1, and sheath gas (N2) flow rate at 12 L min−1.

3.1 Comparison of this method and the reversed-phase method
3.1.1 Comparison of OS standards
In this experiment, six OS standards were analyzed. Table 3 compares the retention times and peak areas of pure and mixing standards. The results indicate that the retention times for all standards remained unchanged. Furthermore, there was no co-elution observed between the pure standards and mixing standards of small-molecular-weight iOSs, such as and . The peak area ratios of pure standards to mixing standards were 1.00 and 0.96, respectively. However, co-elution exists for the long-chain alkane OSs (, , and ), with peak area ratios of 0.57, 0.60, and 0.67, respectively. The mixing standards reduced the signal by almost half, possibly due to a retention time of approximately 0.5 min, falling within the column deadtime.
Table 3Comparison of retention time and peak area in MS between pure standards and mixing standards.
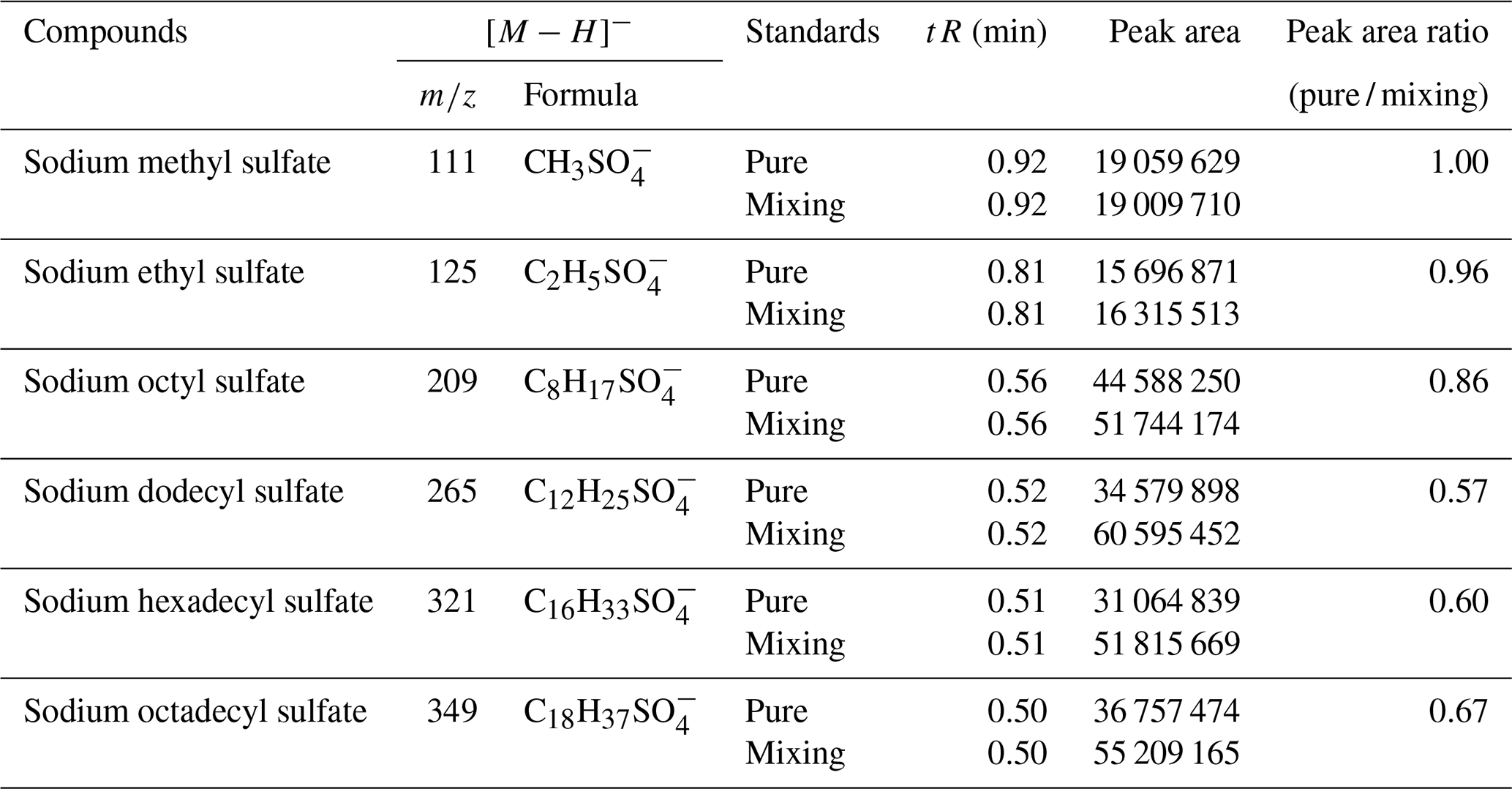
The ratio of the standards with a retention time of 0.8–1 min is close to 1, showing that, even though some of the standards closely elute, this does not affect the instrument response, suggesting no matrix effect. However, the long-chain OSs, which elute in the dead volume, have a large matrix effect, meaning that the small amount of retention in this method is much better than the absence of retention in the reversed-phase method. This observation suggests that the analytical effectiveness of this method on iOSs with high polarity surpasses that of long-chain-alkane OSs.
3.1.2 Comparison of iOSs in ambient sample
The separation of typical OSs, such as ( 215) and ( 199), was notably enhanced using this method, as illustrated in Fig. 1, which compares the separation with the previous reversed-phase column. Specifically for ( 215), the separation of six peaks by this method is superior to reversed-phase chromatography, in which these IEPOX-derived OS isomers co-elute in two peaks (Stone et al., 2012). The resolution of isomers is significant because methyltetrol sulfates have generated the greatest OS signals in prior field studies (Froyd et al., 2010; Lin et al., 2013) and may prove useful in elucidating different OS formation pathways.

Figure 1Comparison of the effects of separation of 199 () and 215 () using the previous method and this work.
