the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 08 Oct 2018
| 08 Oct 2018
Size-resolved online chemical analysis of nanoaerosol particles: a thermal desorption differential mobility analyzer coupled to a chemical ionization time-of-flight mass spectrometer
Andrea C. Wagner
Anton Bergen
Sophia Brilke
Claudia Fuchs
Markus Ernst
Jesica Hoker
Martin Heinritzi
Mario Simon
Bertram Bühner
Joachim Curtius
Andreas Kürten
A new method for size-resolved chemical analysis of nucleation mode aerosol particles (size range from ∼10 to ∼30 nm) is presented. The Thermal Desorption Differential Mobility Analyzer (TD-DMA) uses an online, discontinuous principle. The particles are charged, a specific size is selected by differential mobility analysis and they are collected on a filament by electrostatic precipitation. Subsequently, the sampled mass is evaporated in a clean carrier gas and analyzed by a chemical ionization mass spectrometer. Gas-phase measurements are performed with the same mass spectrometer during the sampling of particles. The characterization shows reproducible results, with a particle size resolution of 1.19 and the transmission efficiency for 15 nm particles being slightly above 50 %. The signal from the evaporation of a test substance can be detected starting from 0.01 ng and shows a linear response in the mass spectrometer. Instrument operation in the range of pg m−3 is demonstrated by an example measurement of 15 nm particles produced by nucleation from dimethylamine, sulfuric acid and water.
- Article
(1575 KB) - Full-text XML
- BibTeX
- EndNote





Aerosol particles play an important role in the earth's climate. They influence the radiative budget directly by scattering and absorbing solar radiation, and indirectly by changing cloud properties such as albedo and lifetime. In climate models, these interactions are still the largest source of uncertainty (Core Writing Team et al., 2007; Fuzzi et al., 2015). Aerosol particles also affect air quality and human health. Depending on their size and shape, they can protrude deep into the lungs (Kreyling et al., 2006) and even enter the bloodstream (Nel, 2005), causing problems such as ischemic stroke (Wellenius et al., 2012), premature mortality (Lelieveld et al., 2015) and many others (e.g., Pope and Dockery, 2006; Donaldson and Borm, 2006).
A large fraction of atmospheric aerosol particles originates from new particle formation (Merikanto et al., 2009; Dunne et al., 2016). Several major formation mechanisms have already been subject of intense research (Hallquist et al., 2009; Zhang et al., 2012; Kulmala et al., 2014; Kirkby et al., 2016), yet various reaction pathways and processes remain unknown. The newly formed particles need to grow to a certain size (∼50–100 nm) to act as cloud condensation nuclei (e.g., Hallquist et al., 2009; Riccobono et al., 2012; Vehkamäki and Riipinen, 2012; Tröstl et al., 2016; Lehtipalo et al., 2016). In order to better understand the nucleation and subsequent growth to cloud condensation nuclei (CCN), the condensing vapors, the freshly nucleated small particles, as well as the larger particles need to be chemically speciated and quantified.
For the analysis of atmospherically relevant nucleation precursors, chemical ionization mass spectrometry is quite commonly used. Depending on the target analyte, the primary ion is chosen. Prominent examples include the proton transfer reaction technique using hydronium ions (Hansel et al., 1995; Graus et al., 2010; Breitenlechner et al., 2017), successfully targeting volatile organic compounds or negatively charged nitrate ions for the detection of sulfuric acid (Eisele and Tanner, 1993; Kürten et al., 2011; Jokinen et al., 2012), the class of extremely low volatile organic compounds (ELVOCs) (Ehn et al., 2014) and clusters of sulfuric acid and dimethylamine (Kürten et al., 2014).
Table 1Instruments capable of chemical analysis of sub-30 nm particles. For interpretation of the parameters, see the discussion Sect. 6.2, and for references to later work on the individual instruments, see the introduction Sect. 1.
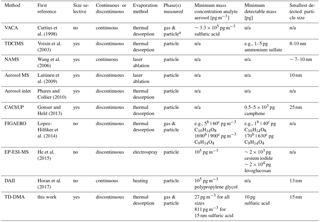
a Not separated, b chamber study, c ambient study; n/a: not applicable or not specified.
Particles that have already grown to larger sizes are frequently analyzed by the well-established technologies of the aerosol mass spectrometer (AMS) (Jayne et al., 2000; Zhang et al., 2011) and single particle mass spectrometry (Noble and Prather, 2000; Bzdek et al., 2012). The chemical composition of particles, however, changes with size, and obtaining detailed chemical information about smaller particles is very challenging as they have an extremely low mass.
To close the gap between the measurement of gas-phase and larger particles, a number of interesting techniques have emerged in the past few years. Only a few instruments are capable of analyzing sub-30 nm particles. They can be distinguished based on the following main criteria (see Table 1): (1) a discontinuous principle to enrich the analyte vs. continuous measurements, (2) size-resolved methods vs. integral sampling methods, (3) particle evaporation method, e.g., by thermal desorption or laser evaporation, and (4) ability to analyze gas and particle phase vs. only particle phase.
The nanoaerosol mass spectrometer (NAMS) (Wang et al., 2006) is a single particle mass spectrometer for small particles and uses a continuous principle. It consists of an aerodynamic lens, ion guide and quadrupole ion trap with laser evaporization. The Volatile Aerosol Component Analyzer (VACA) (Curtius et al., 1998; Arnold et al., 1998; Curtius and Arnold, 2001) is able to continuously measure sulfuric acid in sub-30 nm particles in aircraft plumes, where gas-phase concentrations are much lower than particle-phase concentrations. The thermal desorption chemical ionization mass spectrometer (TDCIMS) (Voisin et al., 2003; Smith et al., 2004; McMurry et al., 2009) is a size-resolved method collecting particles electrostatically and using thermal desorption to measure the particle-phase composition. It has provided important insights to several chemical systems including aerosol from marine environments (Lawler et al., 2014), and has even been able to perform size-resolved measurements for extremely small particle sizes of 8–10 nm (Smith et al., 2010). The aerosol mass spectrometer from Laitinen et al. (2009) precipitates particles on a platinum surface and emits them by laser ablation. It has been used for organic compounds in the size range of 10 to 50 nm (Laitinen et al., 2011). Two other size selective techniques using thermal desorption are the devices by Phares and Collier (2010) and the CaCHUP by Gonser and Held (2013). The Filter Inlet for Gases and AEROsols (FIGAERO) (Lopez-Hilfiker et al., 2014) is a bulk-phase filter sampler using thermal desorption. Although it does not provide size-resolved information, it is able to measure both gas and particle phase and thus to investigate partitioning effects (Lopez-Hilfiker et al., 2015). A new technique for the discontinuous particle-phase analysis is the Electrostatical Precipitation Electrospray Mass Spectrometer (EP-ESI-MS) from He et al. (2015). Particles are charged and electrostatically collected. Thereafter, the material is brought to the gas phase and softly ionized at the same time by using the collection surface as an electrospray tip. The Droplet Assisted Inlet Ionization (DAII) (Horan et al., 2017) condenses water on particles and rapidly evaporates them in a heated inlet. It is a continuous method and has proven the ability to measure test particles as small as 13 nm. A more detailed discussion is given in Sect. 6.2.
