the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 06 Jan 2025
| 06 Jan 2025
Deployment and evaluation of an NH4+∕ H3O+ reagent ion switching chemical ionization mass spectrometer for the detection of reduced and oxygenated gas-phase organic compounds
Cort L. Zang
Megan D. Willis
Reactive organic carbon (ROC) is diverse in its speciation, functionalization, and volatility, with varying implications for ozone production and secondary organic aerosol formation and growth. Chemical ionization mass spectrometry (CIMS) approaches can provide in situ ROC observations, and the CIMS reagent ion controls the detectable ROC species. To expand the range of detectable ROC, we describe a method for switching between the reagent ions and H3O+ in a Vocus chemical ionization time-of-flight mass spectrometer (Vocus-CI-ToFMS). We describe optimization of ion–molecule reactor conditions for both reagent ions, at the same temperature, and compare the ability of and H3O+ to detect a variety of volatile organic compounds (VOCs) and semi-volatile and intermediate-volatility organic compounds (SVOCs and IVOCs), including oxygenates and organic sulfur compounds. Sensitivities are comparable to other similar instruments (up to ∼5 counts s−1 ), with detection limits on the order of 1–10 s of pptv (1 s integration time). We report a method for characterizing and filtering periods of hysteresis following each reagent ion switch and compare use of reagent ions, persistent ambient ions, and a deuterated internal standard for diagnosing this hysteresis. We deploy reagent ion switching in a rural pine forest in central Colorado, US, and use our ambient measurements to compare the capabilities of and H3O+ in the same instrument, without interferences from variation in instrument and inlet designs. We find that H3O+ optimally detects reduced ROC species with high volatility, while improves detection of functionalized ROC compounds, including organic nitrates and oxygenated SVOCs and IVOCs that are readily fragmented by H3O+.
- Article
(4944 KB) - Full-text XML
-
Supplement
(4954 KB) - BibTeX
- EndNote




Tropospheric aerosol formation, oxidant reactivity, and ozone production are driven by the molecularly diverse pool of atmospheric reactive organic carbon (ROC; all organic species excluding methane) (Heald and Kroll, 2020). Speciation of atmospheric ROC is an ongoing analytical challenge (e.g., Goldstein and Galbally, 2007; Hunter et al., 2017), especially at time resolutions relevant to atmospheric mixing and chemistry. While reduced volatile organic compounds (VOCs, with saturation vapor concentration, C*, µg m−3) are an important fraction of ROC, functionalized species with lower volatility (SVOCs and IVOCs with C* between 0.3 and 3×106 µg m−3) are major contributors to ozone production and aerosol formation (e.g., Xu et al., 2021; Heald and Kroll, 2020; Bianchi et al., 2019; Donahue et al., 2011). For example, near-comprehensive measurements of ROC at a forested site showed that SVOCs and IVOCs contribute approximately one-third of ⋅OH reactivity and potential secondary organic aerosol production (Hunter et al., 2017). Further, semi-volatile and oxygenated VOCs contribute to marine secondary aerosol formation (Burkart et al., 2017; Mungall et al., 2017; Croft et al., 2019, 2021), and oxygenated species such as furans contribute significantly to ⋅OH reactivity and aerosol production in wildfire plumes (Xu et al., 2021). In many urban environments, volatile chemical products and other classes of IVOCs make a growing contribution to aerosol and ozone production (Coggon et al., 2021; Zhao et al., 2014b). However, owing to limitations in analytical techniques, as well as partitioning to inlet and instrument surfaces (Deming et al., 2019; Pagonis et al., 2019), oxygenated and otherwise functionalized SVOCs and IVOCs are often unmeasured.
Chemical ionization mass spectrometry (CIMS) represents a family of analytical techniques applied to detect and characterize organic and inorganic trace gases in whole air at high time resolution (e.g., Zhang et al., 2023; Yuan et al., 2017; Huey, 2007). The choice of reagent ion determines the scope of the measurement in terms of ROC functionality and chemistry, while instrument construction and design impacts the range of detectable species in terms of volatility and reactivity (e.g., Riva et al., 2019; Krechmer et al., 2018). A range of reagent ions are in common use and are selective toward specific ROC classes. Oxygenated, multi-functional organic gases can be detected as negative ions using iodide (I− or ) (Lee et al., 2018, 2014), acetate () (Brophy and Farmer, 2015; Roberts et al., 2010), CF3O− (Crounse et al., 2013, 2006), sulfur hexafluoride () (Nah et al., 2018; Huey, 2007), and nitrate () and bromide (Br−) (Rissanen et al., 2019; Bianchi et al., 2019) reagent ions. Highly oxygenated organic species (with µg m−3), together with low-volatility inorganic species (e.g., H2SO4), can be detected with nitrate and bromide ionization at ambient pressure (Bianchi et al., 2019; Rissanen et al., 2019; Riva et al., 2019). CF3O− effectively detects organic peroxides (Crounse et al., 2013, 2006), while I−, Br−, , and detect a range of polar and acidic gases (Riva et al., 2019; Lee et al., 2018, 2014). Reduced VOCs, small oxygenated VOCs (e.g., methanol, ethanol, acetone, acetaldehyde), and reduced sulfur compounds (e.g., dimethyl sulfide and methanethiol) are readily detected as positive ions via proton transfer with hydronium (H3O+) reagent ions (e.g., Kilgour et al., 2022; Pagonis et al., 2019; Krechmer et al., 2018; Yuan et al., 2017). NO+ and allow detection of reduced VOCs, with proton affinities below that of water, that are generally not detectable with H3O+ (Jordan et al., 2009; Smith and Spanel, 2005). While H3O+ can detect functionalized VOCs, fragmentation is common and complicates the interpretation of mass spectra from complex samples (e.g., Coggon et al., 2024; Kilgour et al., 2024; Li et al., 2021; Pagonis et al., 2019; Yuan et al., 2017). To overcome some of the limitations induced by H3O+ ionization, fast separation techniques have been coupled to proton transfer instruments (Claflin et al., 2021; Stockwell et al., 2021; Vermeuel et al., 2023b; Coggon et al., 2024; Kilgour et al., 2024), and other positive-polarity reagent ions have been applied to functionalized ROC. Benzene () reagent ions detect dimethyl sulfide, monoterpenes, and sesquiterpenes with reduced fragmentation and higher selectivity compared to H3O+ (Kim et al., 2016). Water clusters (i.e., (H2O)nH+) can detect a small subset of species detected by H3O+, such as dimethyl sulfide, with high selectivity (Blomquist et al., 2010). An array of oxygenated, multi-functional compounds in the intermediate to semi-volatile range can be detected using ammonium reagent ions (e.g., Xu et al., 2022; Khare et al., 2022; Muller et al., 2020; Hansel et al., 2018), which provide some overlap in the fractions of ROC detected by negative-polarity reagent ions such as I− and CF3O−.
Compared to other positive-polarity reagent ions, ammonium () adduction ionization is selective toward a wider range of multi-functional oxygenated compounds, such as carbonyls, alcohols, ethers, furans, and siloxanes (Xu et al., 2022; Khare et al., 2022; Muller et al., 2020; Zhang et al., 2019; Zaytsev et al., 2019a; Berndt et al., 2019). Ambient observations have recently shown that ionization can detect organic nitrates (Xu et al., 2022), while laboratory studies have demonstrated detection of organic peroxides (Zhou et al., 2018) and peroxy radicals (Hansel et al., 2018). Selected ion flow tube mass spectrometry (SIFT-MS) studies show that ions form the strongest associations with carbonyl groups, relative to other oxygenates (e.g., alcohols and ethers) (Adams 2003). However, the conditions under which reagent ions form and ion–molecule reactions occur determine the dominant reagent ion and ionization mechanism, which in turn controls the scope of detectable compounds and associated sensitivity. Possible reagent ions include (where X= H2O or NH3 and n=0, 1, 2, etc.). In practice, multiple reagent ions can be present, with providing optimal sensitivity to oxygenated compounds (Xu et al., 2022). Reactions with neutral analytes occur through ligand switching (Reaction 1), where the evaporation of X promotes softer adduct formation compared to alone (i.e., n=0) (Xu et al., 2022; Canaval et al., 2019; Adams et al., 2003).
Given an analyte, A, with larger affinity (i.e., the negative enthalpy of the reaction: ; e.g., Xu et al. (2022)) than X, the ionization reaction (1) is exothermic (Adams et al., 2003). Therefore, ligand switching reactions will proceed efficiently at or near the collision limit, with little importance of the reverse reaction unless reaction timescales are long or the reaction is endothermic or only slightly exothermic (Xu et al., 2022; Zaytsev et al., 2019a). Reaction (1) is exothermic for the majority of oxygenates and multi-functional compounds (Xu et al., 2022; Adams et al., 2003; Canaval et al., 2019; Zaytsev et al., 2019a) (Table S3, Hunter and Lias, 2023; Meot-Ner (Mautner) and Lias, 2023); however, ion–molecule reactor (IMR) conditions must be selected to promote pure ion chemistry, optimize sensitivity, and minimize fragmentation.
Many CIMS reagent ions provide access to complementary fractions of ambient ROC, and combining multiple reagent ions in a single CIMS instrument can provide several advantages. First, reagent ion switching can maintain the benefits of selectivity afforded by specific reagent ions while expanding the number of detectable compounds. For example, H3O+ and are combined in proton transfer reaction (PTR) and SIFT-MS instruments to expand detection capabilities to a broad range of alkanes, alkenes, aromatics, and some oxygenated species (e.g., Agarwal et al., 2014; Smith and Spanel, 2005). Further, the combination of nitrate and bromide reagent ions allows detection of a wide range of highly oxygenated molecules along with hydroperoxyl radicals, iodine compounds, and sulfuric acid (He et al., 2023; Rissanen et al., 2019). Iodide, acetate, and water cluster reagent ions have been combined in laboratory studies, using repeated experiments rather than active reagent switching, to expand the range of detectable ROC (Aljawhary et al., 2013) and inform development of reaction mechanisms (e.g., Zhao et al., 2014a). Thus, combining reagent ions can decrease the potentially large number of instruments required to characterize a broad range of ROC classes (e.g., Heald and Kroll, 2020). Second, combining reagent ions in a single instrument allows for direct comparison between the fractions of ROC detected by each chemical ionization reagent (e.g., Zaytsev et al., 2019a). For example, rapid switching between iodide and the acid-selective acetate reagent ion informs detection of organic acids with iodide (e.g., Brophy and Farmer, 2015). Switching between and H3O+ has benefits for measuring both reduced VOCs and their early generation oxidation products (Zaytsev et al., 2019a, b), while also allowing a direct comparison between the subsets of ROC detected by each reagent ion without the complications associated with differing instrument and inlet design (e.g., Riva et al., 2019).
Ambient atmospheric observations with adduct ionization CIMS have focused primarily on urban environments, where ion chemistry allows detection of oxygenated VOCs from volatile chemical products (Xu et al., 2022; Khare et al., 2022). reagent ion switching has so far been limited to laboratory experiments demonstrating feasibility of switching (Muller et al., 2020) and application following laboratory oxidation of VOCs and oxygenated VOCs (Zaytsev et al., 2019a, b). We characterize /H3O+ reagent ion switching using a Vocus chemical ionization time-of-flight mass spectrometer (Vocus-CI-ToFMS) using both laboratory standards and deployment at a rural forested site. We describe selection of ideal IMR conditions for reagent ion switching, at the same temperature, with a focus on sensitivity, fragmentation, and prominence of competing ionization pathways. Using ambient reagent ion switching data, we describe an approach to filter periods of impure ion chemistry, and once filtered, ambient observations allow us to directly compare the fractions of ambient ROC detected by H3O+ and . Our observations demonstrate that is able to detect oxygenated and multi-functional biogenic ROC with both reduced fragmentation and higher selectivity compared to H3O+, illustrating a highly complementary set of CIMS reagent ions.
2.1 Instrument description
The Vocus-CI-ToFMS (Vocus S, Tofwerk AG and Aerodyne Research Inc.) is described in detail elsewhere (Krechmer et al., 2018), with a brief description supplied here. Two features differentiate the Vocus-CI-ToFMS from other chemical ionization or proton transfer reaction time-of-flight mass spectrometers. First, the Vocus-CI-ToFMS is equipped with a focusing ion–molecule reactor (fIMR) which consists of a radio-frequency-only quadrupole oriented around a 10 cm long resistive glass tube (Krechmer et al., 2018). The fIMR focuses ions toward the centerline, reducing ion losses to the walls and promoting ion transmission into a quadrupole high-pass mass filter (big segmented quadrupole, BSQ). Second, polyether ether ketone (PEEK) tubing is used to establish flow restriction between ambient pressure and the fIMR. The use of PEEK at the instrument inlet reduces interactions between sampled air and more absorptive surfaces which impact transmission of SVOCs and IVOCs into the fIMR (Deming et al., 2019). These modifications to the Vocus-CI-ToFMS improve the ability to detect both reduced and oxidized ROC (Riva et al., 2019). The Vocus-CI-ToFMS used in this study has a mass resolving power of ∼ 5000 m dm−1, a mass range of ∼ 50–500 , and a 25 kHz ToF extraction frequency and is equipped with a multi-port reagent ion injection, current-regulated discharge ion source. Details about instrument voltages are available in Table S1 in the Supplement.
When using H3O+ ionization, we inject 20 cm3 min−1 (STP) from the headspace above ultra-high-purity water (MilliporeSigma, OmniSolv LC-MS) under vacuum into the discharge ion source. When switching to ionization, we further add a flow from the headspace above a ∼ 1 w/w % solution of ammonium hydroxide (Oakwood Products Inc., trace metals grade) in water to the ion source. Additionally, when switching between reagent ions, the voltages and pressure in the fIMR and the ion optics are adjusted to complement each reagent ion, taking into account sensitivity, fragmentation, and purity of ionization chemistry; this is discussed in Sect. 3.1. A change between ionization modes results in hysteresis where the ion chemistry is impure. The filtering of hysteretic periods is discussed in Sect. 3.4.
2.2 Reactor pressure and voltage gradient
The fIMR collision energy can be controlled in part by adjusting the conditions that impact the velocity, free path, and thermal energy of ions: axial voltage gradient, pressure, and temperature. The temperature must remain constant during reagent ion switching to allow for switching on 15 min timescales. This restricts control of collisional energy to adjustments of the fIMR voltage gradient and pressure. To understand the impact that these parameters have on ion chemistry, we introduce a constant flow of dilute calibration standard while systematically changing the fIMR voltage gradient and pressure. We change the fIMR pressure in 0.1 mbar increments and hold it constant while we increase the fIMR front voltage by 10 V steps. With , we characterized from 2.5 to 3.5 mbar and from 45 to 65 V cm−1 (60–120 townsends (Td)). For H3O+, we characterized from 1.5 to 2.5 mbar and from 45 to 65 V cm−1 (80–200 Td).
2.3 Sensitivity, detection limit, and fragmentation with standards
We calibrated 23 analytes from multi-component standardized gas cylinders (Apel-Riemer Environmental Inc.) to report sensitivities (counts s−1 ) and detection limits (3σ of the background with 1 s integration). The 23 analytes come from three separate multi-component cylinders where the composition was selected to avoid interferences from fragments (Table S2). Backgrounds were obtained using a zero air generator (Sabio model 1001). We investigate fragmentation of molecular ions using single-component samples of trans-2-hexen-1-ol (96.5 %, Acros Organics, lot A0340603), β-cyclocitral (92.3 %, Thermo Fisher, lot 10237632), 2-hexenal (97.5 %, Oakwood Products Inc., lot 098868J07I), 2-hexanone (100 %, Oakwood Products Inc., lot 098350R22K), and 2-methyl-3-buten-2-ol (99.5 %, Oakwood Products Inc., lot 051281K14H). To calculate a molecular ion fraction, we average 15 s of 2 Hz data and fit peaks corresponding to molecular ions (i.e., for H3O+ and for ), we identify fragments, and clusters then divide the molecular ion signal by the sum of all related peaks. The fIMR conditions for H3O+ ionization during these experiments were 2.2 mbar and 60 °C, with a voltage gradient of 67.5 V cm−1 (140 Td) and a BSQ amplitude of 270 V. Using a 60 °C reaction chamber with H3O+ is lower than commonly reported in the literature (∼ 80–100 °C) (e.g., Vermeuel et al., 2023a; Coggon et al., 2024); this choice arises from fIMR temperature constraints for (Xu et al., 2022) and is discussed in more detail in Sect. 3.1. For ionization the fIMR settings were 3.1 mbar and 60 °C, with a voltage gradient of 60 V cm−1 (90 Td) and a BSQ amplitude of 250 V. The BSQ frequency was 2.2 MHz, and the fIMR amplitude and frequency were 500 V and 1.6 MHz, respectively (Table S1).