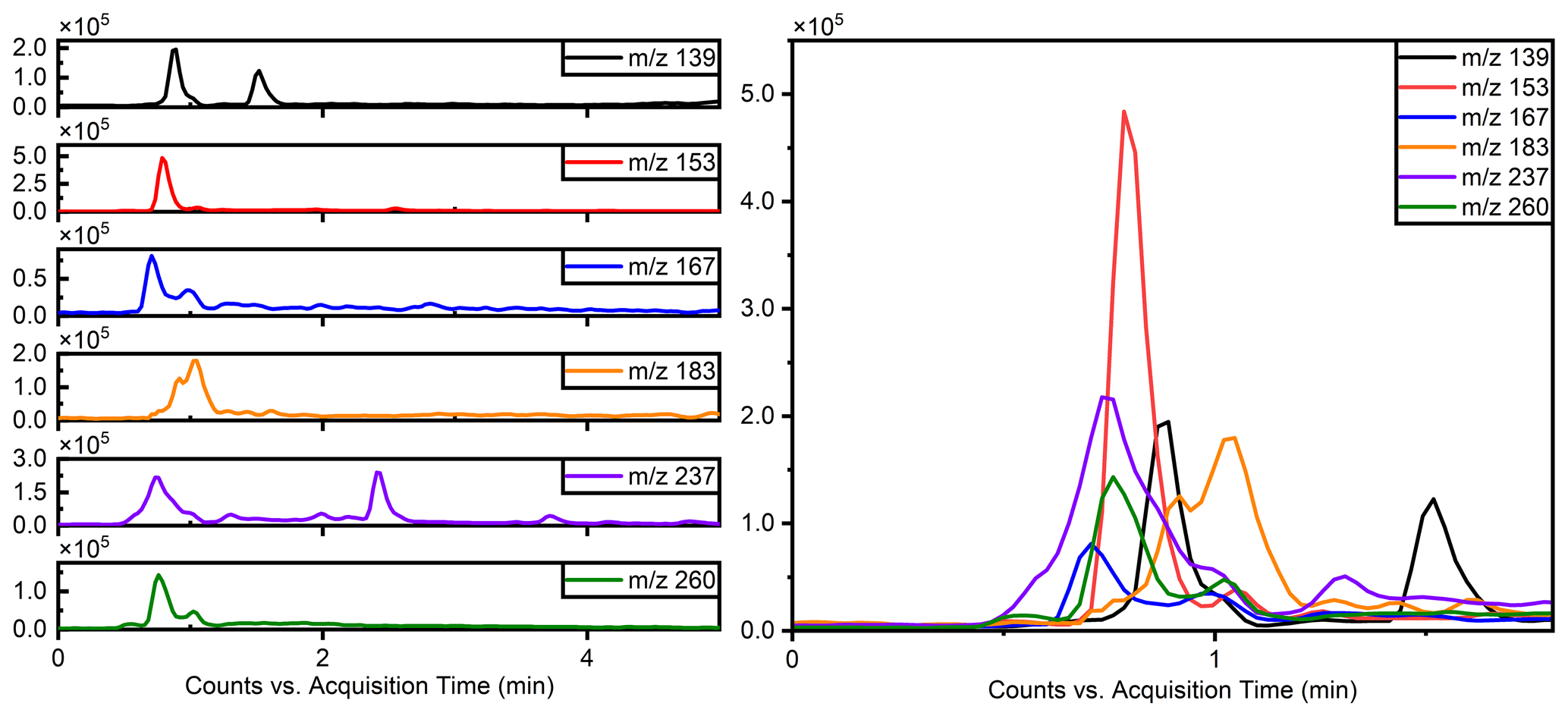
Due to co-eluting effects, the retention time for 139, 153, 155, 167, and 169 under the traditional method was 1.30 min (Stone et al., 2012). However, in employing the HILIC method, significant shifts in retention times were observed. Specifically, retention times for 139 were 0.83 and 1.58 min; were 0.79 and 0.82 min for 153; and were 10.48, 0.69, and 1.00 and 1.46 min for 155, 167, and 169, respectively. Additionally, Fig. 2 displays chromatograms of iOSs with retention times of less than 1 min – while some co-elution persists, their retention times do not precisely overlap. This observation underscores the method's potential for effectively separating lower-molecular-weight and highly polar OSs.
3.2 Linearity of the standard
In this experiment, the sodium octyl-d17 sulfate standard solution (300 µL; 5.3 µg mL−1) was an internal standard, and six commercially available OS standards were employed. Table 4 and Fig. 3 present the linearity for different standards. The standard curves of various compounds were evaluated for their correlation coefficients (r), resulting in values ranging from 0.993 to 0.9991, the resulting slope (k) ranging from 0.966–1.882, and the Pearson significance test (p) indicating values ≤ 0.002. Notably, the standard curve for sodium octyl sulfate ( 209, ) exhibited an r of 0.9991, with a k of 0.966, indicating that the semi-quantification of structurally similar compounds using sodium octyl sulfate as the standard was more precise when sodium octyl-d17 sulfate was used as the internal standard.
Table 4The linear of standards. k is the slope of correlation, r is the correlation coefficient, and p is the Pearson significance test.
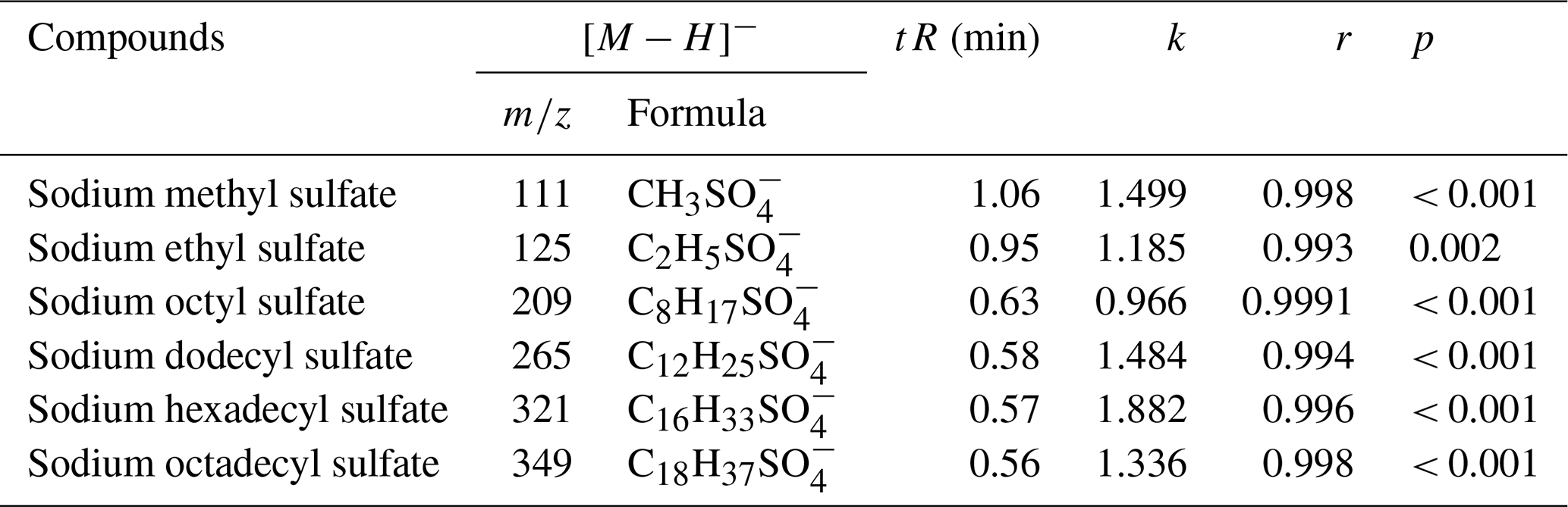

Figure 3Correlations between concentration ratios and area ratios of standards to the internal standard. r is the correlation coefficient.
3.3 UPLC/ESI–MS/MS instrument detection limits and method detection limits
To ensure the effectiveness of this method in monitoring the target compounds in field environmental samples, the standard deviation (SD) was computed by repeatedly injecting the standard sample with the lowest concentration five times in succession. The calculation used the standard curve of Fig. 3.
The instrumental detection limits (IDLs) were established at the 95 % confidence interval, calculated as 3 times SD divided by k. In this experiment, with a sampling volume of 271.2 m3 and considering the entire laboratory analysis process, the method detection limits (MDLs) for these compounds were determined, calculated following Eqs. (1) and (2):
where the area of a sampling filter (82 mm diameter) for OS analysis (S1) was 52.78 cm2 and the total area of a sampling filter (S2) was 411.84 cm2. The total air volume of 4 h sampling at a flow rate of 1.13 m3 min−1 (V0) was 271.2 m3; the solution volume in the vial for UPLC-MS analysis (V1) was 300 µL, which was same as the internal standard added; and the air volume responding to the filter analyzed (V2) was 34.76 m3.