In this study, we present our novel device for the online chemical analysis of nucleation and Aitken mode particles. The Thermal Desorption – Differential Mobility Analyzer (TD-DMA) is capable of measuring in a size-resolved and integral setting. It allows taking gas-phase measurements with the same mass spectrometer. As a mobile interface, it can be combined with different mass spectrometers or other real-time gas-phase analyzers.
Here, the instrument is described (Sect. 2), followed by a detailed characterization (Sect. 3) comprising the DMA unit's transmission efficiency, the particle collection efficiency, the filament temperature and the reproducibility of the evaporation process. The measurement procedure (Sect. 4) is specified, including determination of the background and an exemplary measurement of 15 nm particles. Lastly, the signal is quantified (Sect. 5) and the results are discussed (Sect. 6), closing with a short summary (Sect. 7).
The TD-DMA is designed for size-resolved chemical analysis of nanometer sized aerosol particles. The particles are charged, a specific size is selected and they are electrostatically collected on a filament. Subsequently, the sampled mass is evaporated in a clean carrier gas to be analyzed by a detector, e.g., a mass spectrometer. In this way, the particle phase is efficiently separated from the gas phase and the concentration is enhanced to meet the detection limit of the analyzer. The coupling between TD-DMA and detector allows for gas-phase measurements during particle sample collection periods. This enables an existing gas-phase analyzer to measure the particle phase as well without the requirements of cost and space for a second analyzer. Only a small fraction of the gas-phase measurement time is lost when it is interrupted by the short evaporation period for the particles. The chemical composition of gas and particle phase can be compared directly without the need to consider instrumental differences in, e.g., ionization and characteristics of the mass spectrometer, as would be the case when using two separate instruments. This modular concept allows obtaining a broad picture of both particle and gas-phase chemical composition and to observe the condensation, reactive uptake and partitioning of the analyzed substances.
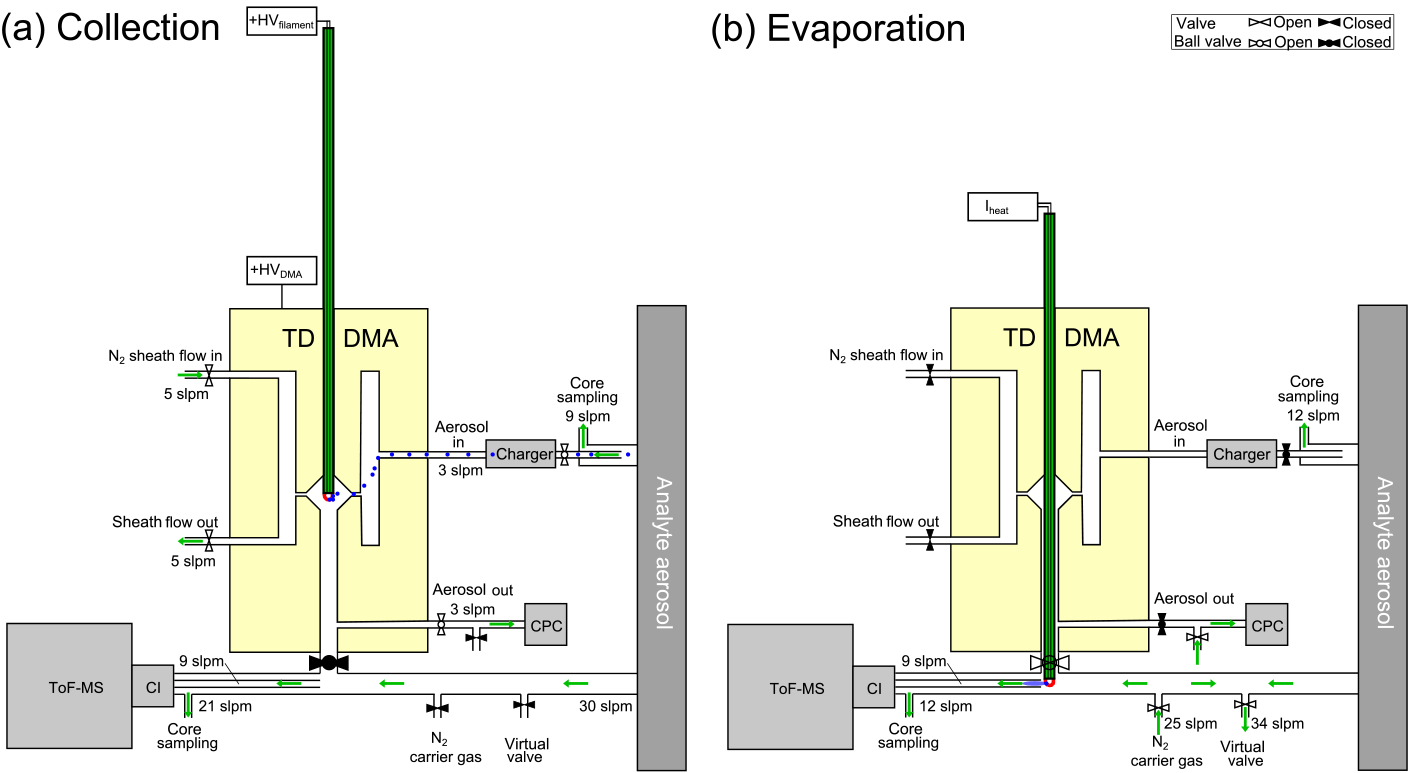
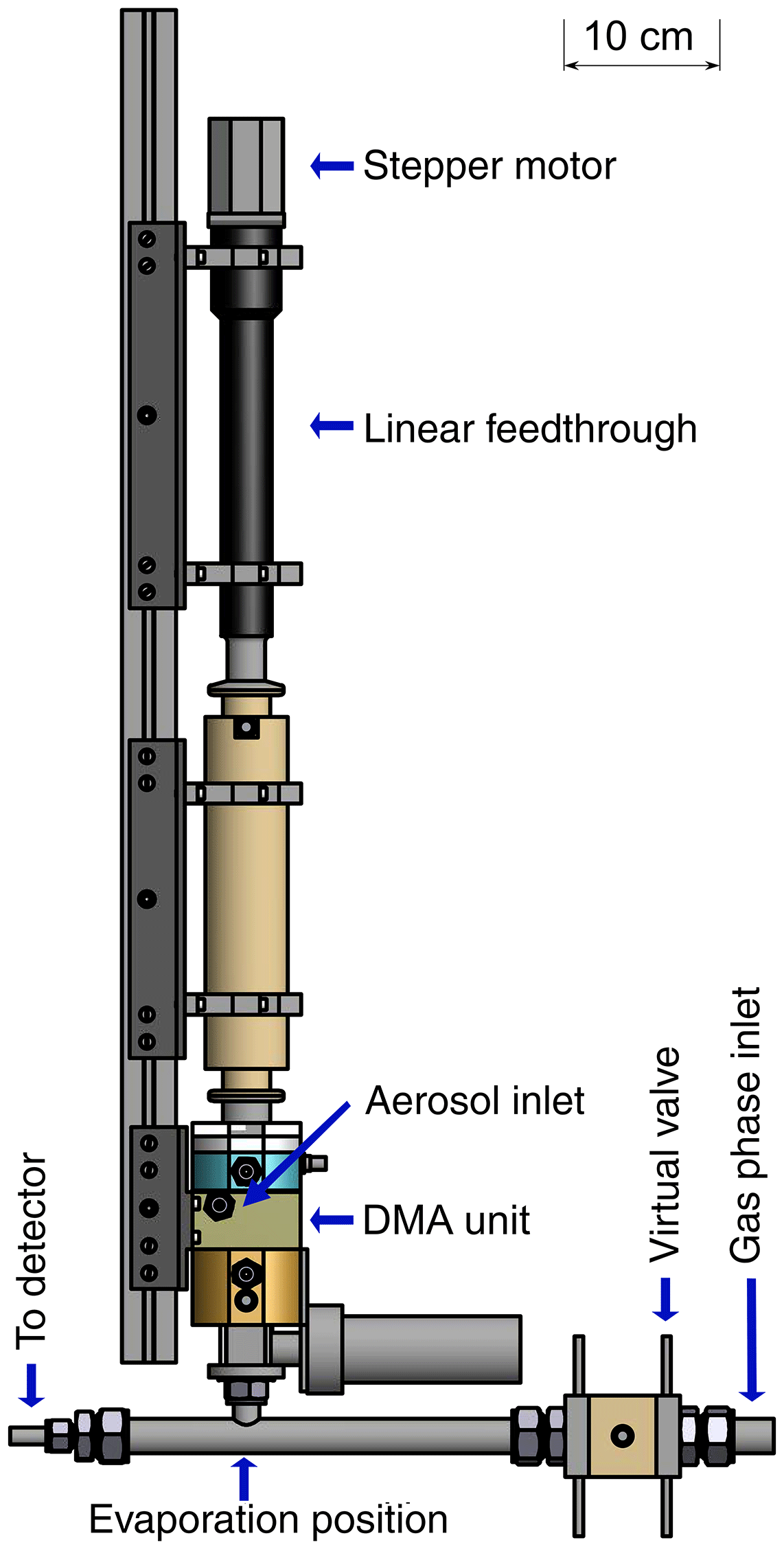
Figure 1Measurement setup of the TD-DMA with step (a) collection, and (b) evaporation. Filled valves correspond to closed, unfilled ones to open. In step (a), particles (blue) from the aerosol sample are charged, size selected and collected on a filament (red) inside the DMA unit (yellow). In step (b), the filament is moved in front of a mass spectrometer and the collected material is evaporated and analyzed.
The TD-DMA is a stand-alone instrument that can be attached to any existing technique suitable for real-time chemical analysis of gas-phase compounds. In this case, individual compounds relevant for nucleation and early growth of atmospheric nanoaerosol particles should be measured. For this we used a chemical ionization atmospheric pressure interface time-of-flight (CI-APi-TOF) mass spectrometer with negative nitrate primary ions generated by a corona ion source (Kürten et al., 2011). This technique is specialized for the detection of sulfuric acid, amines and highly oxygenated organic molecules (HOMs) (Kürten et al., 2014; Simon et al., 2016; Kirkby et al., 2016), and calibrated for its internal transmission efficiency (Heinritzi et al., 2016) as well as for its detection efficiency regarding the sulfuric acid concentration (Kürten et al., 2012). For the gas-phase measurement, the removal of aerosol particles by, e.g., a filter is not required. As the inlet line and ion source are at the same temperature as the analyte aerosol, and the gas composition surrounding the particles is unchanged, the particles do not evaporate significantly on their way to the mass spectrometer and thus are not detected during gas-phase measurements. On the contrary, applying a particle filter would influence the gas-phase measurement negatively as a significant fraction of the gas-phase analyte, especially sulfuric acid or highly oxygenated organics of low volatility, would adsorb on the filter.