2.4 Signal response to ambient relative humidity
To test the effect of relative humidity on sensitivity, we varied the ratio of wet and dry flows (controlled with mass flow controllers, MKS Instruments model 1179C Mass-Flo) to achieve a range of relative humidities. The relative humidity was measured inline (Omega Engineering model HX71-V1). Downstream of the relative humidity measurement 10 cm3 min−1 (STP) of a certified gas standard (Apel-Riemer Environmental Inc. and Airgas for dimethyl sulfide) was added to the humidified flow before being introduced into the Vocus-CI-ToFMS. Relative humidity ranged between 15 % and 85 % during the experiments. Measurements alternated between elevated relative humidity and dry conditions, such that each measurement at elevated humidity could be directly compared to a dry (0 % RH) measurement immediately before.
2.5 Observations at the Manitou Experimental Forest Observatory
We deployed a Vocus-CI-ToFMS at the Manitou Experimental Forest Observatory (MEFO) from 3 to 24 September 2021. MEFO is a rural ponderosa pine forest at middle elevation (∼ 2300 m), located ∼ 40 km northwest of Colorado Springs and ∼ 70 km southwest of Denver (39.1006° N, 105.0942° W). The ROC composition at this site is well characterized, with emissions dominated by local biogenic sources (Hunter et al., 2017; Vermeuel et al., 2023a; Riches et al., 2024; Link et al., 2024). A full description of the field site can be found in Ortega et al. (2014). The Vocus-CI-ToFMS sample inlet was ∼ 4 m of 0.25 in. outer diameter perfluoroalkoxy (PFA) tubing situated ∼ 4 m above ground. The inlet flow was ∼ 3.8 L min−1 (i.e., 2.9 L min−1 (STP)) pulled by a flow-restricted bypass pump, resulting in a laminar flow inlet (Reynolds number of ∼ 1150) and corresponding to a residence time of ∼ 0.7 s. The inlet likely produced wall loss of oxygenated ROC, and while the extent was not quantified, minimizing the inlet inner diameter and maximizing the flow rate, while maintaining laminar flow, serve to minimize inlet losses and tubing delays (Pagonis et al., 2017). The Vocus-CI-ToFMS sub-sampled 93–100 cm3 min−1 (i.e., 71–77 cm3 min−1 (STP)) perpendicular to the main inlet flow, which helps prevent ambient aerosol clogging the capillary inlet interface compared to a linear sub-sampling assembly (Jensen et al., 2023). We performed bi-hourly, 3 min instrument backgrounds with ultra zero air (Airgas, UZA grade) followed by a 1 min, single-point calibration with a certified calibrant mixture (Apel-Riemer Environmental Inc.) for both reagent ions.
The Vocus-CI-ToFMS switched between and H3O+ ionization on 15 min time intervals during the deployment in MEFO. The fIMR conditions for H3O+ ionization were 2.5 mbar and 60 °C, with a voltage gradient of 62 V cm−1 and a BSQ amplitude of 350 V. The fIMR conditions for ionization were 3.1 mbar and 60 °C, with a voltage gradient of 65 V cm−1 and a BSQ amplitude of 250 V. The fIMR settings correspond to values of 114 and 96 Td for H3O+ and , respectively. The BSQ frequency was 2.2 MHz, and the fIMR amplitude and frequency were 450 V and 1.3 MHz, respectively (Table S1). The fIMR parameters for both reagent ions were informed by experiments detailed in Sect. 3.1.
2.6 ARTofMELT expedition on icebreaker (I/B) Oden
We deployed the Vocus-CI-ToFMS aboard the Swedish I/B Oden from 7 May to 15 June 2023 as part of the “Atmospheric rivers and the onset of ice melt” (ARTofMELT) measurement expedition. The cruise on I/B Oden took place within the pack ice and marginal ice zone between Svalbard and Greenland in the Fram Strait. The Vocus-CI-ToFMS switched between and H3O+ ionization on 15 min time intervals during the expedition. The Vocus-CI-ToFMS was mounted to the floor of a sea container on I/B Oden's fourth deck using metal plates and five high-deflection vibration isolation feet (Barry Controls model 2K2-BA-90) and was mounted to an open wall using two wire rope isolators (Enidine model WR6-850-10-E).
Ambient air was sampled through a 0.375 in. outer diameter PFA tube ∼15 m in length. The entire length of tubing was insulated and heated to 30 °C in three separately controlled sections. The Vocus-CI-ToFMS inlet flow was driven by a vacuum pump (Agilent IDP-7 dry scroll) regulated by a mass flow controller (15 L min−1 (STP)). A deuterated internal standard (containing dimethylsulfide-d3, acetone-d6, 2-hexanone-d4, and mesitylene-d12) was injected near the top of the Vocus-CI-ToFMS inlet through a 0.125 in. outer diameter PFA tube at a rate of 5 cm3 min−1 (STP) into the total inlet flow of 15 L min−1 (STP), yielding ∼333 pptv from the nominally 1 ppm standardized cylinder (Apel-Riemer Environmental Inc.). The inlet assembly was affixed to a metal pipe extending from the top of the sea container toward the bow of the ship at a ∼40° angle ∼3 m above the top of the container or 25 m above the water/ice surface. The use of internal standards during this expedition allows us to analyze reagent ion hysteresis (Sect. 3.4) using a persistent and known set of ions. The fIMR conditions for H3O+ ionization during ARTofMELT were 2.2 mbar, 60 °C, a voltage gradient of 67.5 V cm−1 (140 Td), and a 270 V BSQ amplitude. For ionization the fIMR settings were 3.1 mbar, 60 °C, a 60 V cm−1 (90 Td) voltage gradient, and a 250 V BSQ amplitude. The BSQ frequency was 2.2 MHz, and the fIMR amplitude and frequency were 500 V and 1.6 MHz, respectively (Table S1). We reserve further analysis of the ARTofMELT data set for future work.
2.7 Data analysis
Raw mass spectral data were collected with Acquility (version 2.3.18) and TofDaq (version 1.99) (Tofwerk AG) and processed in Tofware (version 3.2.5, Tofwerk AG and Aerodyne Research Inc.). For MEFO data, the time resolution was pre-averaged from 1 to 0.1 Hz. For ARTofMELT data, the time resolution was pre-averaged from 2 to 1 Hz. All data were mass-calibrated, baseline-subtracted, and peak-fit in Tofware. Time-integrated high-resolution ion signals were exported for further analysis in Python (version 3.9.12). Responses in the scans in Sect. 3.1 were interpolated using a linear interpolation on a triangular grid (using matplotlib.tri.LinearTriInterpolator). C* values are estimated using EPI Suite (US EPA, 2023).
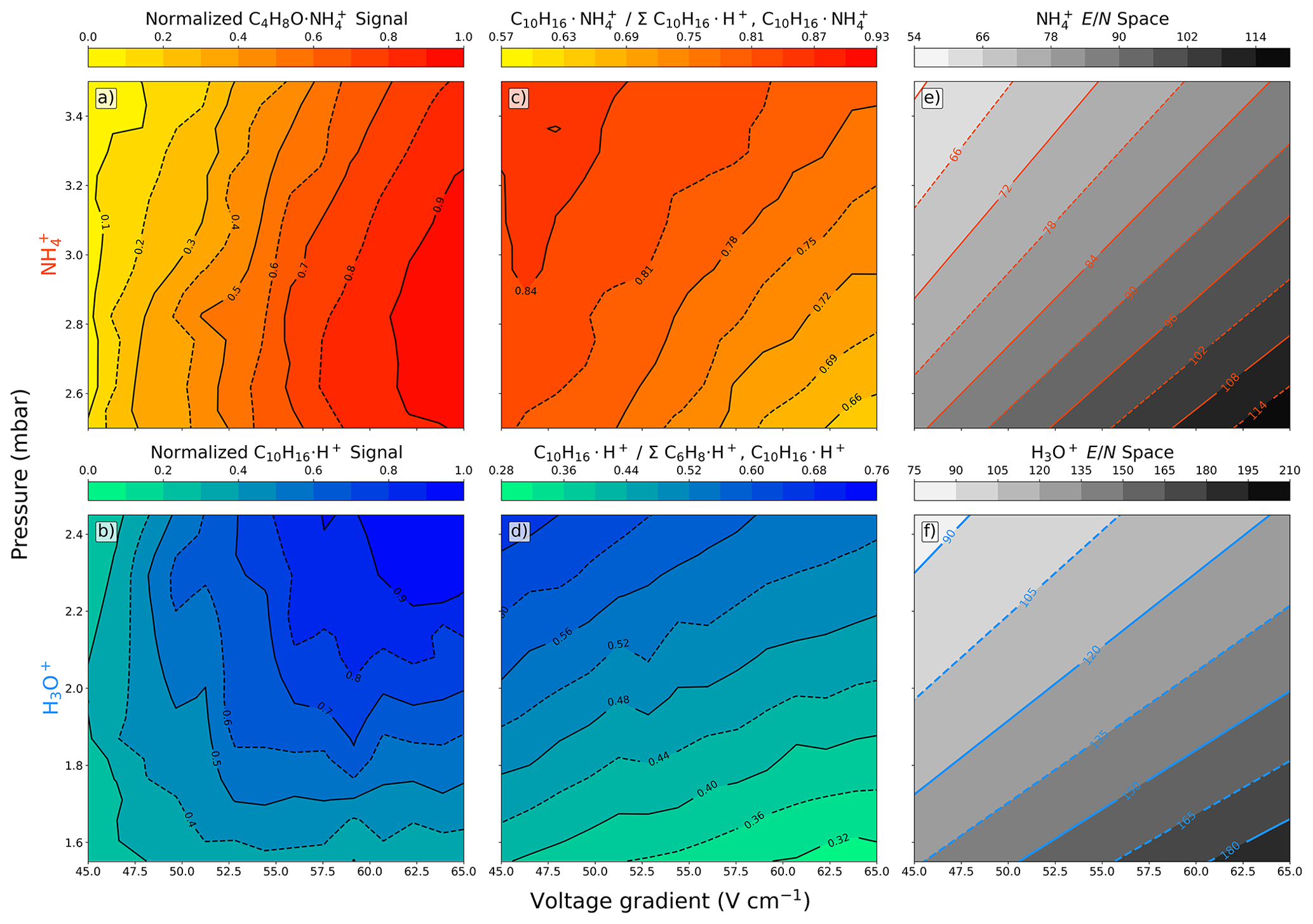
Figure 1(a–d) Contour plots of fIMR pressure and voltage gradient scans with a constant concentration of analyte (10 ppbv) introduced into the Vocus-CI-ToFMS. Normalized signal intensity for (a) methyl ethyl ketone measured with () and (b) α-pinene measured with H3O+ (). (c) Fractional contribution of the molecular ion () to the total α-pinene signal (i.e., the sum of the proton transfer product () and the molecular ion). (d) Fractional contribution of the H3O+ molecular ion () to the total α-pinene signal (i.e., the sum of the α-pinene fragment () and the molecular ion). Contour plots of calculated values over the scanned space for both (e) and (f) H3O+.
3.1 Ion–molecule reactor pressure and voltage gradient
H3O+ and ionization operate optimally at differing combinations of fIMR pressure, voltage gradient, and temperature (Xu et al., 2022; Gouw and Warneke, 2007), all of which impact the reduced electric field () of the fIMR:
where T is the temperature (K), ΔV is the difference between the front and back voltage (V), limr is the fIMR length (m), kB is the Boltzmann constant (J K−1), and p is pressure (Pa or J m−3). has units of townsends (Td, 1 Td =1 V cm2) and describes ion velocity and collisional energy. High values promote increased fragmentation and reduced clustering, while low values promote cluster formation and reduced fragmentation. We analyzed relative sensitivity, fragmentation, and prevalence of ionization pathways while varying the fIMR pressure and voltage gradient with a constant temperature of 60°C (Fig. 1) to inform our selection of fIMR settings. Because fIMR temperature takes tens of minutes to stabilize, we selected a constant 60 °C fIMR temperature to promote clusters in ionization. This is lower than most H3O+ fIMR temperatures (∼ 80–100 °C); however, the effect of lower fIMR temperature on the reagent ion distribution can be mitigated by adjusting other fIMR settings.
Selecting H3O+ ionization fIMR parameters requires balancing fragmentation and sensitivity. We observe a large increase (> 60 % at 2.4 mbar) in the sensitivity to the molecular ion () with increased fIMR voltage gradient (Fig. 1b), which arises from three main factors. First, reduced residence time with increased voltage gradient (163 or 113 µs at 45 or 65 V cm−1, respectively, with pressure and temperature of 2.4 mbar and 333.15 K) could increase ion transmission through the fIMR. Second, at low protonated water clusters contribute to the ionization of α-pinene. The production of protonated water clusters is evident from the reduced benzene sensitivity at lower (Fig. A1) (Gouw and Warneke, 2007). Water clusters have a higher proton affinity (i.e., the negative enthalpy of the reaction: ) compared to water (Hunter and Lias, 1998), and α-pinene has a higher proton affinity than both water and the first water cluster (i.e., (H2O)2), making ionization reactions with both exothermic. In contrast, benzene has a proton affinity higher than water but lower than the first cluster, making the ionization reaction of benzene with (H2O)2H+ endothermic and unlikely. Therefore, the formation of water clusters will reduce the sensitivity to benzene while increasing the sensitivity to α-pinene. Third, the known H3O+ α-pinene fragment, , has a larger contribution to the total α-pinene signal at higher (Fig. 1d), which suggests that part of the increased sensitivity to at higher pressures is also attributable to reduced loss to fragmentation. The combined responses of transmission, fragmentation, and proton affinity to changes in voltage and pressure with H3O+ ionization result in a sensitivity that does not follow directly (Fig. 1b, 1f).
Selecting ionization fIMR parameters requires balancing between signal intensity and purity of ion chemistry. Similar to H3O+, with we observe increased signal intensity of the methyl ethyl ketone molecular ion () at higher voltage gradients (Fig. 1a). In contrast to α-pinene detection with H3O+, sensitivity to is not impacted by fragmentation and changing reagent ion proton affinity across the range in voltage gradient. The sensitivity to the ion is highest at high , but the change in sensitivity is mostly dependent on the voltage gradient and is less impacted by the fIMR pressure (Fig. 1a). We observe a similar trend for other oxygenated ROC species (Fig. A2). The vertical contours in the sensitivity contrasted with the L-shaped contouring in the H3O+ sensitivity (Fig. 1a, b) support the three-factor dependence for H3O+ on transmission, fragmentation, and proton affinity and point to transmission as the major factor impacting sensitivity with ionization. However, at high we observe impurities in the ionization chemistry (i.e., proton transfer products () occurring for α-pinene) with ionization (Fig. 1c). Proton transfer ionization under is undesirable because it provides lower selectivity, leads to higher fragmentation rates compared to the ligand switching mechanism, and complicates interpretation of the mass spectrum. α-Pinene has a lower ammonium affinity than H2O (Canaval et al., 2019), which makes the ligand switching reaction (1) endothermic and thus dependent on increased collisional energy at higher voltage gradients (Xu et al., 2022). We observe a larger contribution of the proton transfer product at higher , which is consistent with electric-field-induced production of through internal proton transfer (Xu et al., 2022). Alternatively, it is also possible that production is enhanced by declustering of the ions to form , which is more likely to undergo proton transfer reactions directly, without the need for internal proton transfer, due to the lower proton affinity of NH3 compared to α-pinene (Canaval et al., 2019). Regardless of the mechanism, formation of proton transfer products is ideally avoided, and we find that their production is minimized at low (Fig. 1c). We note that the formation of secondary clusters (i.e., and ) is negligible over the entire investigated space (Figs. S1, S2). The sensitivity to methyl ethyl ketone and prevalence of undesirable reaction products () are optimal under opposing conditions in the voltage-gradient–pressure space, such that optimal fIMR parameters for require a compromise between sensitivity and purity of ionization chemistry.