Table 5IDLs: instrumental detection limits (µg mL−1). MDLs: method detection limits (ng m−3). M: sample concentration (µg mL−1), total sampling five times. SD: standard deviation.
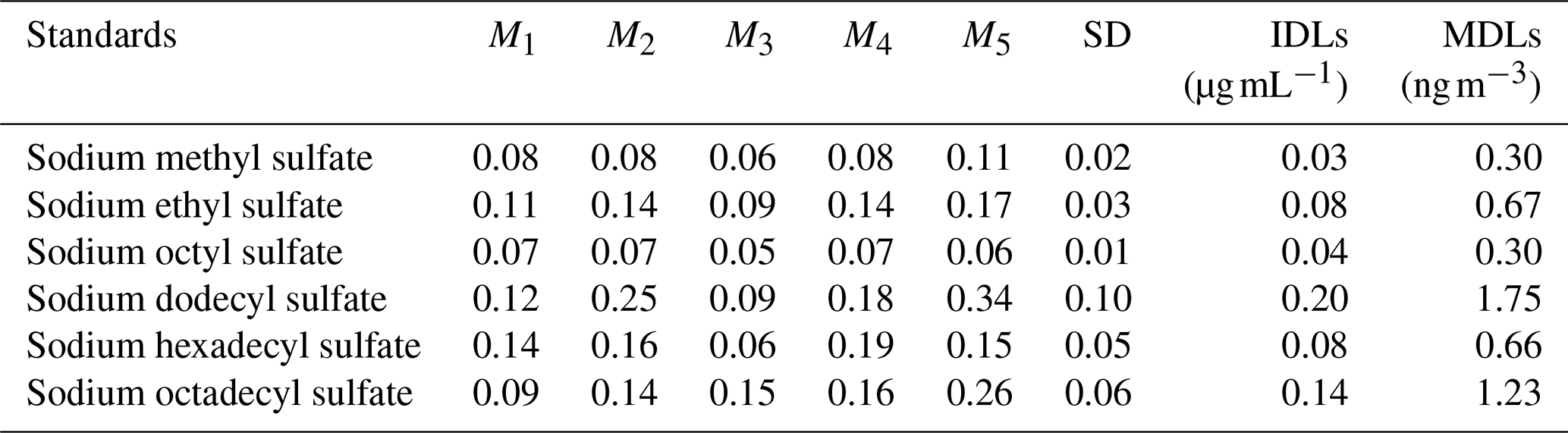
The MDLs of each standard are depicted in Table 5. Of the various standard samples analyzed, the compound with the highest method detection limit was sodium dodecyl sulfate, which measured 1.75 ng m−3. This finding underscores the method's remarkable sensitivity in detecting OSs in environmental aerosols, thereby affirming its effective detection capability.
3.4 Parallelism and recovery
In this experiment, a matrix spike experiment was conducted. Approximately 300 µL of a mixed solution, containing all the standards at a concentration of around 5 µg mL−1, was injected onto a 47 mm blank quartz membrane. This procedure was repeated in parallel five times, and a sample without the mixed solution served as a laboratory blank, adding up to a total of six sample groups for pretreatment analysis. The total quantity of each substance in the treated sample and the content of each substance in the untreated sample were computed, thereby enabling the calculation of the recovery rate for each compound. As demonstrated in Table 6, the recovery rates for various compounds fell within the range of 60.2 %–145 %. These high recovery rates indicate minimal loss of the target compounds during the analysis, which is favorable for accurate detection.
Table 6The recovery and RSD of standards. M: sample recovery (%).
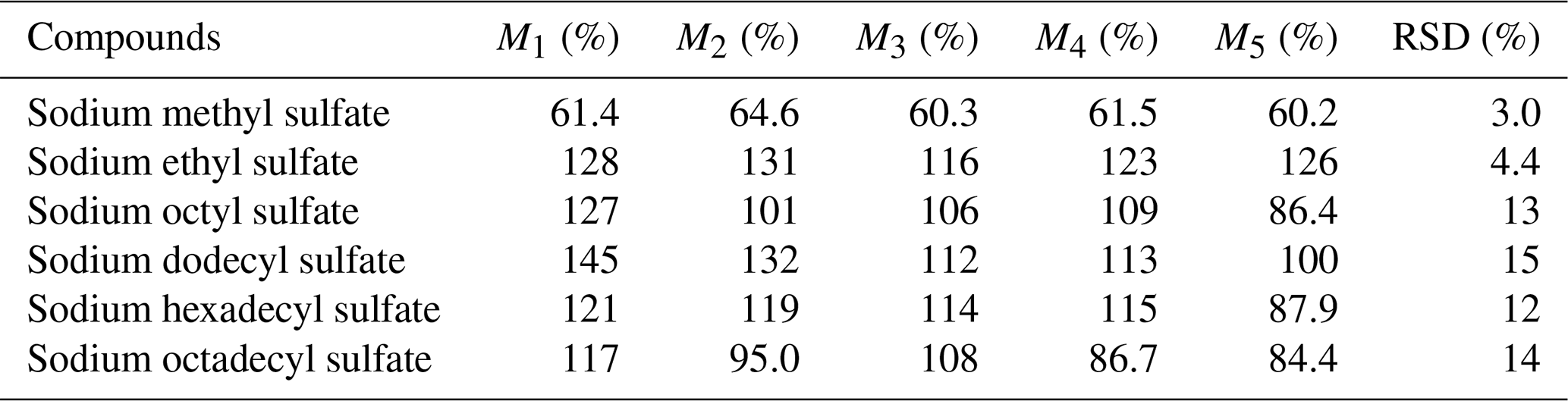
Moreover, it is noteworthy that the relative standard deviations (RSDs) for these standards did not surpass 15 %, underscoring the small relative error and highlighting the experiment's reproducibility. The RSDs of the small molecule were all less than 4.4 %, but the RSDs for long-chain-alkane OSs are all higher than 10 %, indicating that this experiment is favorable for the detection of iOSs. The stability of the analysis process ensures that the results obtained are reliable.