2.1 Setup and measurement procedure
Before a more detailed technical description of the TD-DMA is given (Sect. 2.2), an overview of the measurement procedure shall be provided. The measurement procedure is discontinuous and is performed in two steps as follows: the first step is the collection of particles (see Fig. 1a). The analyte aerosol passes through a charger, where a charge equilibrium is established. A core sampling system, where the analyte is isokinetically sampled from the center of a larger tube, is used to reduce losses in the inlet line (e.g., Wimmer et al., 2015). The aerosol then enters the DMA unit of the TD-DMA. The gas phase of the analyte aerosol is removed by the pure nitrogen sheath gas, which is controlled in a non-recirculating way. Charged particles are attracted towards the central electrode and a well-defined size enters the selection slit. A platinum filament is placed inside the central electrode. It is only exposed to pure nitrogen from the sheath gas and samples the preselected particles by electrostatic precipitation. The position of the filament inside the central electrode implements the collection of particles directly after classification and thereby minimizes losses. While the sampling takes place, the mass spectrometer is used to measure the gas-phase chemical composition of the analyte aerosol through a separate sampling line. It is also possible to collect the whole size distribution by turning off the DMA's sheath flow (see also Sects. 4 and 6).
Once enough particle mass is collected on the filament, the second step begins (see Fig. 1b). The inlet line of the mass spectrometer is flushed with ultrapure nitrogen. Another outlet in the inlet line works as a virtual valve that flushes out the excess nitrogen added to the sampling line while maintaining a flow of air into the sampling line. This prevents contamination of the reservoir where the sample gas is taken from and also keeps the sample gas flow rate constant, which is important for chamber or flow tube experiments. The filament is then moved into the mass spectrometer's inlet line. An electrical current is sent through the filament, which is thereby heated. The sampled particles evaporate and the vapor is analyzed by the mass spectrometer.
2.2 Instrument details
The main part of the TD-DMA system is the in-house developed DMA unit, where the particle size selection and collection takes place. The charger is a soft X-ray diffusion charger (TSI 3088; Tigges et al., 2015). A linear feedthrough (MDC vacuum, 152.4 mm maximum travel distance) and a stepper motor are used to translate a ceramic rod with the collection filament from its collection position to its evaporation position and back. An in-house built electronic control unit allows the precise control of all parameters, i.e., flow rates, valve positions, high voltages and the heating current for the filament. In the collection mode, the unit supplies the DMA central electrode and filament with the required positive high voltages via safe high-voltage (SHV) connectors and cables. In the evaporation mode, the filament is operated with a defined heating current that can be ramped as a function of time to increase the evaporation temperature stepwise (up to about 600 ∘C). The mass flow controllers and magnetic valves enable software controlled adjustment of the flows for the aerosol, sheath gas, carrier gas and virtual valve. Further magnetic valves are used to completely shut-off or redirect flows when necessary, e.g., during the evaporation. Two automated ball valves (Grotec OSE-M) allow for separation of the TD-DMA from the gas-phase measurement during sampling mode as well as closing the aerosol inlet during evaporation mode (Fig. 1). The stepper motor moving the linear feedthrough and filament is also controlled by the electronic unit. PC control of the electronics is realized by means of USB data acquisition boards (Meilhaus 1608 and 3103) and a LabVIEW software program.