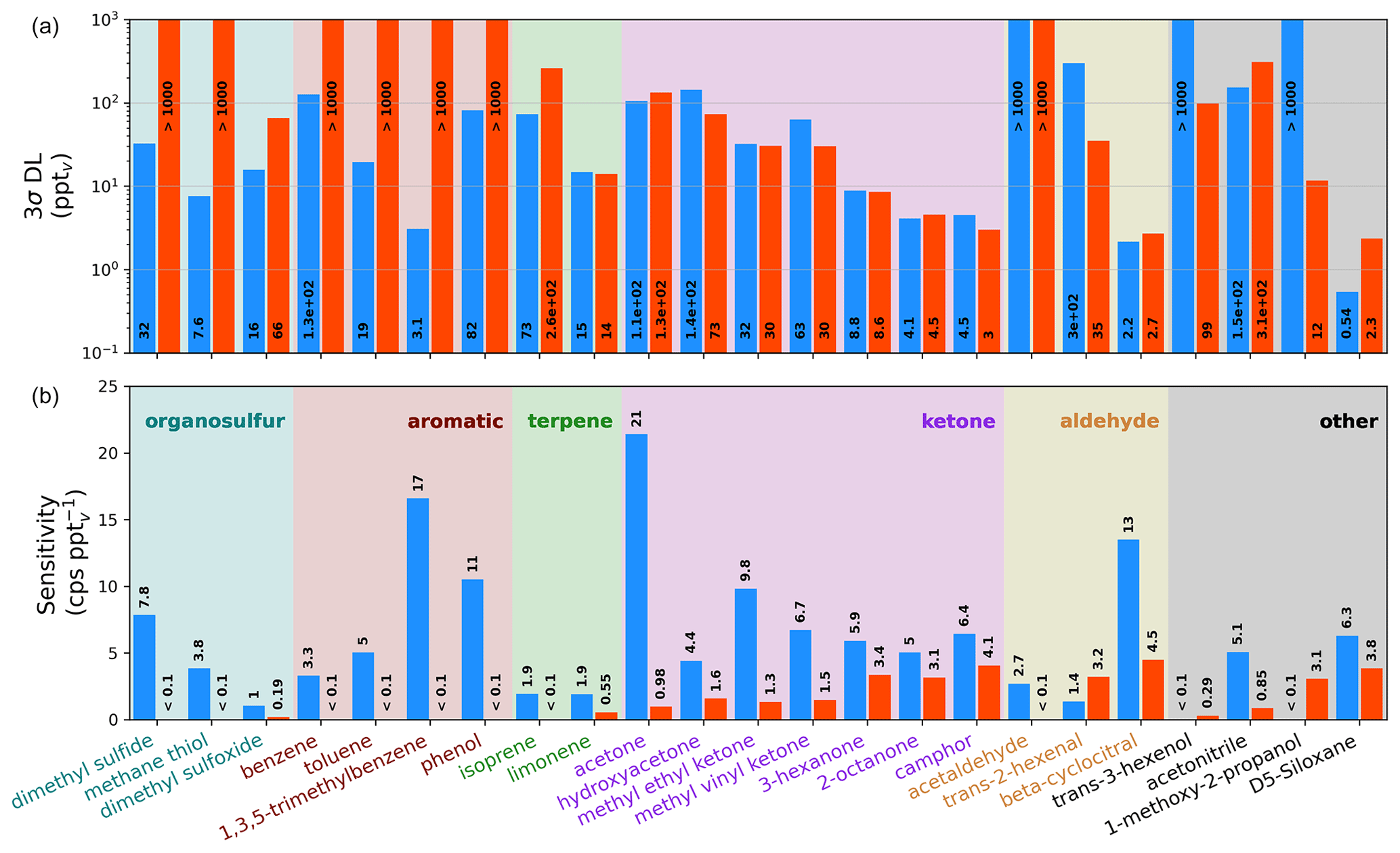
Figure 2Sensitivities (b) and detection limits (a, log y axis) for the Vocus-CI-ToFMS with ionization (orange) and H3O+ ionization (blue) for 23 analytes from standardized gas cylinders, grouped by functional group/compound type. Sensitivities and detection limits (DLs) are calculated for the molecular ion only (i.e., for or for H3O+), and mass spectral fragments are not included. DLs are calculated as 3σ over a 600 s background at 1 Hz.
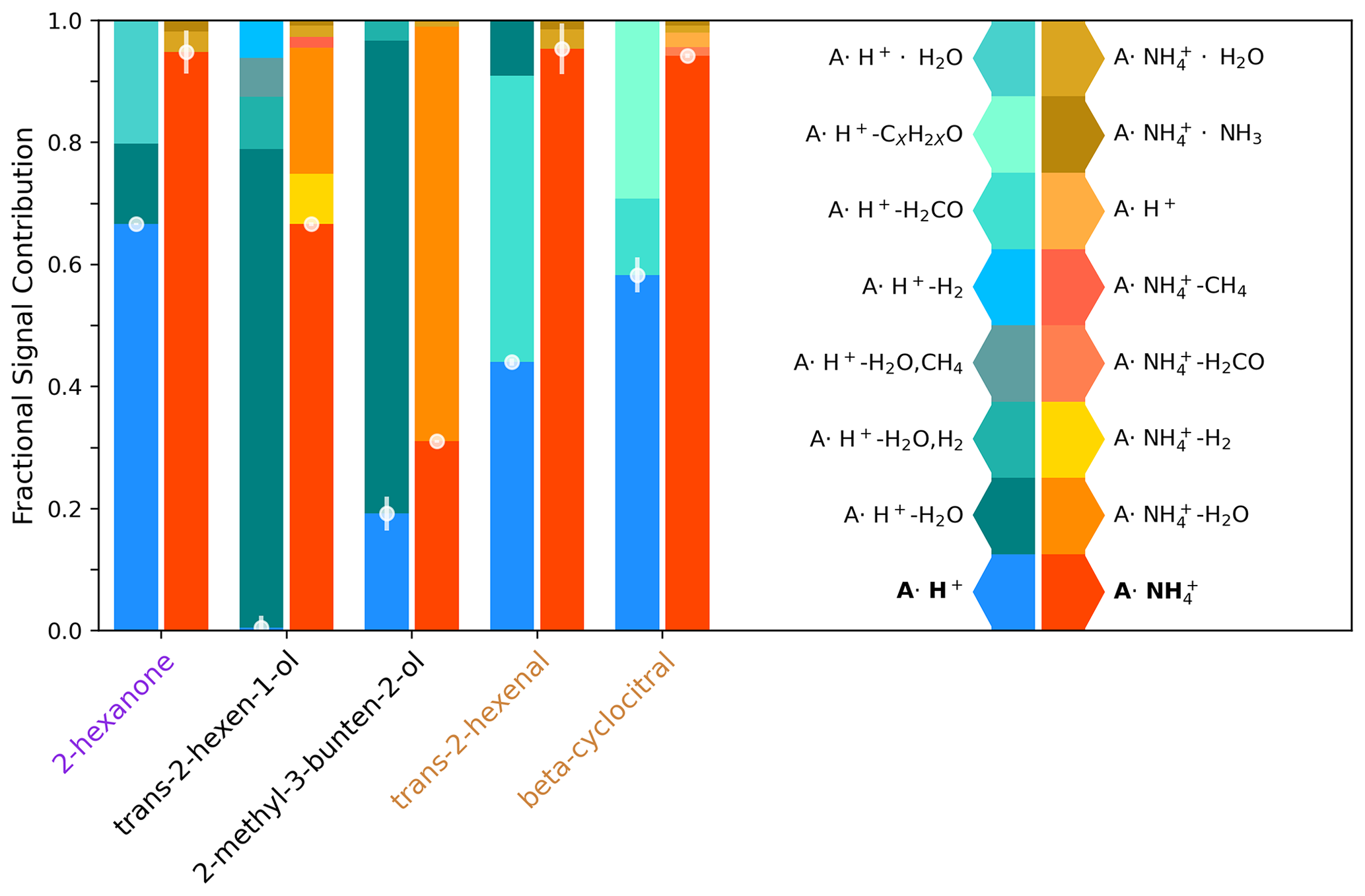
Figure 3Molecular ion fraction and the contribution of various non-molecular ions for five analytes using H3O+ ionization (blue/left) and ionization (orange/right). The contribution of the molecular ion is highlighted with a scatter plot, and uncertainty bars are the standard deviation across 30 mass spectra at 2 Hz. Data represented in this figure are shown in Table S6.
3.2 Sensitivities, detection limits, and fragmentation
Direct calibrations demonstrate the selectivity of relative to H3O+ ionization for a range of carbonyls, hydrocarbons, alcohols, and organic sulfur compounds (Fig. 2). We observe that H3O+ is capable of detecting nearly every compound in this set of analytes from standardized gas cylinders at the molecular ion, which demonstrates the utility of H3O+ as a general reagent ion that allows for detection of reduced and some oxidized species. In contrast, ionization is more selective toward oxygenates, including saturated and unsaturated ketones, unsaturated aldehydes, and the multi-functional 1-methoxy-2-propanol. 's selectivity toward oxygenates demonstrates its utility for expanding the range of compounds detectable with a single instrument, as well as for supporting the identification of molecular ions and fragments detected simultaneously with H3O+. In addition, does not detect aromatics, small alkenes, and reduced sulfur compounds that H3O+ detects well, demonstrating the complementary nature of these reagent ions. While Fig. 2 suggests that H3O+ detects dimethyl sulfoxide (DMSO), an oxidation product of dimethyl sulfide (Barnes et al., 2006), with a detection limit (DL) of 16 pptv, this DL is optimistic because DMSO peak separation is hindered by isobaric ions of protonated benzene and a protonated water cluster of acetic acid. We therefore expect that DMSO cannot be detected with H3O+ in the Vocus-CI-ToFMS at concentrations relevant to the marine boundary layer (i.e., <100 pptv, Putaud et al., 1999; Sciare et al., 2000; Legrand et al., 2001; and Nowak et al., 2001).
For the compounds detected with both ionization modes, sensitivities and detection limits for H3O+ and are in the same order of magnitude (Figs. 2, A3). detects the subset of ketones and the unsaturated aldehydes shown in Fig. 2 with a lower or similar DL to H3O+. 1-methoxy-2-propanol, trans-3-hexenol, and D5-siloxane suggest that has a greater ability than H3O+ to detect oxygenated and functionalized compounds, but this is not broadly apparent across the families of compounds we calibrated directly (Fig. 2). This likely arises because the analytes shown in Fig. 2 are limited to compounds amendable to gas cylinder calibration and are therefore biased toward VOCs, SVOCs, and IVOCs with minimal oxygenation and relatively high volatility (i.e., C* > 9×104 µg m−3; Table S4). Despite the compromises in the fIMR temperature made to allow for the switching system (Sect. 3.1) the sensitivities for ionization reported here are similar to the sensitivities reported in recent literature (Khare et al., 2022; Xu et al., 2022) (Table S5).
Compared to H3O+ ionization, ionization reduces molecular ion fragmentation for functionalized compounds (Fig. 3). We use a molecular ion fraction (Fig. 3; ratio of the molecular ion signal to the total signal from the molecular ion, fragments, and clusters) to analyze the contributions of molecular ions and mass spectral fragments from both and H3O+ ionization for a series of analytes complementary to those calibrated with standardized gas cylinders (Fig. 2). Alcohols fragment substantially using both reagent ions, with H3O+ and trans-2-hexenol fragments almost completely away from the molecular ion resulting in a near-zero molecular ion fraction. This is consistent with the negligible sensitivity to the similarly structured trans-3-hexenol molecular ion (Fig. 2) with H3O+ owing to fragmentation (e.g., Pagonis et al., 2019). In contrast, ionization detects trans-2-hexenol with a molecular ion fraction of 0.67. For the tertiary alcohol, 2-methyl-3-buten-2-ol (2,3,2-MBO), we observe substantial fragmentation with both ionization modes but a higher molecular ion fraction under (i.e., 0.31 with and 0.19 with H3O+). The ketone and aldehydes sampled only fragmented substantially under H3O+ ionization, while retains the molecular structure, leading to a high molecular ion fraction. This is consistent with the lower detection limit for the majority of ketones we examined (with the exceptions of acetone and 2-octanone) and the aldehyde trans-2-hexenal (Fig. 2). Our observations suggest that reduced fragmentation has a larger impact on detection capability of the two reagent ions for more highly oxidized compounds with multiple functional groups. This is observed for 1-methoxy-2-propanol, which is readily detected with but not with H3O+; the detection of oxidized ROC is discussed further in Sect. 3.5. Overall, these observations demonstrate the high selectivity of ionization for oxygenates and the benefits of reduced fragmentation with . Coupling the detection of reduced ROC and organic sulfur from H3O+ with the detection of oxygenates from expands the fraction of atmospheric ROC that we can detect with a single instrument.

Figure 4Signal dependence on sample relative humidity for (a, orange) and H3O+ (b, blue). Measurements were made at relative humidities of 15 %, 30 %, 50 %, 70 %, and 85 %; points are offset from these values for visibility. Ethanol, benzene, m-xylene, and 1,2,4-trimethylbenzene are excluded for due to low signal. Acetonitrile and ethanol are omitted for H3O+ due to low transmission through the BSQ. Methyl ethyl ketone is omitted for H3O+ due to interference of the reagent ion cluster . The error bars represent propagated relative deviations in dry and humidified signals.
3.3 Impact of sample relative humidity
Previous studies have characterized the significant humidity dependence of sensitivity in various CIMS instruments to understand and correct for changing ambient humidity (e.g., Warneke et al., 2001; Gouw and Warneke, 2007; Kari et al., 2018; Zaytsev et al., 2019a). Humidity-driven changes in reagent ion chemistry, and therefore sensitivity, are generally small in the Vocus-CI-ToFMS due to the large flow of water vapor (i.e., 20 cm3 min−1 (STP)) injected into the ion source (Krechmer et al., 2018; Khare et al., 2022). Varying sample humidity with constant analyte concentration demonstrates low humidity dependence with both and H3O+ ionization across a range of reduced and oxygenated ROC (Fig. 4). We note an approximately 10 % increase in the sensitivity to nitriles and oxygenates, while alkene sensitivities remain unchanged up to 85 % RH. We also observe a slight (5 %–10 %) increase in sensitivity with humidity for oxygenated species with H3O+, while alkene sensitivities are less affected. The low humidity dependence of the Vocus-CI-ToFMS has been demonstrated previously for H3O+ for a variety of analytes (Krechmer et al., 2018; Kilgour et al., 2022; Li et al., 2024) and for a select number of small oxygenates, alkenes, and acetonitrile with (Khare et al., 2022; Xu et al., 2022). We demonstrate the low dependence of sensitivity on sample humidity with ionization under different instrumental conditions and for a selection of analytes including oxygenated alkenes and siloxanes (Fig. 4).