3.5 Empirical approach to estimate error in sample analysis
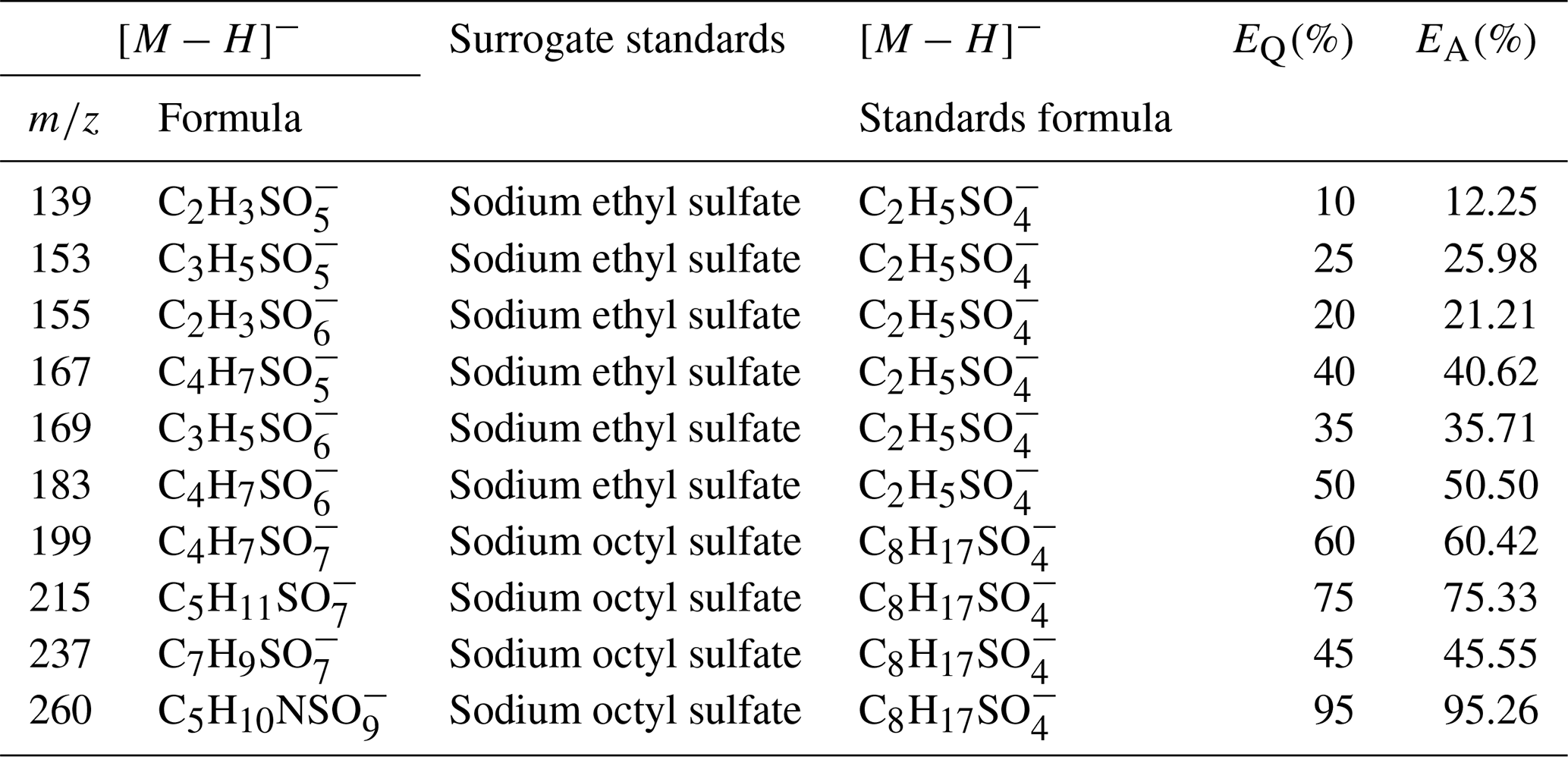
Stone et al. (2012) developed an empirical approach to estimate the error resulting from surrogate quantification (EQ) based on a homologous series of atmospherically relevant compounds. They estimated the relative error introduced by each carbon atom (En), oxygenated functional group (Ef), and alkenes (Ed) to be 15 %, 10 %, and 60 %, respectively. The errors introduced by surrogate quantification are considered additive and are calculated using the equations below. Furthermore, the error in sample analysis (EA) can be estimated through the error propagation of field blank (EFB), spike recovery (ER), relative differences (ED), and the surrogate quantification (EQ) calculated following Eq. (3). The error in sample analysis (EA) is calculated following Eq. (4):
where Δn represents the difference in the number of carbon atoms between a surrogate and an analyte, Δf is the difference in oxygen-containing functional groups between a surrogate and an analyte, and Δd is the difference in alkene functionality between a surrogate and an analyte. As shown in Table 7, the EQ ranged from 10 % to 95 % for the OSs when using sodium ethyl sulfate and sodium octyl sulfate as the surrogates. The EQ values were compared to the previous surrogate with camphorsulfonic acid, with 215 % and 230 % reduced to 75 % and 60 % for 215 and 199, respectively (Zhang et al., 2022), and the EA ranged from 12.25 %–95.26 % for these iOS products. For 215 and 199, the EA is 73.33 % and 60.42 %, respectively.
3.6 MS2 of iOSs
In this experiment, the semi-quantitative determination of iOSs was carried out using sodium octyl-d17 sulfate as the internal standard and using sodium ethyl sulfate and sodium octyl sulfate as the standards. Semi-quantitative analytical methods were employed to monitor the characteristic product ions of OSs (Stone et al., 2009), namely ( 97) and ( 96). MS2 was utilized as a means of identifying OSs and performing semi-quantitative analysis when actual standards were not available.
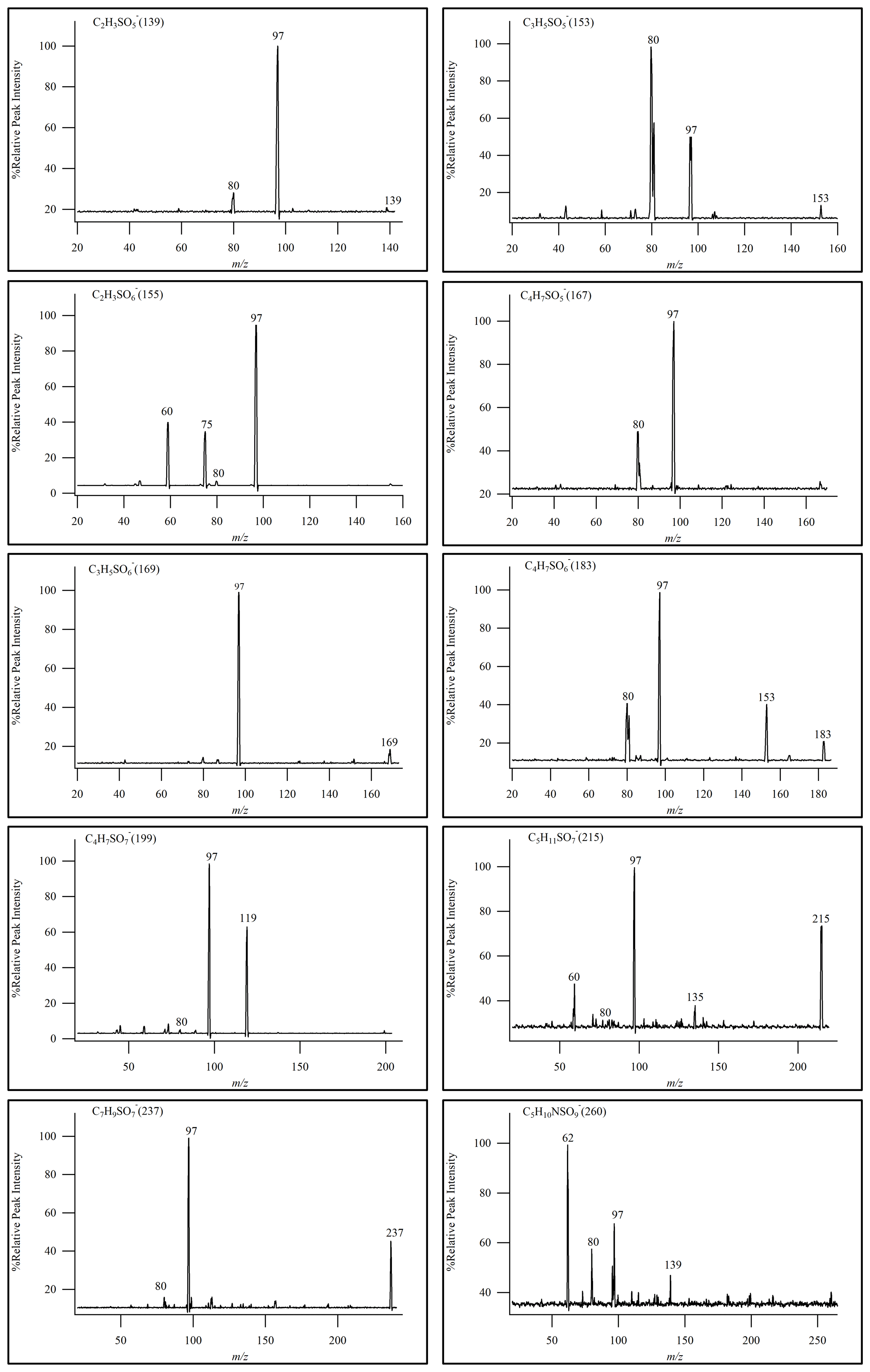
Given the wide array of polar compounds present in field samples and the substantial variations between samples, the final qualitative and quantitative analyses were carried out in full-scan mode. This approach ensured the most comprehensive component analysis results. By evaluating the relative signal intensity using HILIC–TQD, it was possible to identify certain OSs. As shown in Fig. 4, we identified a total of 10 OSs by daughter-ion-scanning mode. In Fig. 4, only one isomer's MS2 is listed for reference.