The DMA unit (see Fig. 2) is designed to collect a maximum amount of particulate mass for a defined diameter. Its characteristics are determined by the dimensions as well as the sheath and aerosol flow rates. The amount of collected particles is maximized when the transmission efficiency is high. Also, increasing the aerosol flow rate leads to an increase in collected mass for constant transmission efficiency. Therefore, we chose the maximum possible aerosol flow rate for the dimensions of the DMA unit, 3 standard liters per minute (slpm). As the overall number of selected particles is higher when the size resolution of the DMA is low, a low size resolution and thus low sheath flow rate is beneficial in this case. However, the size resolution still needs to be high enough to warrant size selection and at values of the sheath flow rate that are too low, the DMA will not work anymore. With a sheath flow of 5 slpm, as much particle mass as possible is collected while still allowing sufficient size selection. The DMA has a cylindrical geometry (Reischl et al., 1997; Chen et al., 1998) with a positive high voltage on the central electrode (1) and ground potential on the outer electrode. The DMA's inner radius is 15 mm, the outer radius is 20 mm and the classification length is 15 mm. The collection of particles on the filament takes place inside the central electrode (2). In this way, losses are minimized as the transfer lines are very short and the particles do not need to overcome an additional voltage step (from high voltage to ground potential) because they do not need to leave the DMA (Franchin et al., 2016). In order to position the filament, the central electrode has a through-hole in its center (3, 4) through which a ceramic rod is inserted. The rod carries two thick copper wires with a short piece of platinum wire spot-welded to both of their ends. The electrical resistance of the thin platinum wire is high compared to that of the remaining system. Thus, when a current is applied, a voltage drop occurs mainly across the filament. The deposited energy leads to heating and desorption of the particle material. Electronic relays suitable for high-voltage operation are used to disconnect the wires from the current source during collection mode; in this mode a third relay is used to apply a positive high voltage to the wires for electrostatic particle collection. While the opening in the first part (3, towards the linear feedthrough) of the central electrode is just large enough to center the ceramic rod while allowing for its axial movement, the through-hole in the second part (4, towards the DMA exit) is larger to allow moving the filament smoothly towards the sampling line of the mass spectrometer. The outlet is closed by a ball valve during collection mode and is only opened when the filament is positioned in the CI-APi-TOF sampling line. In this mode, all other valves connected to the DMA are closed to avoid gas leaking from the DMA into the sampling line or vice versa. The shape of the collection area (2) is an attempt to keep potential disturbances in the air flow small, while maximizing the distance between the filament and electrode to avoid flashovers and leakage currents due to the electric field. However, leakage currents could not be fully avoided (see Sect. 3.2).
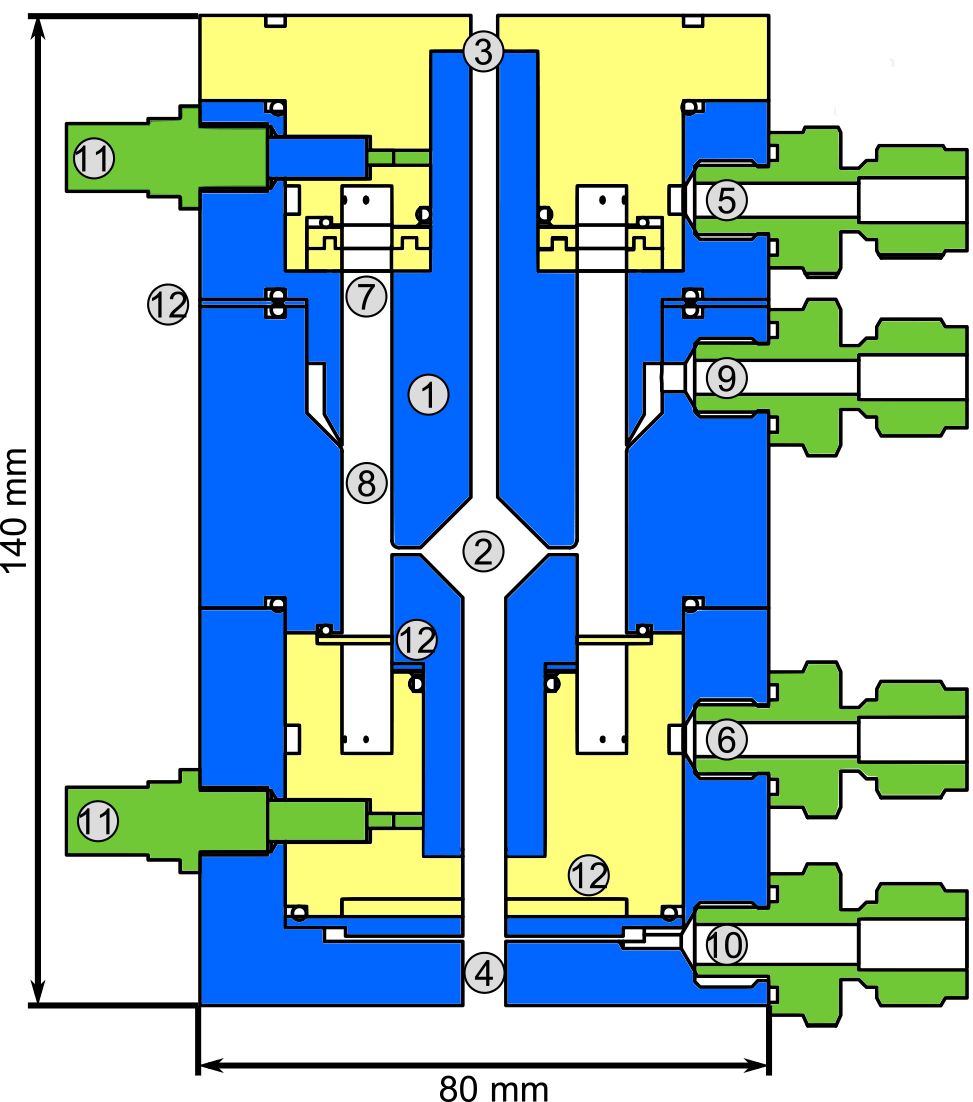
Figure 2Schematic quarter section of the DMA unit of the TD-DMA with (1) central electrode, (2) collection area, (3) inlet for the filament, (4) outlet for the filament, (5) sheath flow inlet, (6) sheath flow outlet, (7) tissue for laminarizing the sheath low, (8) selection area, (9) aerosol inlet, (10) aerosol outlet, (11) SHV supplies and (12) spacer plates. Conductive stainless steel parts are shown in blue, isolating PEEK parts in yellow.