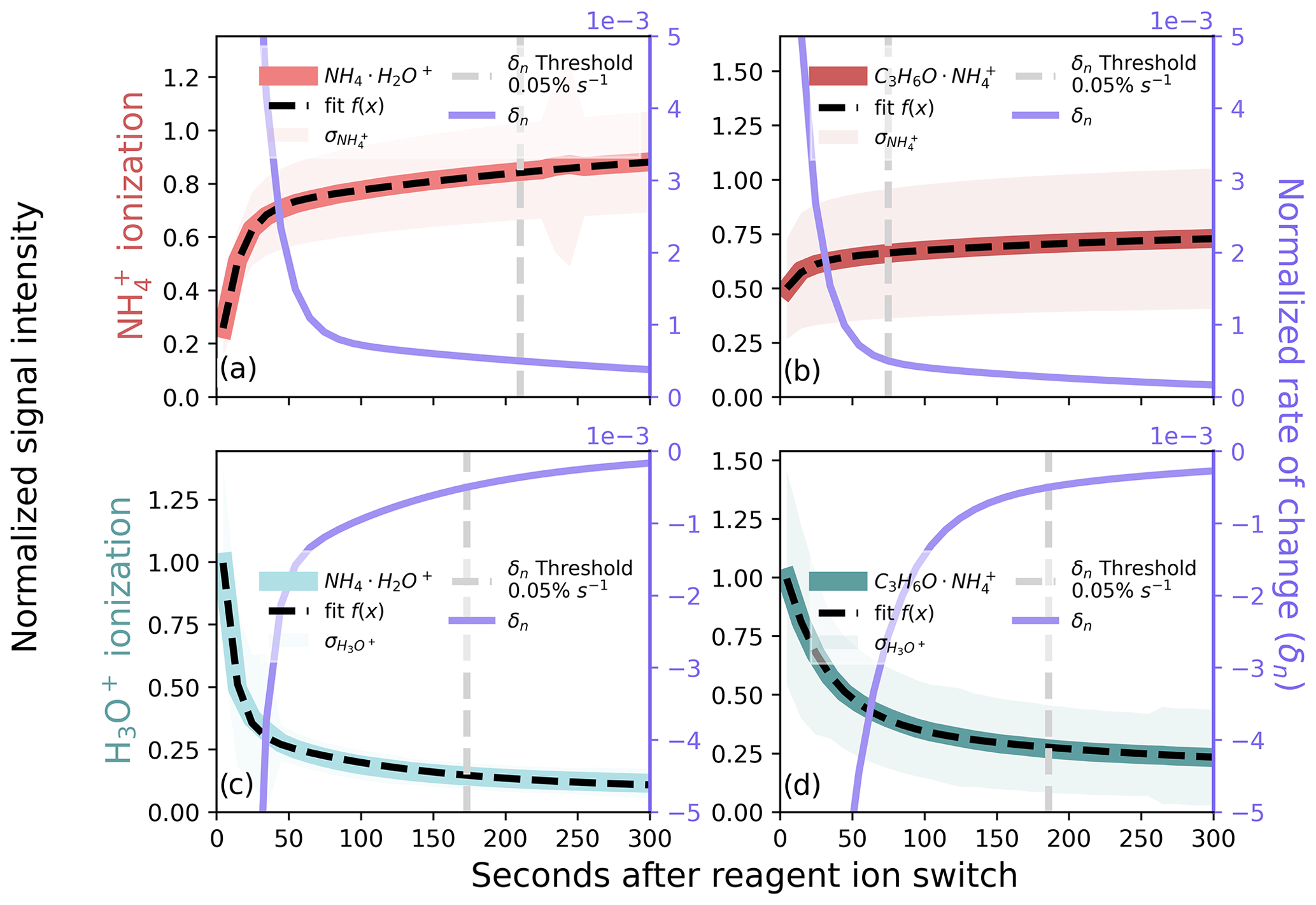
Figure 5Ion signal after a reagent ion switch for (a, b) and H3O+ (c, d) in the MEFO data, showing ions (a, c) and (b, d). We grouped ion signals by the time after a switch and normalized the mean of each group by the maximum, and normalized means were fit with a bi-exponential function. The derivative of the fit (δn) is displayed on the right axes (purple traces) and is used as a metric to filter reagent ion hysteresis. A summary of the amount of data removed as a function of the selected threshold for these ions is shown in Fig. S4.
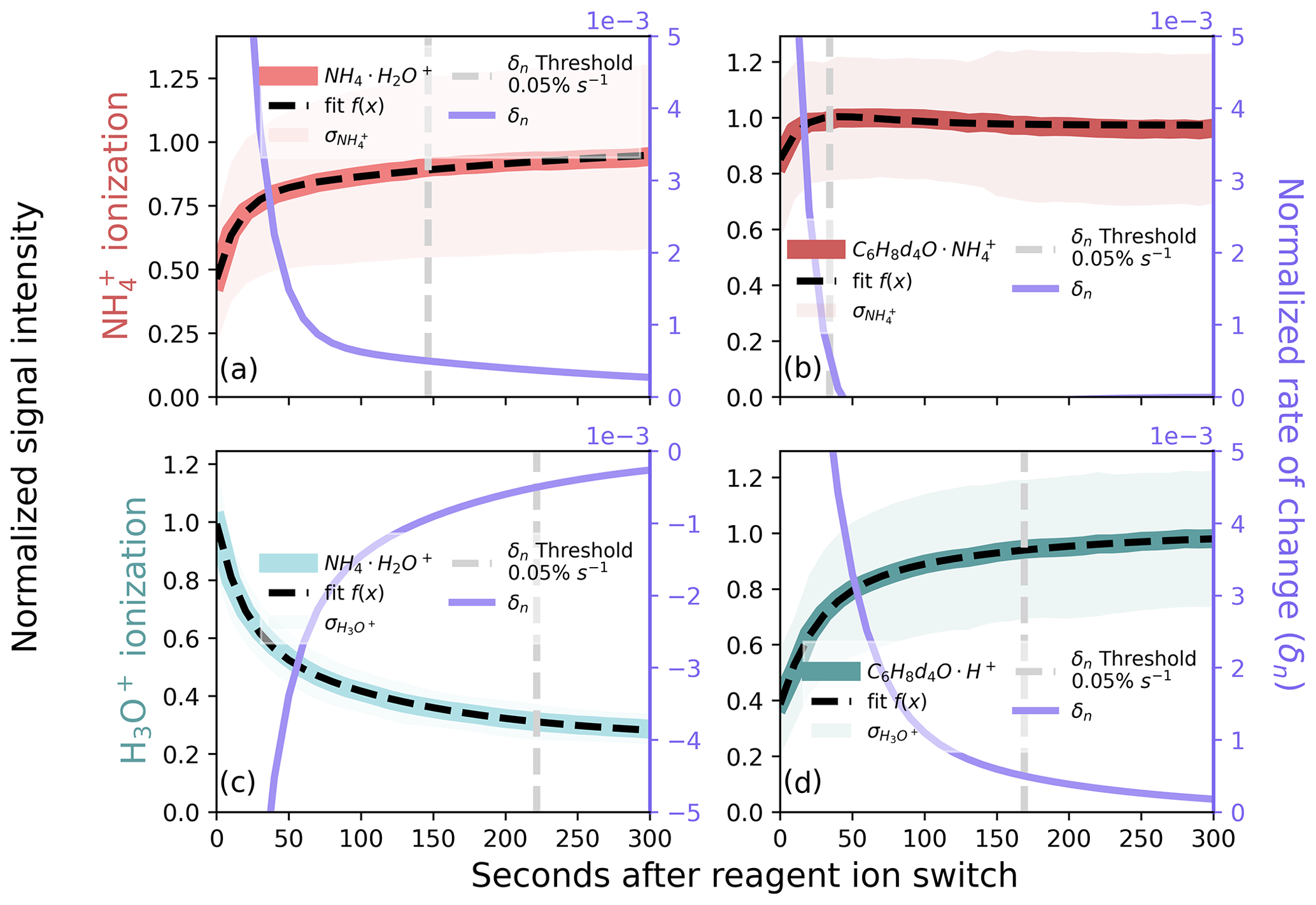
Figure 6Ion signal after a reagent ion switch for (a, b) and H3O+ (c, d) in the ARTofMELT data, showing ions (a, c) and (b) and (d) internal standard ions. We grouped ion signals by the time after a switch and normalized the mean of each group by the maximum, and normalized means were fit with a bi-exponential function. The derivative of the fit (δn) is displayed on the right axes (purple traces) and is used as a metric to filter reagent ion hysteresis. A summary of the amount of data removed as a function of the selected threshold for these ions is shown in Fig. S5.
3.4 Removal of reagent ion hysteresis from switching
Reagent ion chemistry does not stabilize immediately upon switching between and H3O+ ionization. This reagent ion switch requires adjustment of instrument conditions that impact ion chemistry; these include the reagents introduced into the ion source, the fIMR pressure and voltage gradient, and downstream ion optic voltages (Table S1). However, instrument conditions for each reagent ion (Sect. 3.1) are such that analyte detection through the alternate ionization pathway is possible with both and H3O+ ionization (e.g., Zaytsev et al., 2019a). This is in contrast to some other reagent ion pairs (e.g., and I−) where instrument conditions differ drastically, and so hysteresis is not observed (e.g., Brophy and Farmer, 2015). As a result, when and H3O+ are paired in a single instrument, we observe a distinct transitional period of reagent ion hysteresis following each reagent ion switch. The hysteretic period arises from (1) changes in ion transmission due to instrument conditions, such as ion optics and fIMR settings, which are fast (one to tens of seconds), and (2) changes in the reagent ion speciation due to the presence or absence of NH3(g), which is slower (tens of seconds to minutes). Periods of hysteresis must be characterized and removed to ensure stable and consistent measurements without drifting sensitivities over the course of each 15 min measurement period. We accomplish this by monitoring ion stability over a large number of repeated switches.
We compare the utility of three ion types as markers to quantify the timescale of reagent ion switching hysteresis: reagent ion signal, a persistent ambient adduct ion, and known and persistent adduct or proton transfer molecular ions from an internal standard infused in the sampling inlet. We selected ions primarily measured with ionization because the influence of NH3(g) is observed under both H3O+ and ionization modes, whereas the influence of H2O(g) reagent ions is not observed in mode (Fig. S3). This arises because NH3(g) has a higher proton affinity than H2O(g) (Hunter and Lias, 1998), which causes any NH3(g) present in the ion source and fIMR to readily form or at the expense of H3O+ formation.
3.4.1 Filtering hysteresis with reagent and persistent ambient ions
In the absence of a known and persistent signal from an internal standard to diagnose reagent ion hysteresis, we compare the use of (i.e., the prominent ammonium reagent ion signal) and (i.e., a ubiquitous analyte-ammonium molecular ion) under both ionization methods. Both and decay after switching to H3O+ ionization (Fig. 5c, d; NH3(g) depletion in the fIMR) and intensify after switching to ionization (Fig. 5a, b; NH3(g) accumulation in the fIMR). We grouped a total of 558 h of ambient /H3O+ 15 min reagent ion switching observations from MEFO (Sect. 2.5) by time after a reagent ion switch. We normalized the mean signal (in 10 s intervals starting at 5 s into a switch) to the maximum and fit the normalized data with a bi-exponential function (Fig. 5). The bi-exponential function describes changes in both instrument conditions (fast) and equilibration of NH3(g) in the ion source and fIMR that drives reagent ion chemical speciation (slow). We use the derivative of this decay function (δn) to quantify a normalized rate of change in the ion signal as a function of the time after a switch (purple lines in Fig. 5a–d). We use δn to set a threshold for filtering hysteresis, removing data before δn reaches the set threshold. For all ions in Fig. 5, δn changes rapidly in the first ∼ 100 s after a reagent ion switch and slowly approaches but does not reach zero on the measurement timescale (900 s) (Fig. S4), likely due to the timescale for complete NH3(g) equilibration with instrument surfaces.
Monitoring δn has the benefit of being directly related to the abundance of reagent ion; however, both the decay of in H3O+ ionization mode and its initial increase in ionization mode are driven largely by changes in the BSQ mass range. We are able to avoid the impacts of changing BSQ mass range by monitoring an ion with higher . Additionally, using an analyte for filtering reagent ion hysteresis means that we are using a direct measurement of the formation of analyte ions for ionization and a direct measurement of contamination from other reagent ion chemistry with H3O+ ionization. Therefore, in the absence of an internal standard (Sect. 3.4.2), we use the persistent ambient ion to monitor hysteresis. This approach brings two major complications: (1) variable contributions of isomers with potentially disparate sensitivities (e.g., acetone and propionaldehyde) and (2) potentially variable ambient concentrations. A switch-by-switch analysis of hysteresis from MEFO (available as Fig. S6) shows that the 0.05 % s−1 δn cutoffs for calculated in Fig. 5b and d do not capture the majority of the switch-by-switch cutoffs (37 % for and 39 % for H3O+). Therefore, if a persistent ambient ion is used to diagnose hysteresis timescales, this should be done on a switch-by-switch basis. This variability may be associated with ambient variations in the signal which can be avoided by applying our method described in Fig. 5 to a persistent and known signal from an internal standard (Sect. 3.4.2).
The choice of δn threshold represents a compromise between ion chemistry stability and data loss. For both reagent and analyte ions, the amount of data removed becomes very sensitive to a small decrease in the δn threshold below ∼0.05 % s−1 (Fig. S4). During the deployment in MEFO, a 0.05 % s−1 threshold applied to results in the loss of ∼185 s (Fig. 5d) and ∼75 s (Fig. 5b) of data per switch with H3O+ and ionization, respectively. Optimizing ion chemistry stability while preserving data coverage results in the loss of ∼260 s (75 s for and 185 s for H3O+) of data on a reagent switching full cycle (1800 s), corresponding to ∼86 % data retention for 15 min switching intervals.
3.4.2 Filtering hysteresis with reagent and internal standard ions
When an internal standard signal is available, as in the ARTofMELT expedition (Sect. 2.6), reagent ion hysteresis can be more reliably monitored using known unique and persistent molecular ions. We applied the δn thresholding method to a 2-week period from the ARTofMELT data set (from 17 to 31 May 2023). We quantify the timescale of reagent ion hysteresis by monitoring the internal standard signal of 2-hexanone-d4 as with ionization (Fig. 6b) and as with H3O+ ionization (Fig. 6d). For direct comparison to Sect. 3.4.1, we also use the reagent ion under both (Fig. 6a) and H3O+ (Fig. 6c). Notably, in this marine environment the ambient signal is highly variable, precluding its use for filtering reagent ion hysteresis (Fig. S7), further motivating the use of an internal standard to diagnose reagent ion hysteresis. A 0.05 % s−1 threshold applied to 2-hexanone-d4 results in the loss of 34 s from ionization after a switch (Fig. 6b) and 168 s from H3O+ ionization (Fig. 6d). This results in ∼19 % of H3O+ data being removed and ∼ 4 % of ionization from 15 min switching, or ∼89 % total data retention over a full (1800 s) switching cycle. It is worth noting that under both ionization modes, the hysteresis timescale for the is longer (Figs. 6a, c, S8) than the 2-hexanone-d4 internal standard ion. This suggests that a conservative approach to monitoring ion chemistry could be to use the reagent ions for establishing hysteresis timescales. However, the impacts of the BSQ on reagent ions raise concerns of how representative the reagent ion signal is of true composition in the fIMR (Krechmer et al., 2018; Khare et al., 2022; Xu et al., 2022). A switch-by-switch analysis of variation in the hysteresis timescale (available as Fig. S8) demonstrates that the cutoffs calculated in Fig. 6 for 2-hexanone-d4 capture the majority of variability in the hysteresis timescale for independent switches (75 % for and 72 % for H3O+).
Our results using an internal standard for reagent ion hysteresis filtering are qualitatively similar to the analysis above (Sect. 3.4.1) using a persistent ambient ion, though fewer data are removed from ionization. Rather than resulting from use of an ambient or internal standard ion, this difference in hysteresis time is likely the result of having more similar BSQ settings between the two ionization modes during the ARTofMELT campaign (Table S1). The smaller change in BSQ settings results in a faster change in instrument conditions impacting ion transmission. The approach we describe here can be applied easily to other instruments and at different instrument conditions (e.g., fIMR temperature and pressure) to best balance the need for both measurement stability and data coverage. While the use of an internal standard signal is ideal for diagnosing reagent ion switching hysteresis, the choice of both product molecular ions and rate-of-change threshold must be optimized for each application and sampling environment. Overall, our results are qualitatively similar to hysteresis timescales suggested by Zaytsev et al. (2019a) for and H3O+ switching in an Ionicon PTR3: ∼120 s and ∼60 s of data removed after switching to H3O+ and ionization, respectively.