Table 8The compounds in 1648a. M: sample concentration (ng m−3).
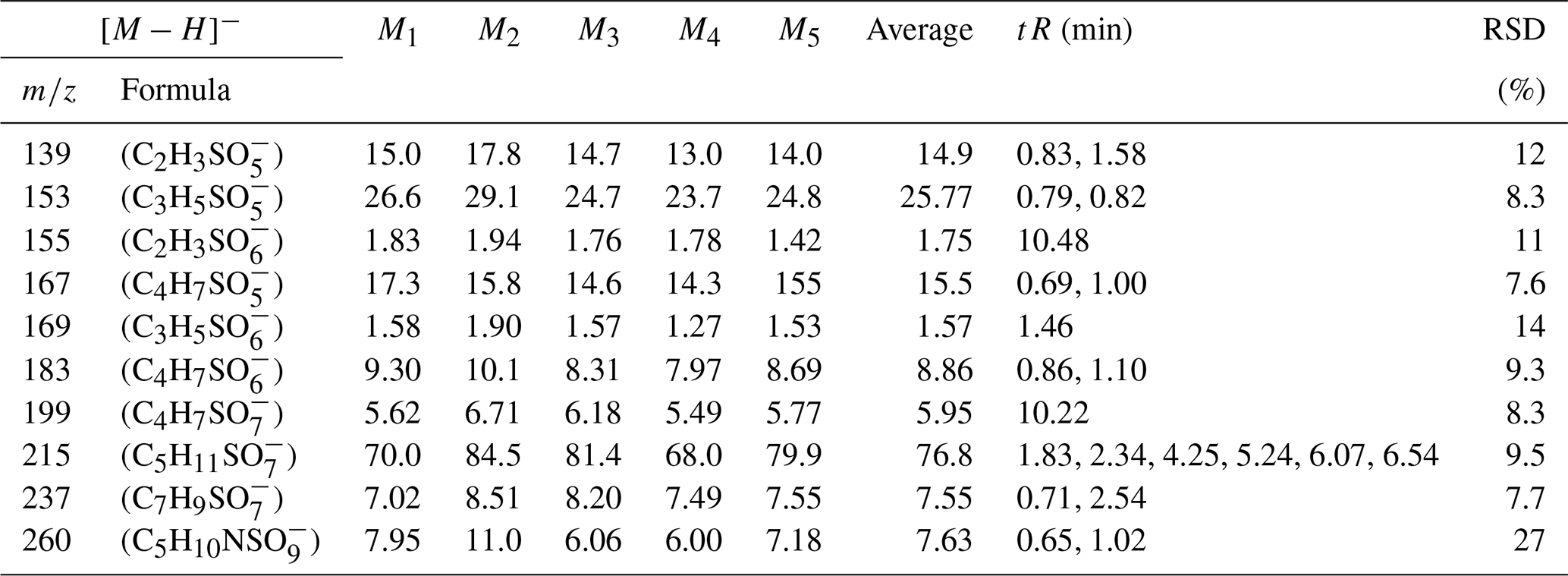
3.7 Measurement of environmental standards
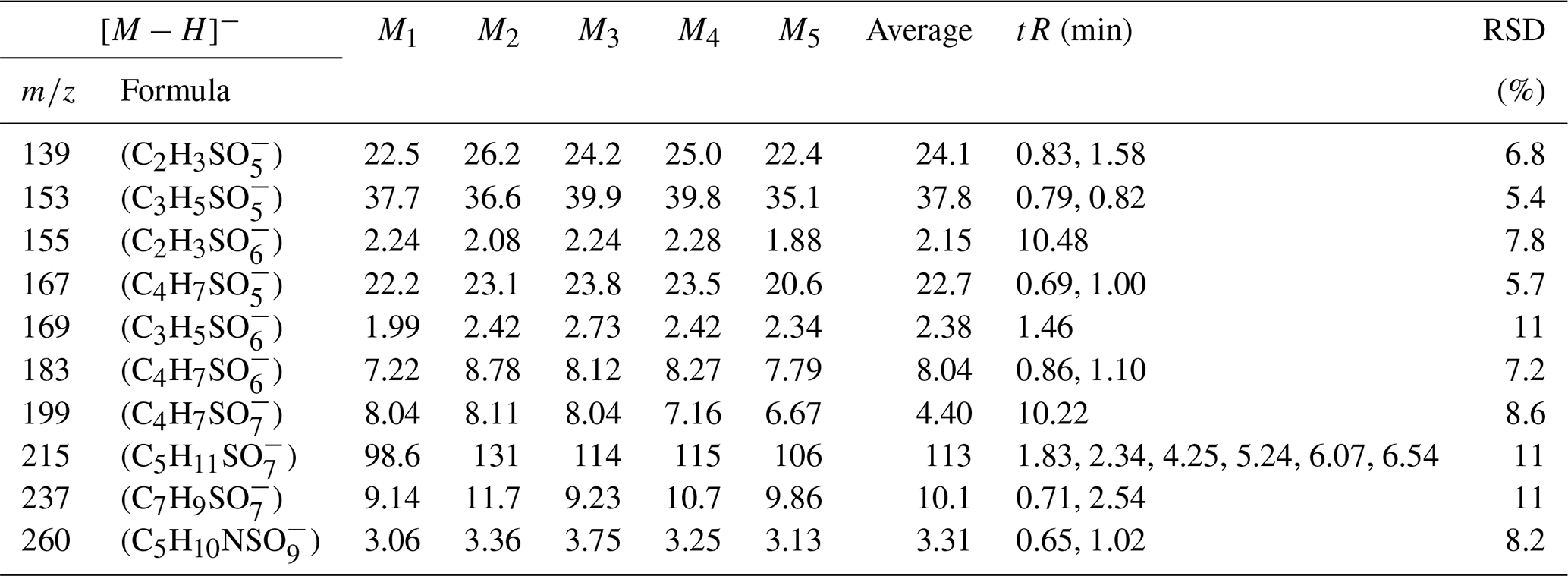
The relatively pristine nature of the standard mixture solution stands in stark contrast to the actual field ambient atmospheric aerosol samples, which are characterized by complex matrices that can significantly influence the analytical results. To comprehensively assess the reliability of this analytical method, we acquired standard particulate matter samples (NIST 1648a and 1649b). We proceeded to analyze the organic components within these samples and determine the content of environmental standard particle samples using the same method. As presented in Tables 8 and 9, the retention times for all iOSs are greater than the deadtime of the column, indicating that the method provides good retention and separation for highly polar iOSs and reveals that the RSD in the analysis of all compounds does not exceed 27 %. This level of deviation falls within the acceptable range for the analysis of organic compounds, affirming the method's suitability for field sample analysis. These results serve as valuable reference points for assessing the organic composition of the atmospheric environment.