The DMA sheath flow is distributed in azimuthal direction by introducing a pressure drop at the inlet (5) and outlet (6) using a circular cavity and a total of 16 small drilled holes. To achieve a laminar sheath flow, which is crucial for accurate size selection, an evenly webbed tissue (7) is placed 20 mm upstream of the selection area (8). The aerosol flow is introduced into the selection area through a narrow slit (9) with a design as recommended by Chen et al. (1998). The aerosol outlet (10) has to be at the side and cannot be on the central axis as this is used for moving the filament to its evaporation position. In order to achieve an evenly distributed sample and outlet flow over the whole cross section of the inner part of the central electrode, a small slit introducing a pressure drop and two outlet connectors (separated by 180∘) are used. The losses in this slit do not matter as the particles are sampled upstream. However, the characterization of the DMA unit (see Sect. 3.1) is performed at the side outlets to ensure the flow conditions are the same as for the actual measurements. Therefore, note that the actual transmission efficiencies at the filament position (i.e., the fraction of particles that reach the filament) are likely higher than determined by the characterization measurements.
A positive high voltage is applied to the central electrode by two SHV connectors (11) as this electrode is split into two parts. Small spacers (12) allow changing the width of the aerosol inlet, the selection slit and the outlet, respectively. These spacers proved to be helpful for finding suitable distances for an appropriate transmission efficiency, resolution and sizing accuracy when first testing the DMA design. The material of the DMA is stainless steel for the conductive parts (blue) and PEEK (polyether ether ketone) for the insulating parts (yellow), as these materials are considered to be chemically inert to the substances present in the aerosol sample. The full instrument (Fig. 10) measures 12 cm × 12 cm × 95 cm and weighs 13 kg.
In this section, the general performance of the TD-DMA is described based on characterization experiments in the laboratory. These measurements are used to derive the sizing accuracy, the DMA's resolution as well as its transmission and collection efficiency. Furthermore, application of defined amounts of sulfuric acid on the filament yields information on the detection limit of the TD-DMA in combination with the CI-APi-TOF and on the linearity between signal and collected mass.
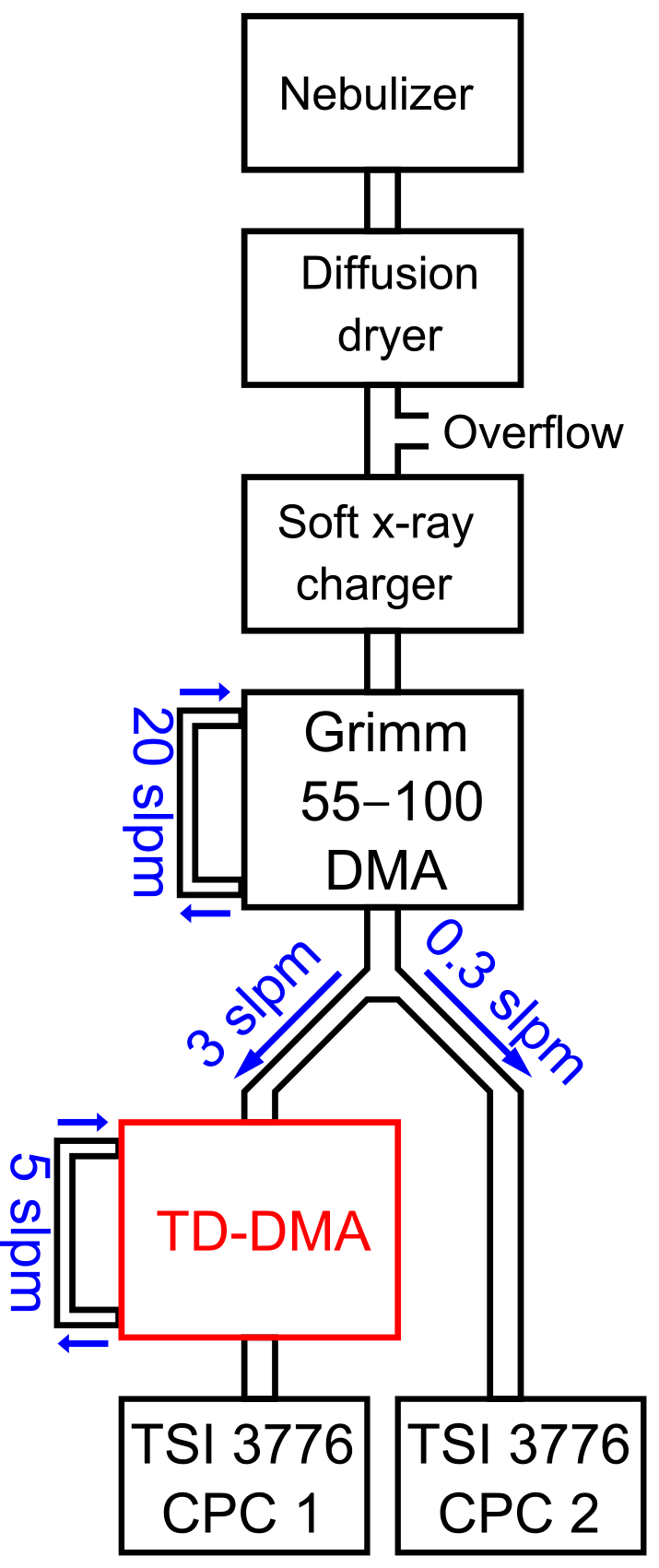
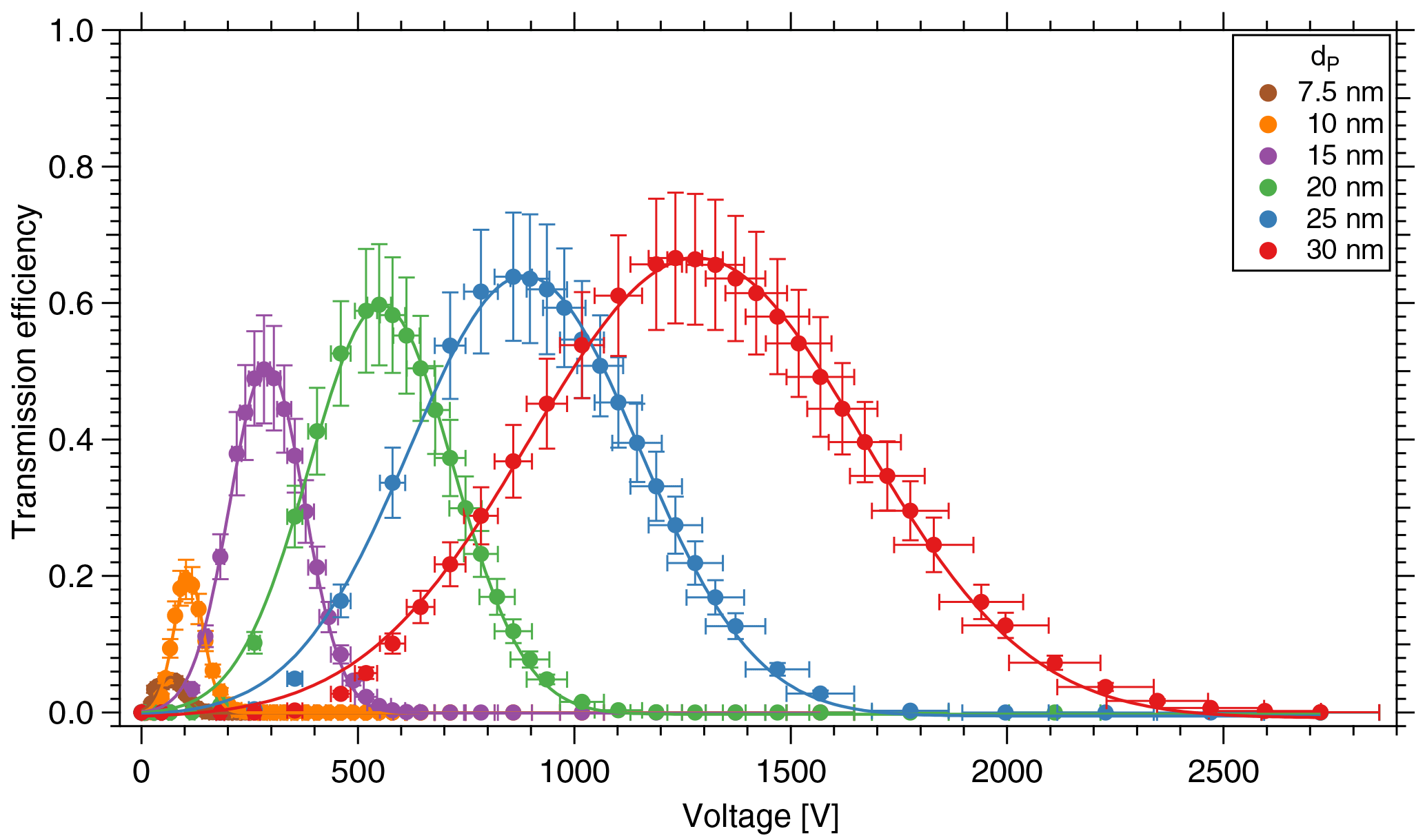
Figure 4Transmission efficiency of the TD-DMA for different diameters. The DMA unit is characterized in a tandem DMA setup.
3.1 DMA unit
The DMA unit is characterized in a tandem DMA setup (Fig. 3). Ammonium nitrate particles are produced in a nebulizer followed by a diffusion dryer. The particles are brought into charge equilibrium by using a soft X-ray charger (TSI model 3088, Tigges et al., 2015). A first DMA (Grimm Aerosol Technik GmbH model 55-100, Jiang et al., 2011) then selects particles of a defined electrical mobility and thus corresponding size (if all particles are singly charged, which is a valid assumption for sub-30 nm particles). This quasi-monodisperse aerosol flow is subsequently split into two flows. One flow enters the