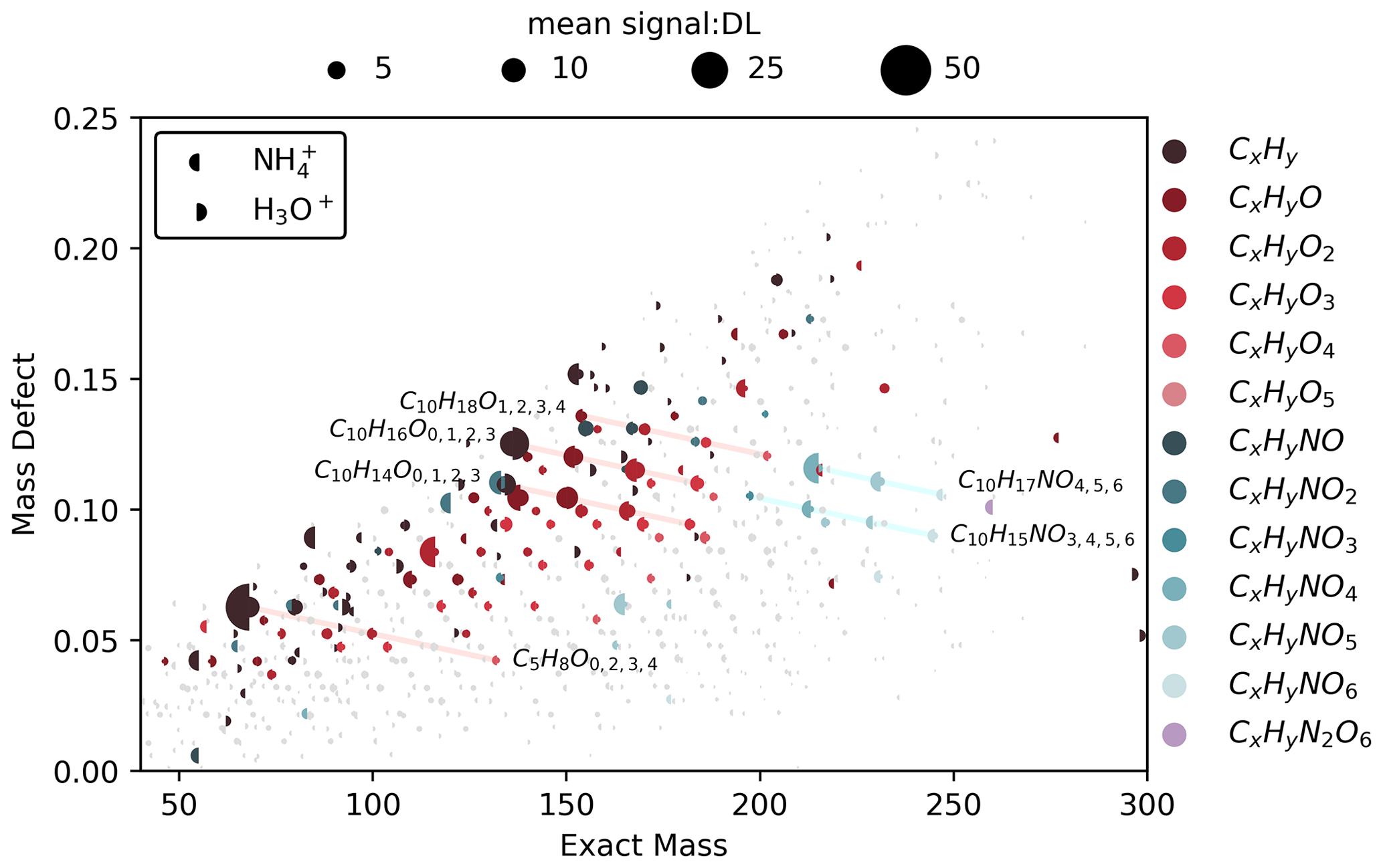
Figure 7Campaign mean mass defect plots for and H3O+ ionization from deployment in MEFO. The mass spectrum is displayed as a left half circle and the H3O+ is displayed as a right half circle. Points are sized by the average signal across the campaign divided by the detection limit signal (DL: 3σ of campaign zero air background). The reagent ion masses have been removed from the ion molecular mass. The top 100 ions in terms of signal-to-DL ratio for both reagent ions have been colored according to their molecular formulae with periods of reagent ion hysteresis removed (Sect. 3.4). A total of 725 ions are shown, and selected ions and their signal-to-DL ratios are displayed in a bar chart format in Fig. S9.
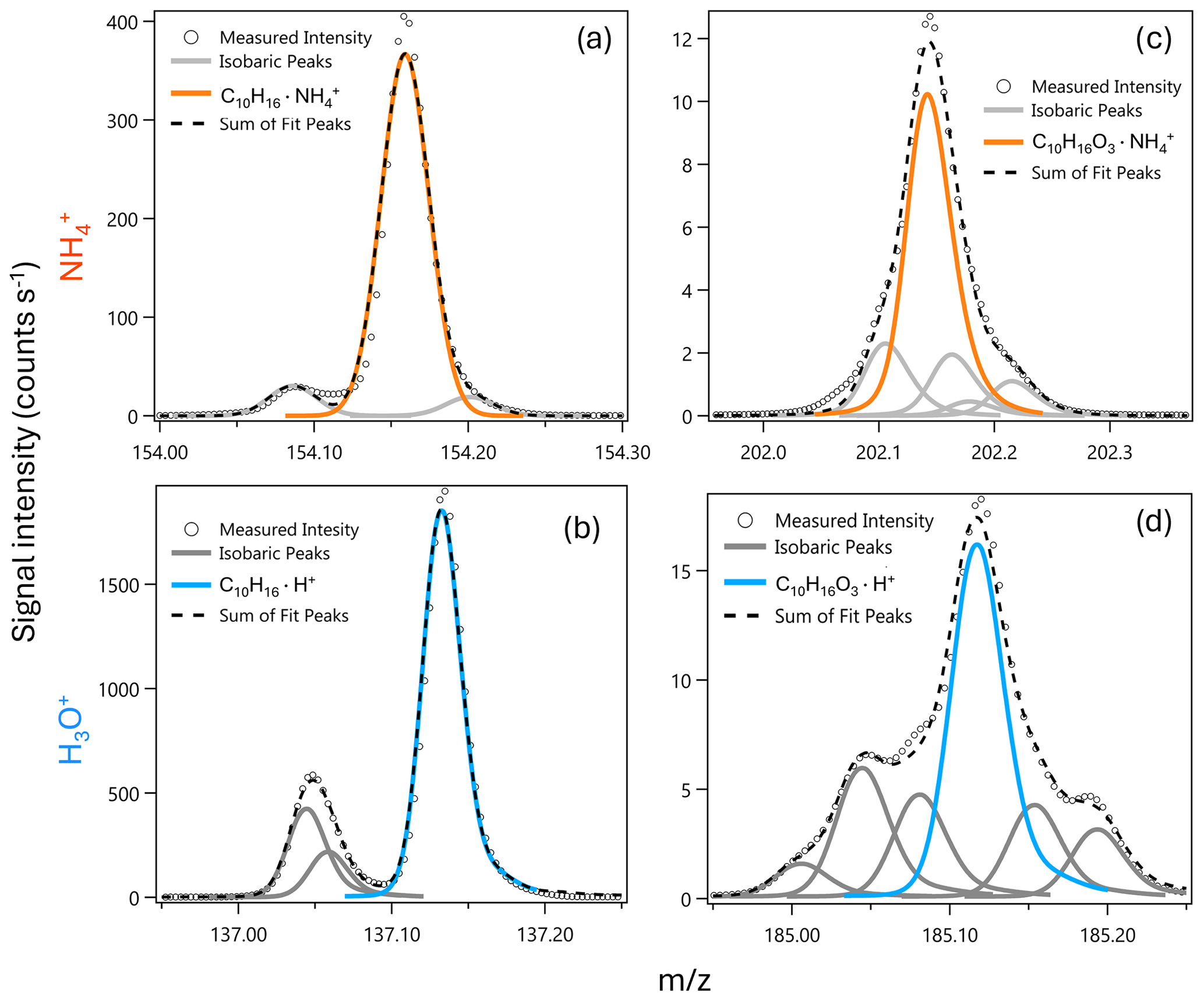
Figure 8Selected campaign average high-resolution mass spectra from MEFO for (a, b) monoterpene (C10H16) and (c, d) monoterpene oxygenate (C10H16O3) molecular ions detected with (a, c, orange) and H3O+ (b, d, blue).
3.5 Reagent ion comparison from ambient measurements in MEFO
We compare the capabilities of and H3O+ reagent ions in a single instrument using ambient observations from MEFO (Sect. 2.5). Switching between H3O+ and on a 15 min timescale over the 21 d deployment allows us to directly compare the two reagent ions in a predominantly biogenic environment (Hunter et al., 2017; Vermeuel et al., 2023a; Riches et al., 2024; Link et al., 2024). A single-instrument approach avoids inlet and instrument design influences on detection that would otherwise complicate a direct reagent ion comparison (e.g., Riva et al., 2019). Previously Zaytsev et al. (2019a) used a switching Ionicon PTR3 with a modified helical tripole reaction chamber to measure products from the ⋅OH-initiated oxidation of 3-methylcatechol. This chamber study demonstrated the sets of compounds detected by each reagent ion and concluded that is able to detect larger, more functionalized molecules, while H3O+ is able to detect smaller organic molecules (Zaytsev et al., 2019a). To facilitate a direct and quantitative reagent ion comparison, we use the ratio of campaign average ambient signal to detection limit signal (i.e., 3σ of the background) for each ion as a measure of the signal-to-noise ratio (Fig. 7). With the assumption that ambient concentrations measured with each reagent ion over the campaign mean are equivalent, a higher signal-to-DL ratio also implies a lower detection limit. This analysis allows us to evaluate the relative capability of each reagent ion without direct calibrations for multifunctional biogenic organic compounds (e.g., Hunter et al., 2017; Vermeuel et al., 2023a; Link et al., 2024).
ionization detects oxygen-containing species with a higher signal-to-noise ratio than H3O+. At MEFO, four series of CxHyOz ions dominate our mass spectrum (highlighted in Fig. 7) – C10H16On, C10H14On, C10H18On, and C5H8On – which represent a mixture of biogenic terpenoid compounds and their early generation oxidation products. The C10H16On and C10H14On series suggest a mixture of primary emissions, such as citral (C10H16O; C* = 1×106 µg m−3) and thymol or carvone (C10H14O; C* = 1–9×105 µg m−3) (McKinney et al., 2011; Kaser et al., 2013; Vermeuel et al., 2023a), and oxidation products of other terpenoids. H3O+ detects the reduced C10H14 species with higher signal-to-noise ratio compared to , with signal-to-DL ratios of 10.5 and 6.00, respectively. Similarly, H3O+ detects C10H16 with a signal-to-DL ratio of 23.5, compared to 14.3 for . Following this C10H16On series, the n=1 ion is detected with a signal-to-DL ratio of 6.31 with H3O+ and 10.8 with . The tendency toward increased signal-to-noise ratio with oxygenation for ionization continues in the C10H14,16On series up to C10H14,16O3 (with C* between 4×102 and 9×103 µg m−3, Table S7). The C10H18On series lacks a C10H18 ion, and the distribution in the x–y scatter between the C10H16 and C10H18O peaks is bimodal (Fig. S10), which suggests multiple paths to form C10H18O ions. These paths are likely (1) primary emissions of C10H18O terpenoid compounds with similar emission profiles to monoterpenes and (2) water clusters formed with monoterpenes ( and ). The C5H8On series represents a combination of fragments, primary emissions, and oxidation products. The C5H8 ion is likely a mixture of isoprene and fragments from larger oxygenates and 2,3,2-MBO with both reagent ions (e.g., Kilgour et al., 2024). Within the C5H8On series, the C5H8 signal-to-DL ratio is larger than expected with ionization; we suspect that this is due partly to fragmentation of other compounds into the C5H8 mass. The sensitivity to isoprene is very low; however, 2,3,2-MBO fragments substantially into C5H8 through dehydration of the tertiary alcohol group (Fig. 3). Fragmentation into C5H8 combined with a low background (Figs. A3, S11) leads to a very large signal-to-DL ratio (Fig. 7). The C5H8O3 and C5H8O4 peaks are likely oxidation products of isoprene and 2,3,2-MBO, while C5H8O2 is likely an isoprene oxidation product (Saunders et al., 2003; Jenkin et al., 2015).
can detect organic nitrates that easily fragment with H3O+ (Aoki et al., 2007; Duncianu et al., 2017) and so often go undetected in ambient measurements with H3O+ ionization (Fig. 7). Organic nitrates ionized with H3O+ fragment to form nitric acid (HNO3) or nitronium ions (), where the loss of HNO3 results in fragmentation into the masses for other oxygenates (Aoki et al., 2007). The two predominant series of organic nitrate ions (C10H15NOn and C10H17NOn, with C* down to tens of µg m−3; Table S7) are generally detected with a higher signal-to-DL ratio with ionization compared to H3O+ ionization, if the ion is detected with H3O+ at all (Fig. 7). The exception is C10H15NO3, which is detected at a higher signal-to-DL ratio (2.13) with H3O+ ionization compared to (0.774); though this ion may arise from dehydration of hydroxy nitrates (i.e., C10H17NO4-H2O), and H3O+ is fragmenting larger organic nitrates into the C10H15NO3 ion. The nitrates we observe are potentially a mixture of carbonyl, hydroxy, and peroxy nitrates derived from the oxidation of monoterpenes (C10H16) and potentially other terpenoid () precursors (Table S8, Fry et al., 2013; Jenkin et al., 2015; Faxon et al., 2018; Bates et al., 2022). Additionally, C5H11NO5 and C5H9NO5 are detected using both reagent ions but both with a higher signal-to-DL ratio with . C5H11NO5 is likely a nitrate from the oxidation 2,3,2-MBO, while both 2,3,2-MBO and isoprene could form the C5H9NO5 ion (Link et al., 2024).
ionization's ability to detect oxygenated compounds with a higher signal-to-noise ratio and lower detection limits than H3O+ arises from two main factors. First, is a softer ionization method compared to H3O+, resulting in less molecular ion fragmentation (Sect. 3.2). This is evident in our ambient data for the known H3O+ monoterpene fragment, , compared to the analogous fragment with ionization, . The fragment is present in ionization mode at a 1 : 10 fragment-to-molecular-ion ratio, compared to a 1 : 1 ratio with H3O+ under our fIMR conditions (Fig. S10). Second, has higher selectivity toward oxygenates compared to H3O+ (Sect. 3.2). Our ambient reagent ion switching observations further demonstrate this selectivity (Figs. 8, A3). The monoterpenes (C10H16) are easily distinguished from isobaric ions with both H3O+ and , but oxygenates (C10H16O3) have multiple isobaric interferences with H3O+ (Fig. 8). Both higher selectivity and reduced fragmentation contribute to fewer isobaric ions with . While our observations demonstrate the utility of for detecting oxidized species that H3O+ ionization struggles to detect (e.g., Yuan et al., 2017; Riva et al., 2019; Pagonis et al., 2019; Coggon et al., 2024), the extent of fragmentation for specific compounds is difficult to diagnose in ambient and complex laboratory mass spectra. Overall, our ambient reagent ion comparison demonstrates quantitatively that is complementary to H3O+, and together these two reagent ions allow improved detection and identification of a range of biogenic reactive organic carbon compounds and their early generation oxidation products.
To expand the range of ROC detectable with a single chemical ionization instrument, we present an approach to combine two positive reagent ions, and H3O+, in a Vocus-CI-ToFMS. To accommodate the need for a constant ion–molecule reactor temperature during switching, we apply an space scanning approach to select fIMR conditions compatible with both reagent ions. We characterize the ability of and H3O+ to detect a range of reduced and oxygenated VOCs, SVOCs, and IVOCs through analysis of laboratory standards and find that H3O+ detects reduced species well and fragments functionalized oxygenates away from the molecular ion, while retains the molecular ion and allows for improved detection of oxygenates. We find that fragmentation generally correlated with , while sensitivity is impacted by a combination of ion transmission, competing ionization pathways, and molecular ion fragmentation. To diagnose and quantify the timescales for reagent ion switching hysteresis, we compare the use of three ion types: reagent ions, a persistent ambient adduct ion, and adduct or proton transfer molecular ions from an internal standard infused in the sampling inlet. Reagent ion signal variability at each switch is driven largely by changes in ion transmission, so it is less representative of ion chemistry, while monitoring a product ion is more directly related to ionization reactions taking place in the fIMR. An internal standard signal provides the ideal means to monitor reagent ion hysteresis with a known and persistent product ion; however, persistent ambient ions and internal standard product ions can produce similar rates of data retention (∼86 %–89 % data retention across a full 1800 s switching cycle with a 0.05 % s−1 rate-of-change threshold). We deploy our reagent ion switching Vocus-CI-ToFMS during a 3-week period at a rural pine forest (Manitou Experimental Forest Observatory) to facilitate a direct and quantitative reagent ion comparison. Our ambient observations demonstrate that detects oxygenated ROC with a higher signal-to-noise ratio and lower DL, including organic nitrates that H3O+ does not detect, while H3O+ detects reduced species that are undetectable with . reagent ion switching takes advantage of the complementary nature of the two reagent ions to expand the range of ROC detectable with a single instrument.