Table 9The compounds in 1649b. M: sample concentration (ng m−3).
3.8 iOSs in ambient PM samples
Concentrations of iOSs quantified in ambient PM2.5 from Guangzhou in October 2018 during the daytime and nighttime are provided in Table 10. Methyltetrol sulfate ( 215, ) is the most prevalent OS known to date (Surratt et al., 2008; Hettiyadura et al., 2015). It is formed through a nucleophilic addition reaction involving an IEPOX ring, catalyzed by sulfuric acid (Surratt et al., 2010). ( 215) exhibited peak retention times of 1.83, 2.34, 4.25, 5.24, 6.07, and 6.54 min and was the most abundant OS measured. On 7 October during the daytime and 7–8 October during the nighttime, its concentrations were 67.3 ng m−3 and 57.9 ng m−3, respectively.
Table 10Ambient concentrations of iOSs measured in PM2.5 in Guangzhou, from 06:00–18:00 LT (UTC+8:00) on 7 October 2018 (daytime) and 18:00–06:00 LT on 7–8 October 2018 (nighttime).

The OS with the formula 260 () is a nitrooxy OS resulting from the photooxidation of isoprene under high-NOx conditions (Gomez-Gonzalez et al., 2008; Surratt et al., 2008). In the course of this experiment, two isomers with an 260 were discovered, with Hettiyadura and colleagues identifying two such isomers in 2019 (Hettiyadura et al., 2019) and Centreville identifying four isomers with 260 (Surratt et al., 2008). 260 exhibits a moderate correlation with methyltetrol sulfate, hinting at isoprene as a likely precursor (Hettiyadura et al., 2019). In this experiment, the concentration of 260 was significantly higher at night than during the day, at 17.5 and 10.2 ng m−3, respectively. Further subsequent experiments could explore the reasons for this diurnal difference in terms of the mechanism of formation of 260.
OS with the formula ( 199, calculated mass: 198.9912) is an oxidation product of isoprene under high-NOx conditions. In this method, the retention time for the peak is 10.22 min, and the concentration of 199 was significantly higher at night than during the day, at 18.1 and 12.5 ng m−3, respectively, suggesting that nighttime chemistry is more conducive to the formation of 199.
In summary, these findings strongly suggest that isoprene serves as the primary and most abundant precursor to OSs. Hettiyadura et al. (2019) demonstrated that during the Atlanta summer, over half of the organic aerosol compounds derived from isoprene are composed of OSs, with methyltetrol sulfate being the predominant constituent. Subsequent experiments can further explore the different formation mechanisms of these iOSs and the reasons for the variations in different isomers.
OSs are a vital component of SOAs. Previously, their measurement using reversed-phase liquid chromatography presented challenges due to a lack of retention and subsequent co-elution with other organic sulfates, small organic acids, polyols, and inorganic ions, resulting in poor separation and matrix effects. In this experiment, we employed HILIC to analyze OSs in the atmospheric environment. HILIC effectively resolved this issue by delaying the elution time of molecules with ionic and polar functional groups, particularly OSs containing carboxyl groups. HILIC retained strongly polar samples that had incomplete or no retention in C18 reverse chromatography, offering a solution to the co-elution problem of OSs with other small compounds in C18 reverse columns, resulting in a robust separation. Specifically for ( 215), the separation of six peaks by this method is superior to reversed-phase chromatography, in which these IEPOX-derived OS isomers co-elute in two peaks.
During this experiment, we conducted iOSs in the atmospheric environment of the Pearl River Delta using HILIC, and our analytical method possessed high sensitivity, enabling effective detection of OSs in environmental aerosols. Each standard exhibited RSD controlled within 15 %, indicating minimal relative errors, high experimental reproducibility, stable analysis procedures, and reliable results. We also simultaneously analyzed two environmental reference standards (NIST 1648a and 1649b), providing some reference for the quantification of atmospheric OSs.
Nonetheless, research on OSs commenced relatively late, and, due to their wide diversity and demanding laboratory synthesis conditions, only a limited number of commercial reference materials are available for quantitative OS analysis. Consequently, the lack of actual standards led us to employ semi-quantitative analysis methods in this experiment, introducing some uncertainty in quantification. Future work should focus on enhancing the quantitative methods for OSs, utilizing actual standards for one-to-one compound quantification, and refining the measurement techniques for OSs. These efforts will contribute to a deeper understanding of SOA precursors, formation mechanisms, and the contribution of OSs to atmospheric aerosols, ultimately guiding research in the field of air pollution prevention and control.
All the code and data are available from Xiang Ding (xiangd@gig.ac.cn) on request.
XD and XMW designed the research. PL, BXL, and YQZ took the measurements. PL analyzed the results. PL, DJB, and XD wrote the paper with contribution from all the co-authors.
The contact author has declared that none of the authors has any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
The authors gratefully acknowledge the financial support provided by the China Scholarship Council (CSC). Ping Liu would also like to thank Jacqui Hamilton, Andrew Rickard, and the Wolfson Atmospheric Chemistry Laboratories at the University of York for hosting her as part of a CSC-funded joint doctoral program placement.
This research has been supported by the Foundation for Innovative Research Groups of the National Natural Science Foundation of China (grant no. 42321003), the National Natural Science Foundation of China (grant no. 42177090), and China Scholarship Council (CSC).
This paper was edited by Meng Gao and reviewed by two anonymous referees.
Bates, K. H. and Jacob, D. J.: A new model mechanism for atmospheric oxidation of isoprene: global effects on oxidants, nitrogen oxides, organic products, and secondary organic aerosol, Atmos. Chem. Phys., 19, 9613–9640, https://doi.org/10.5194/acp-19-9613-2019, 2019.
Brüggemann, M., Poulain, L., Held, A., Stelzer, T., Zuth, C., Richters, S., Mutzel, A., van Pinxteren, D., Iinuma, Y., Katkevica, S., Rabe, R., Herrmann, H., and Hoffmann, T.: Real-time detection of highly oxidized organosulfates and BSOA marker compounds during the F-BEACh 2014 field study, Atmos. Chem. Phys., 17, 1453–1469, https://doi.org/10.5194/acp-17-1453-2017, 2017.
Brüggemann, M., van Pinxteren, D., Wang, Y. C., Yu, J. Z., and Herrmann, H.: Quantification of known and unknown terpenoid organosulfates in PM10 using untargeted LC-HRMS/MS: contrasting summertime rural Germany and the North China Plain, Environ. Chem., 16, 333–346, https://doi.org/10.1071/en19089, 2019.