TD-DMA, after which a CPC (TSI model 3776, Hermann et al., 2007) measures the number concentration N1, while the filament is removed. The other flow is directly led into a CPC which serves as a reference and measures the concentration N2. This method is based on having identical counting efficiencies for both CPCs. Therefore, the CPCs are characterized against each other for each diameter using the same setup but without the TD-DMA, and a corresponding correction factor is applied. The ratio of the two measured concentrations yields the TD-DMA's transmission efficiency τ:
Note that the aerosol coming from the first DMA is not strictly monodisperse; instead the DMA provides a Gaussian-shaped size distribution. As in this case, the resolution of the first DMA is much higher than that of the second DMA, Eq. (1) introduces only a small error; using the methods by Stolzenburg and McMurry (2008) this can be demonstrated for the relevant sizes of the TD-DMA. At a fixed diameter selected for the first DMA, the TD-DMA voltage is varied. This method is repeated for six particle diameters dP, ranging from 7.5 to 30 nm. These measurements yield the transmission efficiency curves (Fig. 4). The error bars for the transmission efficiency are calculated using the statistical error of all individual measurements of the CPCs as well as assuming systematic errors of 10 % as suggested by the manufacturer (TSI Incorporated, 2007). The errors of the voltage set by the TD-DMA control box originate from the high voltage module and are assumed to be 5 %. As the selection slit of the TD-DMA has a non-infinitesimal width, the resulting distribution is not singular but triangular to a first approximation. Particle diffusion distorts it further and results in a Gaussian shaped curve (Stolzenburg and McMurry, 2008). The experimental data points are fit by the Gaussian function
where τmax is the maximum transmission efficiency for each diameter, HWHM is the half width at the half maximum of the curve, and Ucenter is the voltage applied to the central electrode to select the given particle size. In Fig. 5a, the fitted voltage values from the measurement are displayed as a function of the mobility diameter. The theoretical curve is calculated from the dimensions of the DMA according to Stolzenburg and McMurry (2008) and Wang and Flagan (1990). The curve fits best when corrected by a factor of 0.81. The reason for this is unclear, but the factor is constant for all measurements. The maximum transmission efficiency τmax for each diameter is shown in Fig. 5b. For 15 nm particles, it is slightly above 50 %. These values are used to estimate the collected mass when sampling.
The resolution R is defined as the ratio of the electrical mobility Z to the width of the transmission curve at half maximum with respect to electrical mobility (Zhang and Flagan, 1996; Flagan, 2008):
Deriving the electrical mobility from the diameter (Stolzenburg and McMurry, 2008; Hinds, 1999),
the abscissa can be transformed into mobility values, using q the particle charge (for singly charged particles, q equals one elementary charge), CC the Cunningham slip correction factor and η the viscosity of the fluid. The resolution can then be derived using the fitted HWHM of the Gaussian curves. The resolution vs. the diameter is displayed in Fig. 5, lower panel, which shows a maximum value of 1.19.