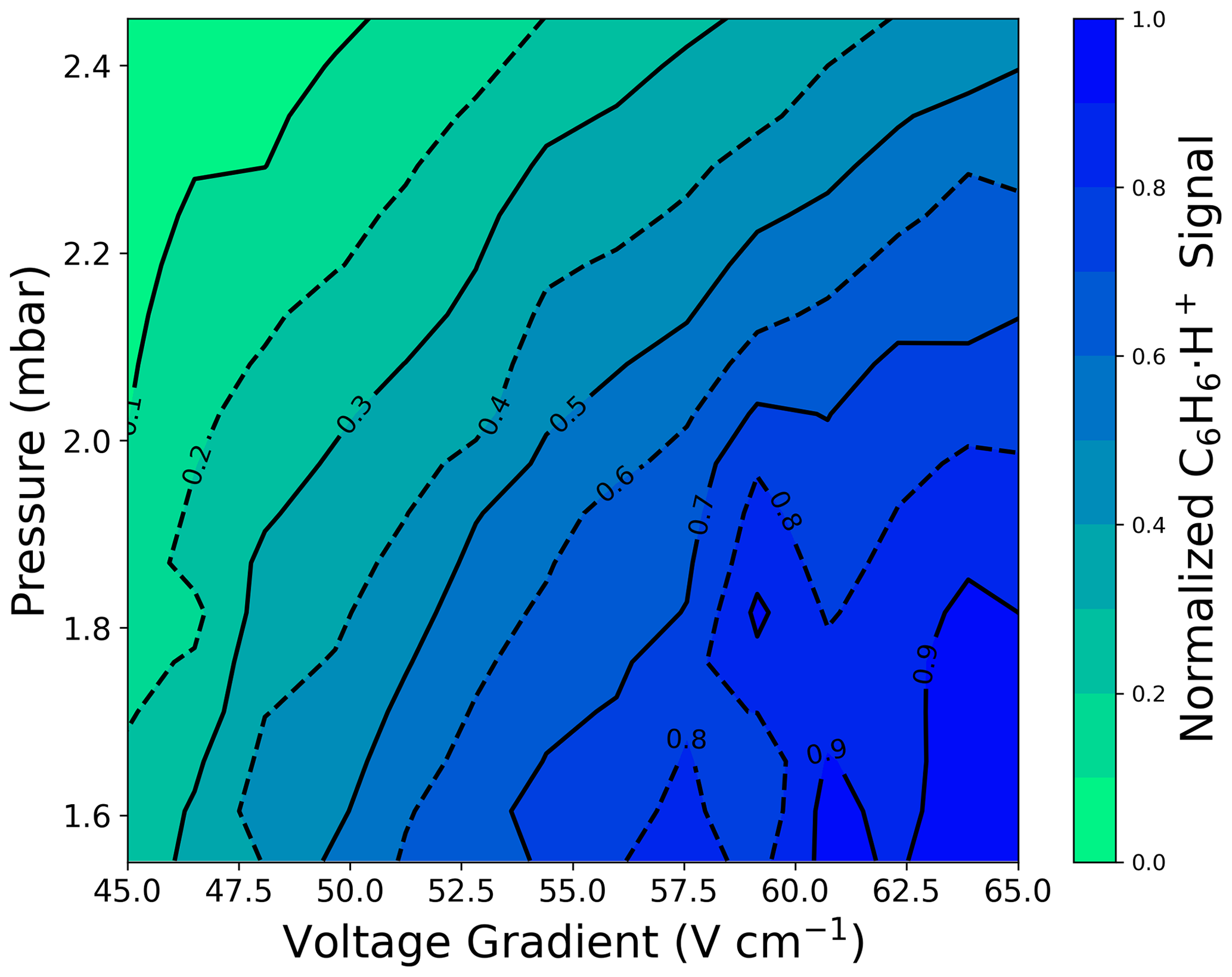
Figure A1Normalized signal intensity for benzene measured with H3O+ () from pressure–voltage-gradient scans.
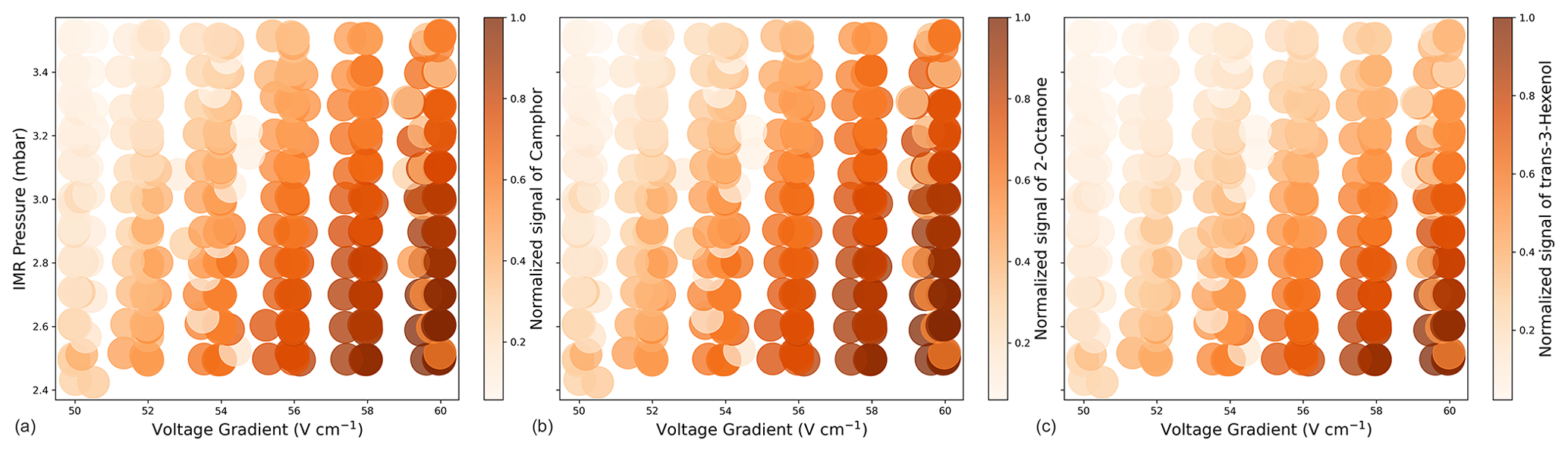
Figure A2Normalized signal intensity for camphor (a), 2-octanone (b), and trans-3-hexenol (c) measured with (, , and , respectively).
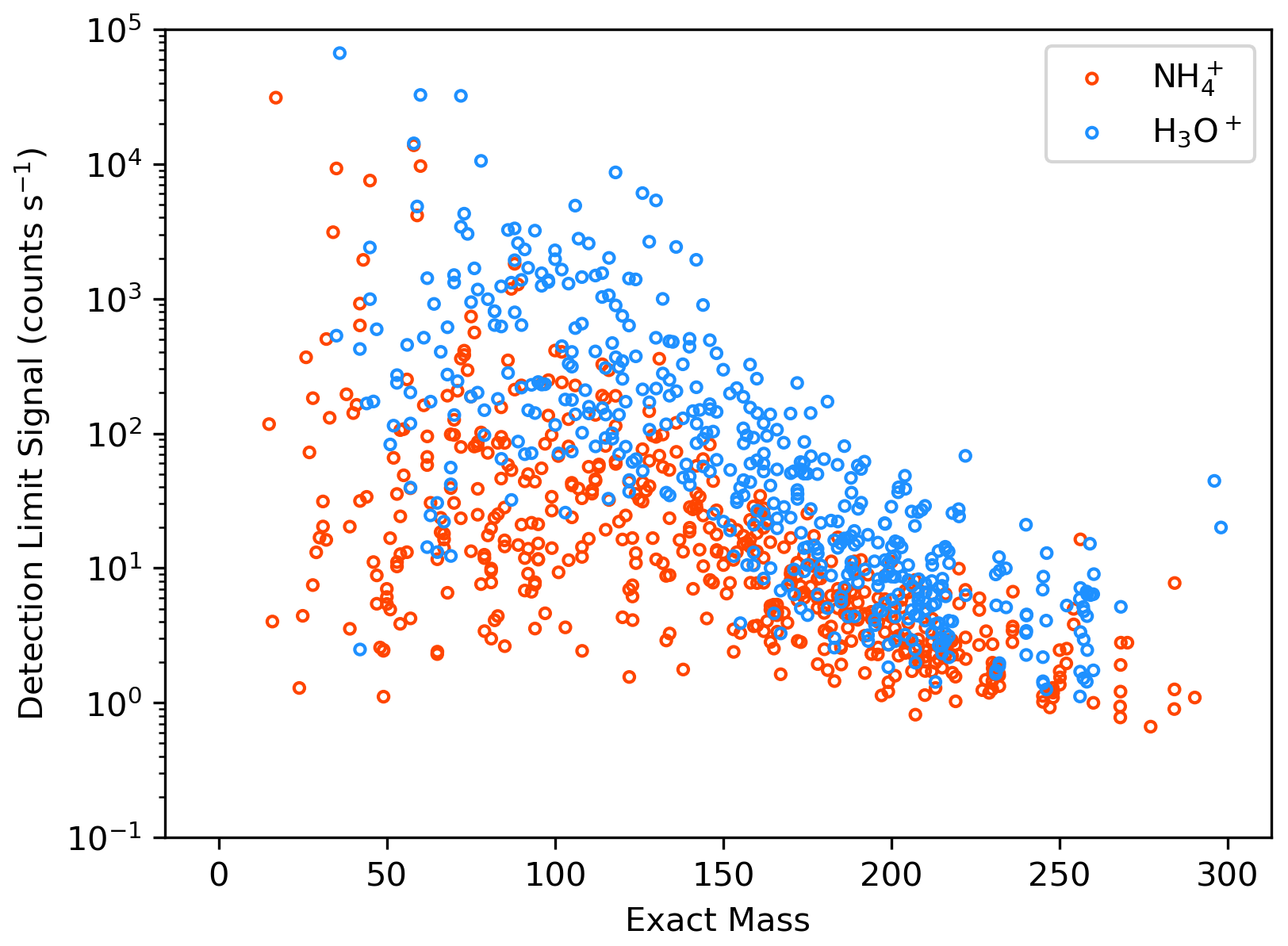
Figure A3Mass-dependent campaign average detection limit signal for ions detected with (orange) and H3O+ (blue). The reagent ion masses have been removed from ion exact masses.
Data and Python code required to regenerate figures are available at https://doi.org/10.7910/DVN/FL0CZM (Zang and Willis, 2024).
The supplement related to this article is available online at: https://doi.org/10.5194/amt-18-17-2025-supplement.
CLZ and MDW designed research. CLZ collected and analyzed field and laboratory data, with significant input from MDW. CLZ and MDW wrote the manuscript.
The contact author has declared that neither of the authors has any competing interests.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
We thank the Flux Closure Study (FluCS) team with researchers from Colorado State University (CSU), University of Minnesota Twin Cities (UM), and Indiana University Bloomington (IU), including Sara Williams (CSU), Mj Riches (CSU), Matson Pothier (CSU), Michael Link (CSU), Lauren Garofalo (CSU), Delphine Farmer (CSU), Michael Vermeuel (UM), Dylan Millet (UM), Emily Reidy (IU), Paige Price (IU), Brandon Bottorff (IU), and Phillip Stevens (IU), for their support with field observations at MEFO. We also acknowledge Tucker Melles (CSU) for supporting field observations at MEFO and ARTofMELT. We thank the National Center for Atmospheric Research (NCAR) for MEFO field site maintenance, as well as Paula Fornwalt and Steve Alton from the United States Forest Service (USFS) for support during field observations. This work is part of the ARTofMELT (Atmospheric rivers and the onset of ice melt) project. The ARTofMELT expedition was supported and organized by the Swedish Polar Research Secretariat (SPRS) on the Swedish research icebreaker Oden in spring 2023 under the SWEDARCTIC program. Support also came from the Swedish Council for Research Infrastructures (grant no. 2021-00153) and the Knut and Alice Wallenberg Foundation (grant no. 2016-0024). The authors are grateful to the co-chief scientists Michael Tjernström and Paul Zieger, to the SPRS coordinator Åsa Lindgren and the SPRS support team, and to captain Mattias Petersson and the crew on I/B Oden.
This research has been supported by the US National Science Foundation Directorate for Geosciences (grant no. AGS-2211153) and Colorado State University.
This paper was edited by Bin Yuan and reviewed by three anonymous referees.