Bryant, D. J., Dixon, W. J., Hopkins, J. R., Dunmore, R. E., Pereira, K. L., Shaw, M., Squires, F. A., Bannan, T. J., Mehra, A., Worrall, S. D., Bacak, A., Coe, H., Percival, C. J., Whalley, L. K., Heard, D. E., Slater, E. J., Ouyang, B., Cui, T., Surratt, J. D., Liu, D., Shi, Z., Harrison, R., Sun, Y., Xu, W., Lewis, A. C., Lee, J. D., Rickard, A. R., and Hamilton, J. F.: Strong anthropogenic control of secondary organic aerosol formation from isoprene in Beijing, Atmos. Chem. Phys., 20, 7531–7552, https://doi.org/10.5194/acp-20-7531-2020, 2020.
Bryant, D. J., Elzein, A., Newland, M., White, E., Swift, S., Watkins, A., Deng, W., Song, W., Wang, S., Zhang, Y., Wang, X., Rickard, A. R., and Hamilton, J. F.: Importance of Oxidants and Temperature in the Formation of Biogenic Organosulfates and Nitrooxy Organosulfates, ACS Earth and Space Chemistry, 5, 2291–2306, https://doi.org/10.1021/acsearthspacechem.1c00204, 2021.
Bryant, D. J., Mayhew, A. W., Pereira, K. L., Budisulistiorini, S. H., Prior, C., Unsworth, W., Topping, D. O., Rickard, A. R., and Hamilton, J. F.: Overcoming the lack of authentic standards for the quantification of biogenic secondary organic aerosol markers, Environmental Science: Atmospheres, 3, 221–229, https://doi.org/10.1039/D2EA00074A, 2023a.
Bryant, D. J., Nelson, B. S., Swift, S. J., Budisulistiorini, S. H., Drysdale, W. S., Vaughan, A. R., Newland, M. J., Hopkins, J. R., Cash, J. M., Langford, B., Nemitz, E., Acton, W. J. F., Hewitt, C. N., Mandal, T., Gurjar, B. R., Shivani, Gadi, R., Lee, J. D., Rickard, A. R., and Hamilton, J. F.: Biogenic and anthropogenic sources of isoprene and monoterpenes and their secondary organic aerosol in Delhi, India, Atmos. Chem. Phys., 23, 61–83, https://doi.org/10.5194/acp-23-61-2023, 2023b.
Cui, T. Q., Zeng, Z. X., dos Santos, E. O., Zhang, Z. F., Chen, Y. Z., Zhang, Y., Rose, C. A., Budisulistiorini, S. H., Collins, L. B., Bodnar, W. M., de Souza, R. A. F., Martin, S. T., Machado, C. M. D., Turpin, B. J., Gold, A., Ault, A. P., and Surratt, J. D.: Development of a hydrophilic interaction liquid chromatography (HILIC) method for the chemical characterization of water-soluble isoprene epoxydiol (IEPOX)-derived secondary organic aerosol, Environ. Sci.-Process Impacts, 20, 1524–1536, https://doi.org/10.1039/c8em00308d, 2018.
Froyd, K. D., Murphy, S. M., Murphy, D. M., de Gouw, J. A., Eddingsaas, N. C., and Wennberg, P. O.: Contribution of isoprene-derived organosulfates to free tropospheric aerosol mass, P. Natl. Acad. Sci. USA, 107, 21360–21365, https://doi.org/10.1073/pnas.1012561107, 2010.
Gao, S., Surratt, J. D., Knipping, E. M., Edgerton, E. S., Shahgholi, M., and Seinfeld, J. H.: Characterization of polar organic components in fine aerosols in the southeastern United States: Identity, origin, and evolution, J. Geophys. Res.-Atmos., 111, D14314, https://doi.org/10.1029/2005jd006601, 2006.
Gomez-Gonzalez, Y., Surratt, J. D., Cuyckens, F., Szmigielski, R., Vermeylen, R., Jaoui, M., Lewandowski, M., Offenberg, J. H., Kleindienst, T. E., Edney, E. O., Blockhuys, F., Van Alsenoy, C., Maenhaut, W., and Claeys, M.: Characterization of organosulfates from the photooxidation of isoprene and unsaturated fatty acids in ambient aerosol using liquid chromatography/(-) electrospray ionization mass spectrometry, J. Mass Spectrom., 43, 371–382, https://doi.org/10.1002/jms.1329, 2008.
Hansen, A. M. K., Hong, J., Raatikainen, T., Kristensen, K., Ylisirniö, A., Virtanen, A., Petäjä, T., Glasius, M., and Prisle, N. L.: Hygroscopic properties and cloud condensation nuclei activation of limonene-derived organosulfates and their mixtures with ammonium sulfate, Atmos. Chem. Phys., 15, 14071–14089, https://doi.org/10.5194/acp-15-14071-2015, 2015.
He, Q. F., Ding, X., Fu, X. X., Zhang, Y. Q., Wang, J. Q., Liu, Y. X., Tang, M. J., Wang, X. M., and Rudich, Y.: Secondary Organic Aerosol Formation From Isoprene Epoxides in the Pearl River Delta, South China: IEPOX- and HMML-Derived Tracers, J. Geophys. Res.-Atmos., 123, 6999–7012, https://doi.org/10.1029/2017jd028242, 2018.
Hettiyadura, A. P. S., Stone, E. A., Kundu, S., Baker, Z., Geddes, E., Richards, K., and Humphry, T.: Determination of atmospheric organosulfates using HILIC chromatography with MS detection, Atmos. Meas. Tech., 8, 2347–2358, https://doi.org/10.5194/amt-8-2347-2015, 2015.
Hettiyadura, A. P. S., Jayarathne, T., Baumann, K., Goldstein, A. H., de Gouw, J. A., Koss, A., Keutsch, F. N., Skog, K., and Stone, E. A.: Qualitative and quantitative analysis of atmospheric organosulfates in Centreville, Alabama, Atmos. Chem. Phys., 17, 1343–1359, https://doi.org/10.5194/acp-17-1343-2017, 2017.
Hettiyadura, A. P. S., Al-Naiema, I. M., Hughes, D. D., Fang, T., and Stone, E. A.: Organosulfates in Atlanta, Georgia: anthropogenic influences on biogenic secondary organic aerosol formation, Atmos. Chem. Phys., 19, 3191–3206, https://doi.org/10.5194/acp-19-3191-2019, 2019.
Hodzic, A., Kasibhatla, P. S., Jo, D. S., Cappa, C. D., Jimenez, J. L., Madronich, S., and Park, R. J.: Rethinking the global secondary organic aerosol (SOA) budget: stronger production, faster removal, shorter lifetime, Atmos. Chem. Phys., 16, 7917–7941, https://doi.org/10.5194/acp-16-7917-2016, 2016.
Huang, R.-J., Cao, J., Chen, Y., Yang, L., Shen, J., You, Q., Wang, K., Lin, C., Xu, W., Gao, B., Li, Y., Chen, Q., Hoffmann, T., O'Dowd, C. D., Bilde, M., and Glasius, M.: Organosulfates in atmospheric aerosol: synthesis and quantitative analysis of PM2.5 from Xi'an, northwestern China, Atmos. Meas. Tech., 11, 3447–3456, https://doi.org/10.5194/amt-11-3447-2018, 2018.