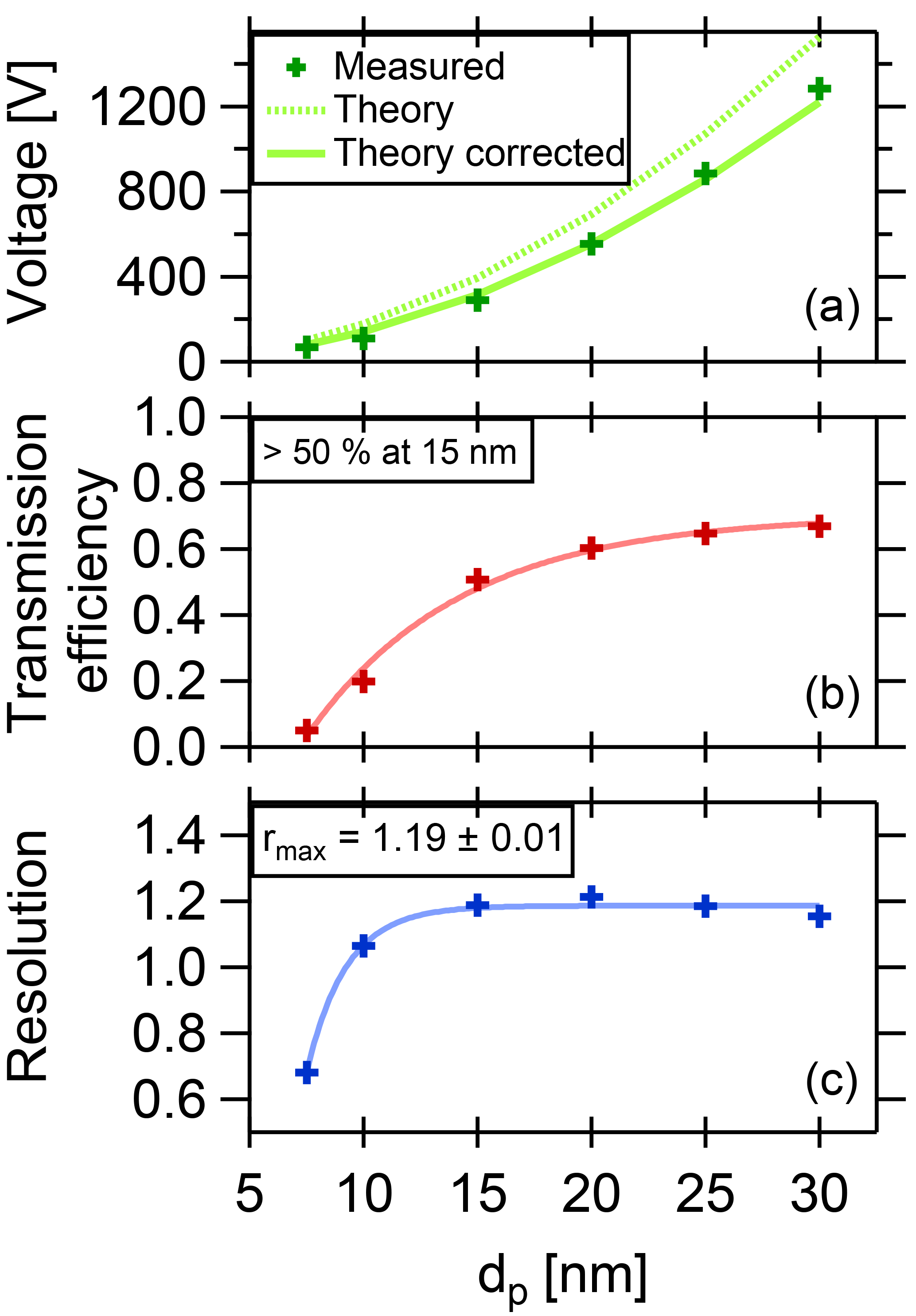
Figure 5Summary of DMA performance parameters. The DMA is optimized for a coarse resolution and high transmission in order to collect a maximum amount of particle mass while still allowing size selection.
With a resolution of 1.19 and a transmission efficiency above 50 % at 15 nm particle size, the performance of the TD-DMA is suitable for atmospheric field studies and aerosol chamber investigations.
3.2 Collection efficiency
The same tandem DMA setup as in Sect. 3.1 is also used to characterize the collection efficiency of the filament as a function of the particle diameter. For these measurements, the first DMA and the TD-DMA are set to select the same particle size. The filament voltage is increased to the maximum possible value without influencing the voltage on the central electrode, which is also monitored continuously. Unfortunately, it is technically not possible to set the filament voltage high enough to collect 100 % of the particles for all tested particle sizes. The reason for this is most likely a leak current along the surface of the ceramic rod and thus a transfer of charge from filament to central electrode. Therefore, the filament voltage is set as high as possible with some margin to avoid an influence on the DMA high voltage.
The ratio of the two measured concentrations corrected with the DMA transmission efficiency equals the fraction of particles that are not collected on the filament. The collection efficiency is thus defined as
with τ(dP) being the transmission efficiency from Sect. 3.1. The results are displayed in Fig. 6. The efficiency at which the selected particles are collected on the filament decreases with increasing size. As the flow velocity and the distance between electrode and filament remain constant during the sampling, the collection efficiency depends only on the particle electrical mobility for a given potential difference between filament and central electrode. However, the collection efficiency is close to unity for all sizes up to 15 nm and above 50 % for sizes up to 30 nm, which can be regarded as the current upper size limit of the TD-DMA. Therefore, it can be claimed that the collection efficiency is sufficiently high for the size range up to 30 nm, and the results can be corrected with the experimentally determined values (see Sect. 5.1, Eq. 11).
3.3 Filament temperature
In order to evaporate the sample in front of the mass spectrometer, the filament is heated. This is achieved by sending an electric current through the wire. Due to the rather high resistance of the filament, power is released and the filament temperature increases. Also, the platinum changes its resistance (RFilament) due to the heating. This feature can be used to estimate the filament temperature in a similar way as in platinum thermometers such as Pt100. Here, it is not crucial to know the temperature with high precision, nevertheless it can be beneficial to determine it, e.g., in order to gain information about the volatilities of substances in an aerosol sample.
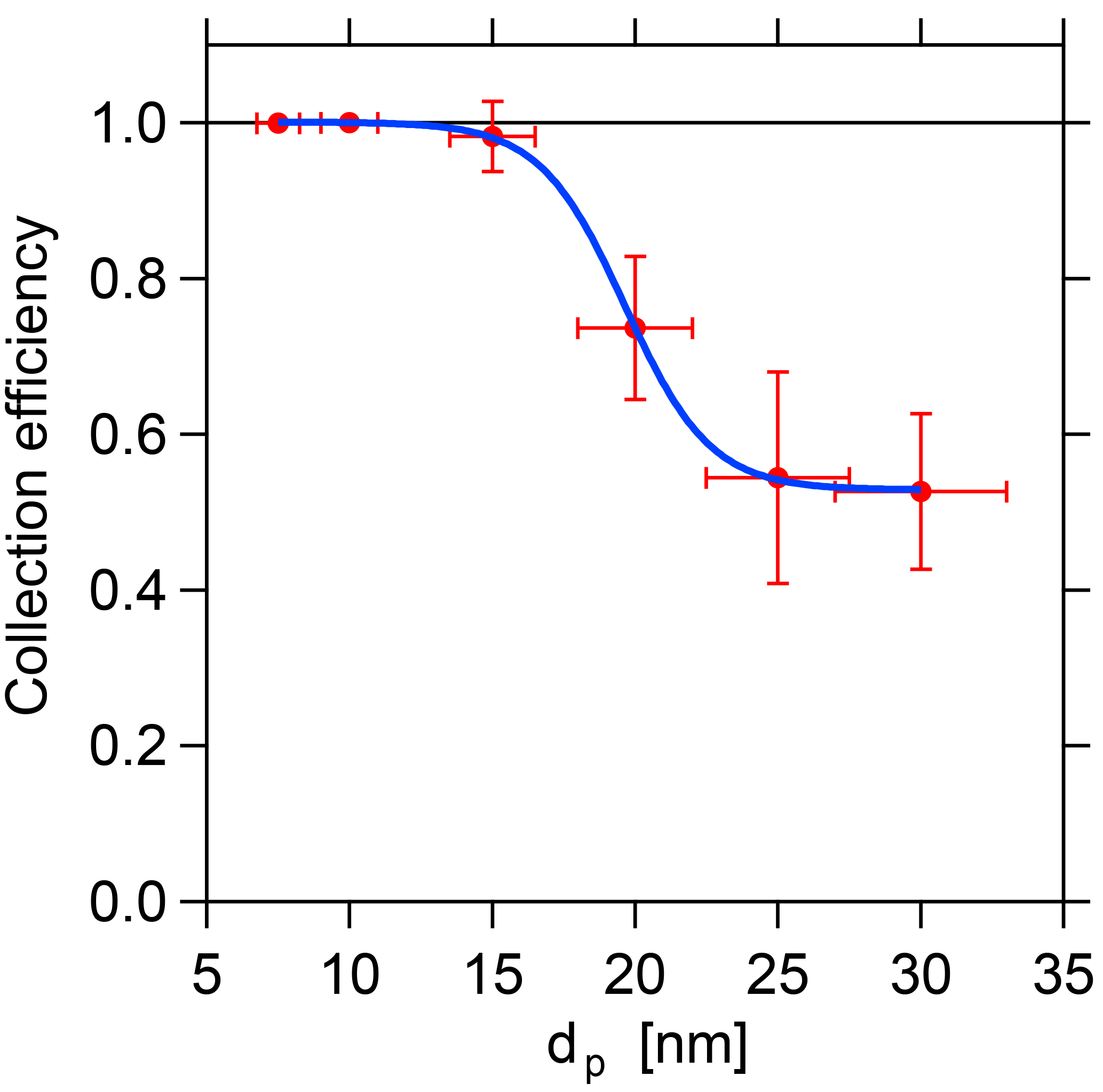
Figure 6Collection efficiency depending on the particle size. Due to the decreased electrical mobility, the required collection voltage increases with particle size, which currently limits the collection efficiency at larger sizes.