Adams, N., Babcock, L., Mostefaoui, T., and Kerns, M.: Selected ion flow tube study of NH association and of product switching reactions with a series of organic molecules, Int. J. Mass Spectrom., 223–224, 459–471, 2003. a, b, c
Agarwal, B., Gonzalez-Mendez, R., Lanza, M., Sulzer, P., Mark, T. D., Thomas, N., and Mayhew, C. A.: Sensitivity and selectivity of switchable reagent ion soft chemical ionization mass spectrometry for the detection of picric acid, J. Phys. Chem. A, 118, 8229–8236, https://doi.org/10.1021/jp5010192, 2014. a
Aljawhary, D., Lee, A. K. Y., and Abbatt, J. P. D.: High-resolution chemical ionization mass spectrometry (ToF-CIMS): application to study SOA composition and processing, Atmos. Meas. Tech., 6, 3211–3224, https://doi.org/10.5194/amt-6-3211-2013, 2013. a
Aoki, N., Inomata, S., and Tanimoto, H.: Detection of C1–C5 alkyl nitrates by proton transfer reaction time-of-flight mass spectrometry, Int. J. Mass Spectrom., 263, 12–21, https://doi.org/10.1016/j.ijms.2006.11.018, 2007. a, b
Barnes, I., Hjorth, J., and Mihalopoulos, N.: Dimethyl Sulfide and Dimethyl Sulfoxide and Their Oxidation in the Atmosphere, Chem. Rev., 106, 940–975, https://doi.org/10.1021/cr020529+, 2006. a
Bates, K. H., Burke, G. J. P., Cope, J. D., and Nguyen, T. B.: Secondary organic aerosol and organic nitrogen yields from the nitrate radical (NO3) oxidation of alpha-pinene from various RO2 fates, Atmos. Chem. Phys., 22, 1467–1482, https://doi.org/10.5194/acp-22-1467-2022, 2022. a
Berndt, T., Scholz, W., Mentler, B., Fischer, L., Hoffmann, E. H., Tilgner, A., Hyttinen, N., Prisle, N. L., Hansel, A., and Herrmann, H.: Fast Peroxy Radical Isomerization and OH Recycling in the Reaction of OH Radicals with Dimethyl Sulfide, J. Phys. Chem. Lett., 10, 6478–6483, https://doi.org/10.1021/acs.jpclett.9b02567, 2019. a
Bianchi, F., Kurten, T., Riva, M., Mohr, C., Rissanen, M. P., Roldin, P., Berndt, T., Crounse, J. D., Wennberg, P. O., Mentel, T. F., Wildt, J., Junninen, H., Jokinen, T., Kulmala, M., Worsnop, D. R., Thornton, J. A., Donahue, N., Kjaergaard, H. G., and Ehn, M.: Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol, Chem. Rev., 119, 3472–3509, https://doi.org/10.1021/acs.chemrev.8b00395, 2019. a, b, c
Blomquist, B. W., Huebert, B. J., Fairall, C. W., and Faloona, I. C.: Determining the sea-air flux of dimethylsulfide by eddy correlation using mass spectrometry, Atmos. Meas. Tech., 3, 1–20, https://doi.org/10.5194/amt-3-1-2010, 2010. a
Brophy, P. and Farmer, D. K.: A switchable reagent ion high resolution time-of-flight chemical ionization mass spectrometer for real-time measurement of gas phase oxidized species: characterization from the 2013 southern oxidant and aerosol study, Atmos. Meas. Tech., 8, 2945–2959, https://doi.org/10.5194/amt-8-2945-2015, 2015. a, b, c
Burkart, J., Hodshire, A. L., Mungall, E. L., Pierce, J. R., Collins, D. B., Ladino, L. A., Lee, A. K. Y., Irish, V., Wentzell, J. J. B., Liggio, J., Papakyriakou, T., Murphy, J., and Abbatt, J.: Organic Condensation and Particle Growth to CCN Sizes in the Summertime Marine Arctic Is Driven by Materials More Semivolatile Than at Continental Sites, Geophys. Res. Lett., 44, 10,725–10,734, https://doi.org/10.1002/2017gl075671, 2017. a
Canaval, E., Hyttinen, N., Schmidbauer, B., Fischer, L., and Hansel, A.: NH Association and Proton Transfer Reactions With a Series of Organic Molecules, Front Chem., 7, 191, https://doi.org/10.3389/fchem.2019.00191, 2019. a, b, c, d
Claflin, M. S., Pagonis, D., Finewax, Z., Handschy, A. V., Day, D. A., Brown, W. L., Jayne, J. T., Worsnop, D. R., Jimenez, J. L., Ziemann, P. J., de Gouw, J., and Lerner, B. M.: An in situ gas chromatograph with automatic detector switching between PTR- and EI-TOF-MS: isomer-resolved measurements of indoor air, Atmos. Meas. Tech., 14, 133–152, https://doi.org/10.5194/amt-14-133-2021, 2021. a
Coggon, M. M., Gkatzelis, G. I., McDonald, B. C., Gilman, J. B., Schwantes, R. H., Abuhassan, N., Aikin, K. C., Arend, M. F., Berkoff, T. A., Brown, S. S., Campos, T. L., Dickerson, R. R., Gronoff, G., Hurley, J. F., Isaacman-VanWertz, G., Koss, A. R., Li, M., McKeen, S. A., Moshary, F., Peischl, J., Pospisilova, V., Ren, X., Wilson, A., Wu, Y., Trainer, M., and Warneke, C.: Volatile chemical product emissions enhance ozone and modulate urban chemistry, P. Natl. Acad. Sci. USA, 118, 32, https://doi.org/10.1073/pnas.2026653118, 2021. a
Coggon, M. M., Stockwell, C. E., Claflin, M. S., Pfannerstill, E. Y., Xu, L., Gilman, J. B., Marcantonio, J., Cao, C., Bates, K., Gkatzelis, G. I., Lamplugh, A., Katz, E. F., Arata, C., Apel, E. C., Hornbrook, R. S., Piel, F., Majluf, F., Blake, D. R., Wisthaler, A., Canagaratna, M., Lerner, B. M., Goldstein, A. H., Mak, J. E., and Warneke, C.: Identifying and correcting interferences to PTR-ToF-MS measurements of isoprene and other urban volatile organic compounds, Atmos. Meas. Tech., 17, 801–825, https://doi.org/10.5194/amt-17-801-2024, 2024. a, b, c, d
Croft, B., Martin, R. V., Leaitch, W. R., Burkart, J., Chang, R. Y.-W., Collins, D. B., Hayes, P. L., Hodshire, A. L., Huang, L., Kodros, J. K., Moravek, A., Mungall, E. L., Murphy, J. G., Sharma, S., Tremblay, S., Wentworth, G. R., Willis, M. D., Abbatt, J. P. D., and Pierce, J. R.: Arctic marine secondary organic aerosol contributes significantly to summertime particle size distributions in the Canadian Arctic Archipelago, Atmos. Chem. Phys., 19, 2787–2812, https://doi.org/10.5194/acp-19-2787-2019, 2019. a
Croft, B., Martin, R. V., Moore, R. H., Ziemba, L. D., Crosbie, E. C., Liu, H., Russell, L. M., Saliba, G., Wisthaler, A., Müller, M., Schiller, A., Galí, M., Chang, R. Y.-W., McDuffie, E. E., Bilsback, K. R., and Pierce, J. R.: Factors controlling marine aerosol size distributions and their climate effects over the northwest Atlantic Ocean region, Atmos. Chem. Phys., 21, 1889–1916, https://doi.org/10.5194/acp-21-1889-2021, 2021. a
Crounse, J. D., McKinney, K. A., Kwan, A. J., and Wennberg, P. O.: Measurement of gas-phase hydroperoxides by chemical ionization mass spectrometry, Anal. Chem., 78, 6726–6732, https://doi.org/10.1021/ac0604235, 2006. a, b
Crounse, J. D., Nielsen, L. B., Jorgensen, S., Kjaergaard, H. G., and Wennberg, P. O.: Autoxidation of Organic Compounds in the Atmosphere, J. Phys. Chem. Lett., 4, 3513–3520, https://doi.org/10.1021/jz4019207, 2013. a, b
Deming, B. L., Pagonis, D., Liu, X., Day, D. A., Talukdar, R., Krechmer, J. E., de Gouw, J. A., Jimenez, J. L., and Ziemann, P. J.: Measurements of delays of gas-phase compounds in a wide variety of tubing materials due to gas–wall interactions, Atmos. Meas. Tech., 12, 3453–3461, https://doi.org/10.5194/amt-12-3453-2019, 2019. a, b
Donahue, N. M., Epstein, S. A., Pandis, S. N., and Robinson, A. L.: A two-dimensional volatility basis set: 1. organic-aerosol mixing thermodynamics, Atmos. Chem. Phys., 11, 3303–3318, https://doi.org/10.5194/acp-11-3303-2011, 2011. a
Duncianu, M., David, M., Kartigueyane, S., Cirtog, M., Doussin, J.-F., and Picquet-Varrault, B.: Measurement of alkyl and multifunctional organic nitrates by proton-transfer-reaction mass spectrometry, Atmos. Meas. Tech., 10, 1445–1463, https://doi.org/10.5194/amt-10-1445-2017, 2017. a
Hunter, E. P. and Lias, S. G.: Proton Affinity Evaluation, in: NIST Chemistry WebBook, NIST Standard Refernce Database Number 69, edited by: Linstrom, P. J. and Mallard, W. G., National Institude of Standards and Technology, Gaithersburg MD, 20899, https://doi.org/10.18434/T4D303, 2023. a
Faxon, C., Hammes, J., Le Breton, M., Pathak, R. K., and Hallquist, M.: Characterization of organic nitrate constituents of secondary organic aerosol (SOA) from nitrate-radical-initiated oxidation of limonene using high-resolution chemical ionization mass spectrometry, Atmos. Chem. Phys., 18, 5467–5481, https://doi.org/10.5194/acp-18-5467-2018, 2018. a
Fry, J. L., Draper, D. C., Zarzana, K. J., Campuzano-Jost, P., Day, D. A., Jimenez, J. L., Brown, S. S., Cohen, R. C., Kaser, L., Hansel, A., Cappellin, L., Karl, T., Hodzic Roux, A., Turnipseed, A., Cantrell, C., Lefer, B. L., and Grossberg, N.: Observations of gas- and aerosol-phase organic nitrates at BEACHON-RoMBAS 2011, Atmos. Chem. Phys., 13, 8585–8605, https://doi.org/10.5194/acp-13-8585-2013, 2013. a
Goldstein, A. and Galbally, I. E.: Known and Unexplored Organic Constituents in the Earth’s Atmosphere, Environ. Sci. Technol., 1515–1520 pp., https://doi.org/10.1021/es072476p, 2007. a
Gouw, J. D. and Warneke, C.: Measurements of volatile organic compounds in the earth's atmosphere using proton‐transfer‐reaction mass spectrometry, Mass Spectrom. Rev., 26, 223–257, https://doi.org/10.1002/mas.20119, 2007. a, b, c
Hansel, A., Scholz, W., Mentler, B., Fischer, L., and Berndt, T.: Detection of RO2 radicals and other products from cyclohexene ozonolysis with NH and acetate chemical ionization mass spectrometry, Atmos. Environ., 186, 248–255, https://doi.org/10.1016/j.atmosenv.2018.04.023, 2018. a, b
He, X.-C., Shen, J., Iyer, S., Juuti, P., Zhang, J., Koirala, M., Kytökari, M. M., Worsnop, D. R., Rissanen, M., Kulmala, M., Maier, N. M., Mikkilä, J., Sipilä, M., and Kangasluoma, J.: Characterisation of gaseous iodine species detection using the multi-scheme chemical ionisation inlet 2 with bromide and nitrate chemical ionisation methods, Atmos. Meas. Tech., 16, 4461–4487, https://doi.org/10.5194/amt-16-4461-2023, 2023. a
Heald, C. L. and Kroll, J. H.: The fuel of atmospheric chemistry: Toward a complete description of reactive organic carbon, Sci. Adv., 6, eaay8967, https://doi.org/10.1126/sciadv.aay8967, 2020. a, b, c
Huey, L. G.: Measurement of trace atmospheric species by chemical ionization mass spectrometry: speciation of reactive nitrogen and future directions, Mass Spectrom. Rev., 26, 166–84, https://doi.org/10.1002/mas.20118, 2007. a, b
Hunter, E. and Lias, S.: Evaluated Gas Phase Basicities and Proton Affinities of Molecules: An Update, J. Phys. & Chem. Ref. Data (JPCRD), 27, 413–656, https://doi.org/10.1063/1.556018, 1998. a, b
Hunter, J. F., Day, D. A., Palm, B. B., Yatavelli, R. L. N., Chan, A. W. H., Kaser, L., Cappellin, L., Hayes, P. L., Cross, E. S., Carrasquillo, A. J., Campuzano-Jost, P., Stark, H., Zhao, Y., Hohaus, T., Smith, J., Hansel, A., Karl, T., Goldstein, A. H., Guenther, A., Worsnop, D., Thornton, J. A., Heald, C. L., Jimenez, J. L., and Kroll, J. H.: Comprehensive characterization of atmospheric organic carbon at a forested site, Nat. Geosci., 10, 748–753, https://doi.org/10.1038/ngeo3018, 2017. a, b, c, d, e
Jenkin, M. E., Young, J. C., and Rickard, A. R.: The MCM v3.3.1 degradation scheme for isoprene, Atmos. Chem. Phys., 15, 11433–11459, https://doi.org/10.5194/acp-15-11433-2015, 2015. a, b
Jensen, A. R., Koss, A. R., Hales, R. B., and de Gouw, J. A.: Measurements of volatile organic compounds in ambient air by gas-chromatography and real-time Vocus PTR-TOF-MS: calibrations, instrument background corrections, and introducing a PTR Data Toolkit, Atmos. Meas. Tech., 16, 5261–5285, https://doi.org/10.5194/amt-16-5261-2023, 2023. a
Jordan, A., Haidacher, S., Hanel, G., Hartungen, E., Herbig, J., Märk, L., Schottkowsky, R., Seehauser, H., Sulzer, P., and Märk, T. D.: An online ultra-high sensitivity Proton-transfer-reaction mass-spectrometer combined with switchable reagent ion capability (PTR+SRI−MS), Int. J. Mass Spectrom., 286, 32–38, https://doi.org/10.1016/j.ijms.2009.06.006, 2009. a
Kari, E., Miettinen, P., Yli-Pirilä, P., Virtanen, A., and Faiola, C. L.: PTR-ToF-MS product ion distributions and humidity-dependence of biogenic volatile organic compounds, Int. J. Mass Spectrom., 430, 87–97, https://doi.org/10.1016/j.ijms.2018.05.003, 2018. a
Kaser, L., Karl, T., Guenther, A., Graus, M., Schnitzhofer, R., Turnipseed, A., Fischer, L., Harley, P., Madronich, M., Gochis, D., Keutsch, F. N., and Hansel, A.: Undisturbed and disturbed above canopy ponderosa pine emissions: PTR-TOF-MS measurements and MEGAN 2.1 model results, Atmos. Chem. Phys., 13, 11935–11947, https://doi.org/10.5194/acp-13-11935-2013, 2013. a
Khare, P., Krechmer, J. E., Machesky, J. E., Hass-Mitchell, T., Cao, C., Wang, J., Majluf, F., Lopez-Hilfiker, F., Malek, S., Wang, W., Seltzer, K., Pye, H. O. T., Commane, R., McDonald, B. C., Toledo-Crow, R., Mak, J. E., and Gentner, D. R.: Ammonium adduct chemical ionization to investigate anthropogenic oxygenated gas-phase organic compounds in urban air, Atmos. Chem. Phys., 22, 14377–14399, https://doi.org/10.5194/acp-22-14377-2022, 2022. a, b, c, d, e, f, g
Kilgour, D. B., Novak, G. A., Sauer, J. S., Moore, A. N., Dinasquet, J., Amiri, S., Franklin, E. B., Mayer, K., Winter, M., Morris, C. K., Price, T., Malfatti, F., Crocker, D. R., Lee, C., Cappa, C. D., Goldstein, A. H., Prather, K. A., and Bertram, T. H.: Marine gas-phase sulfur emissions during an induced phytoplankton bloom, Atmos. Chem. Phys., 22, 1601–1613, https://doi.org/10.5194/acp-22-1601-2022, 2022. a, b
Kilgour, D. B., Novak, G. A., Claflin, M. S., Lerner, B. M., and Bertram, T. H.: Production of oxygenated volatile organic compounds from the ozonolysis of coastal seawater, Atmos. Chem. Phys., 24, 3729–3742, https://doi.org/10.5194/acp-24-3729-2024, 2024. a, b, c
Kim, M. J., Zoerb, M. C., Campbell, N. R., Zimmermann, K. J., Blomquist, B. W., Huebert, B. J., and Bertram, T. H.: Revisiting benzene cluster cations for the chemical ionization of dimethyl sulfide and select volatile organic compounds, Atmos. Meas. Tech., 9, 1473–1484, https://doi.org/10.5194/amt-9-1473-2016, 2016. a
Krechmer, J., Lopez-Hilfiker, F., Koss, A., Hutterli, M., Stoermer, C., Deming, B., Kimmel, J., Warneke, C., Holzinger, R., Jayne, J., Worsnop, D., Fuhrer, K., Gonin, M., and de Gouw, J.: Evaluation of a New Reagent-Ion Source and Focusing Ion–Molecule Reactor for Use in Proton-Transfer-Reaction Mass Spectrometry, Anal. Chem., 90, 12011–12018, https://doi.org/10.1021/acs.analchem.8b02641, 2018. a, b, c, d, e, f, g