Iinuma, Y., Mueller, C., Boege, O., Gnauk, T., and Herrmann, H.: The formation of organic sulfate esters in the limonene ozonolysis secondary organic aerosol (SOA) under acidic conditions, Atmos. Environ., 41, 5571–5583, https://doi.org/10.1016/j.atmosenv.2007.03.007, 2007.
Kundu, S., Kawamura, K., Andreae, T. W., Hoffer, A., and Andreae, M. O.: Diurnal variation in the water-soluble inorganic ions, organic carbon and isotopic compositions of total carbon and nitrogen in biomass burning aerosols from the LBA-SMOCC campaign in Rondonia, Brazil, J. Aerosol Sci., 41, 118–133, https://doi.org/10.1016/j.jaerosci.2009.08.006, 2010.
Le Breton, M., Wang, Y., Hallquist, Å. M., Pathak, R. K., Zheng, J., Yang, Y., Shang, D., Glasius, M., Bannan, T. J., Liu, Q., Chan, C. K., Percival, C. J., Zhu, W., Lou, S., Topping, D., Wang, Y., Yu, J., Lu, K., Guo, S., Hu, M., and Hallquist, M.: Online gas- and particle-phase measurements of organosulfates, organosulfonates and nitrooxy organosulfates in Beijing utilizing a FIGAERO ToF-CIMS, Atmos. Chem. Phys., 18, 10355–10371, https://doi.org/10.5194/acp-18-10355-2018, 2018.
Lin, Y.-H., Knipping, E. M., Edgerton, E. S., Shaw, S. L., and Surratt, J. D.: Investigating the influences of SO2 and NH3 levels on isoprene-derived secondary organic aerosol formation using conditional sampling approaches, Atmos. Chem. Phys., 13, 8457–8470, https://doi.org/10.5194/acp-13-8457-2013, 2013.
Lukács, H., Gelencsér, A., Hoffer, A., Kiss, G., Horváth, K., and Hartyáni, Z.: Quantitative assessment of organosulfates in size-segregated rural fine aerosol, Atmos. Chem. Phys., 9, 231–238, https://doi.org/10.5194/acp-9-231-2009, 2009.
Olson, C. N., Galloway, M. M., Yu, G., Hedman, C. J., Lockett, M. R., Yoon, T., Stone, E. A., Smith, L. M., and Keutsch, F. N.: Hydroxycarboxylic Acid-Derived Organosulfates: Synthesis, Stability, and Quantification in Ambient Aerosol, Environ. Sci. Technol., 45, 6468–6474, https://doi.org/10.1021/es201039p, 2011.
Riva, M., Da Silva Barbosa, T., Lin, Y.-H., Stone, E. A., Gold, A., and Surratt, J. D.: Chemical characterization of organosulfates in secondary organic aerosol derived from the photooxidation of alkanes, Atmos. Chem. Phys., 16, 11001–11018, https://doi.org/10.5194/acp-16-11001-2016, 2016.
Romero, F. and Oehme, M.: Organosulfates – A new component of humic-like substances in atmospheric aerosols?, J. Atmos. Chem., 52, 283–294, https://doi.org/10.1007/s10874-005-0594-y, 2005.
Shakya, K. M. and Peltier, R. E.: Investigating Missing Sources of Sulfur at Fairbanks, Alaska, Environ. Sci. Technol., 47, 9332–9338, https://doi.org/10.1021/es402020b, 2013.
Shakya, K. M. and Peltier, R. E.: Non-sulfate sulfur in fine aerosols across the United States: Insight for organosulfate prevalence, Atmos. Environ., 100, 159–166, https://doi.org/10.1016/j.atmosenv.2014.10.058, 2015.
Staudt, S., Kundu, S., Lehmler, H.-J., He, X., Cui, T., Lin, Y.-H., Kristensen, K., Glasius, M., Zhang, X., Weber, R. J., Surratt, J. D., and Stone, E. A.: Aromatic organosulfates in atmospheric aerosols: Synthesis, characterization, and abundance, Atmos. Environ., 94, 366–373, https://doi.org/10.1016/j.atmosenv.2014.05.049, 2014.
Stone, E. A., Hedman, C. J., Sheesley, R. J., Shafer, M. M., and Schauer, J. J.: Investigating the chemical nature of humic-like substances (HULIS) in North American atmospheric aerosols by liquid chromatography tandem mass spectrometry, Atmos. Environ., 43, 4205–4213, https://doi.org/10.1016/j.atmosenv.2009.05.030, 2009.
Stone, E. A., Yang, L. M., Yu, L. Y. E., and Rupakheti, M.: Characterization of organosulfates in atmospheric aerosols at Four Asian locations, Atmos. Environ., 47, 323–329, https://doi.org/10.1016/j.atmosenv.2011.10.058, 2012.
Surratt, J. D., Lewandowski, M., Offenberg, J. H., Jaoui, M., Kleindienst, T. E., Edney, E. O., and Seinfeld, J. H.: Effect of acidity on secondary organic aerosol formation from isoprene, Environ. Sci. Technol., 41, 5363–5369, https://doi.org/10.1021/es0704176, 2007a.
Surratt, J. D., Kroll, J. H., Kleindienst, T. E., Edney, E. O., Claeys, M., Sorooshian, A., Ng, N. L., Offenberg, J. H., Lewandowski, M., Jaoui, M., Flagan, R. C., and Seinfeld, J. H.: Evidence for organosulfates in secondary organic aerosol, Environ. Sci. Technol., 41, 517–527, https://doi.org/10.1021/es062081q, 2007b.
Surratt, J. D., Gomez-Gonzalez, Y., Chan, A. W. H., Vermeylen, R., Shahgholi, M., Kleindienst, T. E., Edney, E. O., Offenberg, J. H., Lewandowski, M., Jaoui, M., Maenhaut, W., Claeys, M., Flagan, R. C., and Seinfeld, J. H.: Organosulfate formation in biogenic secondary organic aerosol, J. Phys. Chem. A, 112, 8345–8378, https://doi.org/10.1021/jp802310p, 2008.
Surratt, J. D., Chan, A. W. H., Eddingsaas, N. C., Chan, M. N., Loza, C. L., Kwan, A. J., Hersey, S. P., Flagan, R. C., Wennberg, P. O., and Seinfeld, J. H.: Reactive intermediates revealed in secondary organic aerosol formation from isoprene, P. Natl. Acad. Sci. USA, 107, 6640–6645, https://doi.org/10.1073/pnas.0911114107, 2010.
Tolocka, M. P. and Turpin, B.: Contribution of Organosulfur Compounds to Organic Aerosol Mass, Environ. Sci. Technol., 46, 7978–7983, https://doi.org/10.1021/es300651v, 2012.
Zhang, Y.-Q., Ding, X., He, Q.-F., Wen, T.-X., Wang, J.-Q., Yang, K., Jiang, H., Cheng, Q., Liu, P., Wang, Z.-R., He, Y.-F., Hu, W.-W., Wang, Q.-Y., Xin, J.-Y., Wang, Y.-S., and Wang, X.-M.: Observational Insights into Isoprene Secondary Organic Aerosol Formation through the Epoxide Pathway at Three Urban Sites from Northern to Southern China, Environ. Sci. Technol., 56, 4795–4805, https://doi.org/10.1021/acs.est.1c06974, 2022.