Lee, B. H., Lopez-Hilfiker, F. D., Mohr, C., Kurtén, T., Worsnop, D. R., and Thornton, J. A.: An Iodide-Adduct High-Resolution Time-of-Flight Chemical-Ionization Mass Spectrometer: Application to Atmospheric Inorganic and Organic Compounds, Environ. Sci. Technol., 48, 6309–6317, https://doi.org/10.1021/es500362a, 2014. a, b
Lee, B. H., Lopez-Hilfiker, F. D., Veres, P. R., McDuffie, E. E., Fibiger, D. L., Sparks, T. L., Ebben, C. J., Green, J. R., Schroder, J. C., Campuzano-Jost, P., Iyer, S., D'Ambro, E. L., Schobesberger, S., Brown, S. S., Wooldridge, P. J., Cohen, R. C., Fiddler, M. N., Bililign, S., Jimenez, J. L., Kurtén, T., Weinheimer, A. J., Jaegle, L., and Thornton, J. A.: Flight Deployment of a High-Resolution Time-of-Flight Chemical Ionization Mass Spectrometer: Observations of Reactive Halogen and Nitrogen Oxide Species, J. Geophys. Res.-Atmos., 123, 7670–7686, https://doi.org/10.1029/2017jd028082, 2018. a, b
Legrand, M., Sciare, J., Jourdain, B., and Genthon, C.: Subdaily variations of atmospheric dimethylsulfide, dimethylsulfoxide, methanesulfonate, and non‐sea‐salt sulfate aerosols in the atmospheric boundary layer at Dumont d'Urville (coastal Antarctica) during summer, J. Geophys. Res.-Atmos., 106, 14409–14422, https://doi.org/10.1029/2000JD900840, 2001. a
Li, F., Huang, D. D., Tian, L., Yuan, B., Tan, W., Zhu, L., Ye, P., Worsnop, D., Hoi, K. I., Mok, K. M., and Li, Y. J.: Response of protonated, adduct, and fragmented ions in Vocus proton-transfer-reaction time-of-flight mass spectrometer (PTR-ToF-MS), Atmos. Meas. Tech., 17, 2415–2427, https://doi.org/10.5194/amt-17-2415-2024, 2024. a
Li, H., Canagaratna, M. R., Riva, M., Rantala, P., Zhang, Y., Thomas, S., Heikkinen, L., Flaud, P.-M., Villenave, E., Perraudin, E., Worsnop, D., Kulmala, M., Ehn, M., and Bianchi, F.: Atmospheric organic vapors in two European pine forests measured by a Vocus PTR-TOF: insights into monoterpene and sesquiterpene oxidation processes, Atmos. Chem. Phys., 21, 4123–4147, https://doi.org/10.5194/acp-21-4123-2021, 2021. a
Link, M. F., Pothier, M. A., Vermeuel, M. P., Riches, M., Millet, D. B., and Farmer, D. K.: In-Canopy Chemistry, Emissions, Deposition, and Surface Reactivity Compete to Drive Bidirectional Forest-Atmosphere Exchange of VOC Oxidation Products, ACS ES&T Air, 1, 305–315, https://doi.org/10.1021/acsestair.3c00074, 2024. a, b, c, d
McKinney, K. A., Lee, B. H., Vasta, A., Pho, T. V., and Munger, J. W.: Emissions of isoprenoids and oxygenated biogenic volatile organic compounds from a New England mixed forest, Atmos. Chem. Phys., 11, 4807–4831, https://doi.org/10.5194/acp-11-4807-2011, 2011. a
Meot-Ner (Mautner), M. and Lias, S. G.: Binding Energies Between Ions and Molecules, and The Thermochemistry of Cluster Ions, in: NIST Chemistry WebBook, NIST Standard Reference Database Number 69, edited by: Linstrom, P. J. and Mallard, W. G., National Institude of Standards and Technology, Gaithersburg MD, 20899, https://doi.org/10.18434/T4D303, 2023. a
Muller, M., Piel, F., Gutmann, R., Sulzer, P., Hartungen, E., and Wisthaler, A.: A novel method for producing NH4+ reagent ions in the hollow cathode glow discharge ion source of PTR-MS instruments, Int. J. Mass Spectrom., 447, 116254, https://doi.org/10.1016/j.ijms.2019.116254, 2020. a, b, c
Mungall, E. L., Abbatt, J. P. D., Wentzell, J. J. B., Lee, A. K. Y., Thomas, J. L., Blais, M., Gosselin, M., Miller, L. A., Papakyriakou, T., Willis, M. D., and Liggio, J.: Microlayer source of oxygenated volatile organic compounds in the summertime marine Arctic boundary layer, P. Natl. Acad. Sci. USA, 114, 6203–6208, https://doi.org/10.1073/pnas.1620571114, 2017. a
Nah, T., Ji, Y., Tanner, D. J., Guo, H., Sullivan, A. P., Ng, N. L., Weber, R. J., and Huey, L. G.: Real-time measurements of gas-phase organic acids using SF6− chemical ionization mass spectrometry, Atmos. Meas. Tech., 11, 5087–5104, https://doi.org/10.5194/amt-11-5087-2018, 2018. a
Nowak, J. B., Davis, D. D., Chen, G., Eisele, F. L., Mauldin, R. L., Tanner, D. J., Cantrell, C., Kosciuch, E., Bandy, A., Thornton, D., and Clarke, A.: Airborne observations of DMSO, DMS, and OH at marine tropical latitudes, Geophys. Res. Lett., 28, 2201–2204, https://doi.org/10.1029/2000GL012297, 2001. a
Ortega, J., Turnipseed, A., Guenther, A. B., Karl, T. G., Day, D. A., Gochis, D., Huffman, J. A., Prenni, A. J., Levin, E. J. T., Kreidenweis, S. M., DeMott, P. J., Tobo, Y., Patton, E. G., Hodzic, A., Cui, Y. Y., Harley, P. C., Hornbrook, R. S., Apel, E. C., Monson, R. K., Eller, A. S. D., Greenberg, J. P., Barth, M. C., Campuzano-Jost, P., Palm, B. B., Jimenez, J. L., Aiken, A. C., Dubey, M. K., Geron, C., Offenberg, J., Ryan, M. G., Fornwalt, P. J., Pryor, S. C., Keutsch, F. N., DiGangi, J. P., Chan, A. W. H., Goldstein, A. H., Wolfe, G. M., Kim, S., Kaser, L., Schnitzhofer, R., Hansel, A., Cantrell, C. A., Mauldin, R. L., and Smith, J. N.: Overview of the Manitou Experimental Forest Observatory: site description and selected science results from 2008 to 2013, Atmos. Chem. Phys., 14, 6345–6367, https://doi.org/10.5194/acp-14-6345-2014, 2014. a
Pagonis, D., Krechmer, J. E., de Gouw, J., Jimenez, J. L., and Ziemann, P. J.: Effects of gas–wall partitioning in Teflon tubing and instrumentation on time-resolved measurements of gas-phase organic compounds, Atmos. Meas. Tech., 10, 4687–4696, https://doi.org/10.5194/amt-10-4687-2017, 2017. a
Pagonis, D., Sekimoto, K., and de Gouw, J.: A Library of Proton-Transfer Reactions of H3O(+) Ions Used for Trace Gas Detection, J. Am. Soc. Mass Spectrom., 30, 1330–1335, https://doi.org/10.1007/s13361-019-02209-3, 2019. a, b, c, d, e
Putaud, J., Davison, B., Watts, S., Mihalopoulos, N., Nguyen, B., and Hewitt, C.: Dimethylsulfide and its oxidation products at two sites in Brittany (France), Atmos. Environ., 33, 647–659, https://doi.org/10.1016/S1352-2310(98)00104-6, 1999. a
Riches, M., Berg, T. C., Vermeuel, M. P., Millet, D. B., and Farmer, D. K.: Wildfire Smoke Directly Changes Biogenic Volatile Organic Emissions and Photosynthesis of Ponderosa Pines, Geophys. Res. Lett., 51, e2023GL106667, https://doi.org/10.1029/2023GL106667, 2024. a, b
Rissanen, M. P., Mikkilä, J., Iyer, S., and Hakala, J.: Multi-scheme chemical ionization inlet (MION) for fast switching of reagent ion chemistry in atmospheric pressure chemical ionization mass spectrometry (CIMS) applications, Atmos. Meas. Tech., 12, 6635–6646, https://doi.org/10.5194/amt-12-6635-2019, 2019. a, b, c
Riva, M., Rantala, P., Krechmer, J. E., Peräkylä, O., Zhang, Y., Heikkinen, L., Garmash, O., Yan, C., Kulmala, M., Worsnop, D., and Ehn, M.: Evaluating the performance of five different chemical ionization techniques for detecting gaseous oxygenated organic species, Atmos. Meas. Tech., 12, 2403–2421, https://doi.org/10.5194/amt-12-2403-2019, 2019. a, b, c, d, e, f, g
Roberts, J. M., Veres, P., Warneke, C., Neuman, J. A., Washenfelder, R. A., Brown, S. S., Baasandorj, M., Burkholder, J. B., Burling, I. R., Johnson, T. J., Yokelson, R. J., and de Gouw, J.: Measurement of HONO, HNCO, and other inorganic acids by negative-ion proton-transfer chemical-ionization mass spectrometry (NI-PT-CIMS): application to biomass burning emissions, Atmos. Meas. Tech., 3, 981–990, https://doi.org/10.5194/amt-3-981-2010, 2010. a
Saunders, S. M., Jenkin, M. E., Derwent, R. G., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): tropospheric degradation of non-aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 161–180, https://doi.org/10.5194/acp-3-161-2003, 2003. a
Sciare, J., Kanakidou, M., and Mihalopoulos, N.: Diurnal and seasonal variation of atmospheric dimethylsulfoxide at Amsterdam Island in the southern Indian Ocean, J. Geophys. Res.-Atmos., 105, 17257–17265, https://doi.org/10.1029/1999JD901186, 2000. a
Smith, D. and Spanel, P.: Selected ion flow tube mass spectrometry (SIFT-MS) for on-line trace gas analysis, Mass Spectrom. Rev., 24, 661–700, https://doi.org/10.1002/mas.20033, 2005. a, b
Stockwell, C. E., Coggon, M. M., Gkatzelis, G. I., Ortega, J., McDonald, B. C., Peischl, J., Aikin, K., Gilman, J. B., Trainer, M., and Warneke, C.: Volatile organic compound emissions from solvent- and water-borne coatings – compositional differences and tracer compound identifications, Atmos. Chem. Phys., 21, 6005–6022, https://doi.org/10.5194/acp-21-6005-2021, 2021. a
US EPA: Estimations Programs Interface Suite for Microsoft Windows, United States Environmental Protection Agency, https://www.epa.gov/tsca-screening-tools/epi-suitetm-estimation-program-interface (last access: 9 December 2024), 2023. a
Vermeuel, M. P., Millet, D. B., Farmer, D. K., Pothier, M. A., Link, M. F., Riches, M., Williams, S., and Garofalo, L. A.: Closing the Reactive Carbon Flux Budget: Observations From Dual Mass Spectrometers Over a Coniferous Forest, J. Geophys. Res.-Atmos., 128, e2023JD038753, https://doi.org/10.1029/2023JD038753, 2023a. a, b, c, d, e
Vermeuel, M. P., Novak, G. A., Kilgour, D. B., Claflin, M. S., Lerner, B. M., Trowbridge, A. M., Thom, J., Cleary, P. A., Desai, A. R., and Bertram, T. H.: Observations of biogenic volatile organic compounds over a mixed temperate forest during the summer to autumn transition, Atmos. Chem. Phys., 23, 4123–4148, https://doi.org/10.5194/acp-23-4123-2023, 2023b. a
Warneke, C., Van Der Veen, C., Luxembourg, S., De Gouw, J., and Kok, A.: Measurements of benzene and toluene in ambient air using proton-transfer-reaction mass spectrometry: calibration, humidity dependence, and field intercomparison, Int. J. Mass Spectrom., 207, 167–182, https://doi.org/10.1016/S1387-3806(01)00366-9, 2001. a
Xu, L., Crounse, J. D., Vasquez, K. T., Allen, H., Wennberg, P. O., Bourgeois, I., Brown, S. S., Campuzano-Jost, P., Coggon, M. M., Crawford, J. H., DiGangi, J. P., Diskin, G. S., Fried, A., Gargulinski, E. M., Gilman, J. B., Gkatzelis, G. I., Guo, H., Hair, J. W., Hall, S. R., Halliday, H. A., Hanisco, T. F., Hannun, R. A., Holmes, C. D., Huey, L. G., Jimenez, J. L., Lamplugh, A., Lee, Y. R., Liao, J., Lindaas, J., Neuman, J. A., Nowak, J. B., Peischl, J., Peterson, D. A., Piel, F., Richter, D., Rickly, P. S., Robinson, M. A., Rollins, A. W., Ryerson, T. B., Sekimoto, K., Selimovic, V., Shingler, T., Soja, A. J., St Clair, J. M., Tanner, D. J., Ullmann, K., Veres, P. R., Walega, J., Warneke, C., Washenfelder, R. A., Weibring, P., Wisthaler, A., Wolfe, G. M., Womack, C. C., and Yokelson, R. J.: Ozone chemistry in western U.S. wildfire plumes, Sci. Adv., 7, eabl3648, https://doi.org/10.1126/sciadv.abl3648, 2021. a, b
Xu, L., Coggon, M. M., Stockwell, C. E., Gilman, J. B., Robinson, M. A., Breitenlechner, M., Lamplugh, A., Crounse, J. D., Wennberg, P. O., Neuman, J. A., Novak, G. A., Veres, P. R., Brown, S. S., and Warneke, C.: Chemical ionization mass spectrometry utilizing ammonium ions (NH CIMS) for measurements of organic compounds in the atmosphere, Atmos. Meas. Tech., 15, 7353–7373, https://doi.org/10.5194/amt-15-7353-2022, 2022. a, b, c, d, e, f, g, h, i, j, k, l, m, n, o, p
Yuan, B., Koss, A. R., Warneke, C., Coggon, M., Sekimoto, K., and de Gouw, J. A.: Proton-Transfer-Reaction Mass Spectrometry: Applications in Atmospheric Sciences, Chem. Rev., 117, 13187–13229, https://doi.org/10.1021/acs.chemrev.7b00325, 2017. a, b, c, d
Zang, C. and Willis, M.: Replication Data for: Deployment and evaluation of an NH/H3O+ reagent-ion switching chemical ionization mass spectrometer for the detection of reduced and oxygenated gas-phase organic compounds, Harvard Dataverse [data set], https://doi.org/10.7910/DVN/FL0CZM, 2024. a
Zaytsev, A., Breitenlechner, M., Koss, A. R., Lim, C. Y., Rowe, J. C., Kroll, J. H., and Keutsch, F. N.: Using collision-induced dissociation to constrain sensitivity of ammonia chemical ionization mass spectrometry (NH CIMS) to oxygenated volatile organic compounds, Atmos. Meas. Tech., 12, 1861–1870, https://doi.org/10.5194/amt-12-1861-2019, 2019a. a, b, c, d, e, f, g, h, i, j, k
Zaytsev, A., Koss, A. R., Breitenlechner, M., Krechmer, J. E., Nihill, K. J., Lim, C. Y., Rowe, J. C., Cox, J. L., Moss, J., Roscioli, J. R., Canagaratna, M. R., Worsnop, D. R., Kroll, J. H., and Keutsch, F. N.: Mechanistic study of the formation of ring-retaining and ring-opening products from the oxidation of aromatic compounds under urban atmospheric conditions, Atmos. Chem. Phys., 19, 15117–15129, https://doi.org/10.5194/acp-19-15117-2019, 2019b. a, b
Zhang, Q., Zou, X., Liang, Q., Wang, H., Huang, C., Shen, C., and Chu, Y.: Ammonia-Assisted Proton Transfer Reaction Mass Spectrometry for Detecting Triacetone Triperoxide (TATP) Explosive, J. Am. Soc. Mass Spectrom., 30, 501–508, https://doi.org/10.1007/s13361-018-2108-6, 2019. a
Zhang, W., Xu, L., and Zhang, H.: Recent advances in mass spectrometry techniques for atmospheric chemistry research on molecular‐level, Mass Spectrom. Rev., 43, mas.21857, https://doi.org/10.1002/mas.21857, 2023. a
Zhao, R., Mungall, E. L., Lee, A. K. Y., Aljawhary, D., and Abbatt, J. P. D.: Aqueous-phase photooxidation of levoglucosan – a mechanistic study using aerosol time-of-flight chemical ionization mass spectrometry (Aerosol ToF-CIMS), Atmos. Chem. Phys., 14, 9695–9706, https://doi.org/10.5194/acp-14-9695-2014, 2014a. a
Zhao, Y., Hennigan, C. J., May, A. A., Tkacik, D. S., de Gouw, J. A., Gilman, J. B., Kuster, W. C., Borbon, A., and Robinson, A. L.: Intermediate-volatility organic compounds: a large source of secondary organic aerosol, Environ. Sci. Technol., 48, 13743–13750, https://doi.org/10.1021/es5035188, 2014b. a
Zhou, S., Rivera-Rios, J. C., Keutsch, F. N., and Abbatt, J. P. D.: Identification of organic hydroperoxides and peroxy acids using atmospheric pressure chemical ionization–tandem mass spectrometry (APCI-MS/MS): application to secondary organic aerosol, Atmos. Meas. Tech., 11, 3081–3089, https://doi.org/10.5194/amt-11-3081-2018, 2018. a