The control unit of the TD-DMA monitors the voltage U and current I across the filament. However, the circuit in the electronic control unit as well as the wires connecting the filament also add to the total resistance:
The filament resistance at room temperature (20 ∘C) is 0.4 Ω. Using this number together with the temperature dependent resistance of platinum,
with Ω K−1 and Ω K−2 (International Electrotechnical Commission, 2008), R(0 ∘C) can be derived. Then, by applying very small currents that hardly warm the filament and measuring the magnetic field resulting thereof with a current clamp, one can estimate the resistance of the system as Ω. With this information we can derive the temperature T during all times of the evaporation process by using the feedback voltage U and current I:
During standard operation, the filament is heated up to 350 ∘C for evaporating the sample and up to 600 ∘C for cleaning.
3.4 Sample mass calibration and reproducibility
Sample mass calibration tests relate evaporated mass to mass spectrometer response. This method is used to test the evaporation method regarding its reproducibility and is required for the signal quantification (see Sect. 5.2) and determination of the detection limit. When installing the TD-DMA at the mass spectrometer, e.g., after transport, they are also used to find the optimum filament position in front of the mass spectrometers ion source.
For the sample mass calibration experiments, a defined amount of a test substance is deposited on the filament and evaporated in front of the mass spectrometer. In this case, a solution of sulfuric acid (H2SO4) in water is used. After reaction with the nitrate primary ions, this substance produces different ion signals (peaks) with the main contribution coming from clusters of sulfuric acid with the primary ion monomer () and bisulfate ions (). Sulfuric acid dimers () as well as clusters of sulfuric acid with the primary ion dimer () are also detected, but with a much lower intensity than the former, so that clusters with the primary ion trimer and higher are negligible.
However, for measuring internally or externally mixed particles, it should be noted that these consist of more than one substance; therefore, the signals are spread over many m∕z peaks, which will require more total particle mass to overcome the detection limit. In addition, the ionization efficiency of a compound will affect its detection limit in the same way as in gas-phase measurements.
Figure 7a shows the typical shape of the signals resulting from the desorption process. It shows a sharp increase when a certain temperature is reached followed by an exponential decay reaching background levels within less than 30 s. This indicates that the substance desorbs from the filament within a narrow temperature range and that the temperature distribution of the filament is fairly homogeneous, otherwise the signal would be smeared out more strongly.
For signal analysis, the full spectrum is corrected with the relative transmission efficiency (Heinritzi et al., 2016), which takes into account that the detection efficiency inside the mass spectrometer is a function of ion mass. The individual signals Sj are then normalized by the primary ions' count rates, in this case (m∕z 62), (m∕z 125) and (m∕z 188). Subsequently, the signal is integrated over the evaporation time in order to obtain the total signal resulting of a substance i from the evaporation. Finally, the signals of all relevant peaks j are summed up.
These time integrated signals are displayed in Fig. 7b and c. All data points are corrected with blank measurements obtained by evaporating only the solvent (water), without the test substance. Reproducible results are obtained for a sample mass as small as 0.01 ng. Additionally, the filament is heated without any sample or solvent on it to obtain a zero measurement (see also Sect. 4.1, heating background). This zero measurement plus 3 times its standard deviation defines the lower detection limit. The zero measurement and the measurement points below 0.03 ng, although they show a good reproducibility, are systematically lower than the signals when larger masses are applied, which is not yet understood.

Figure 7Sample mass calibration with (a) typical desorption shape of signals, (c) integrated signal vs. injected mass, and (b) zoom on lower masses with detection limits.
Starting from a deposited mass of 0.03 ng, the data indicates very good linearity, which is an important feature of the TD-DMA combined with a CI-APi-TOF. Furthermore, we can now relate the mass spectrometer signal to a sample mass. For the component used here, the conversion factor k is 42.95 ng (ncps s)−1. For other substances, their individual ionization efficiencies relative to that of the test substance need to be taken into account (relative ionization efficiency RIEi, see also Sect. 5.2) and a mass spectrometer signal relates to a mass of
In conclusion, the reproducibility and linearity of the evaporation process can be clearly stated.
For measuring a nucleation event, the following procedure is desirable: at the beginning of the event, the integral measurement mode is used to analyze the freshly nucleated, smallest particles. When the mode diameter of the particle size distribution reaches around 10 nm, several size-resolved measurements are performed. The sampling time is adjusted so that enough mass is collected for a given particle size and concentration in the aerosol sample. However, due to the minute mass contained in the nanoparticles, such a procedure is not always possible, especially for short or weak nucleation events. In the future, we thus aim to improve the sensitivity, as suggested in Sect. 6. For now, in case of short nucleation events or those with low concentrations, one can choose one size and sample for the whole event or use the integral mode. In either case, different kinds of background measurements are important and thus the different steps during a full measurement cycle are described in the following.
4.1 Background measurements and signal correction
To verify measurements and to distinguish the signal arising from particles from possible other sources, background measurements need to be taken regularly.
-
Mass spectrometer background. To determine this background contribution, the nitrogen carrier gas (the flow applied to the sampling line of the mass spectrometer during the evaporation of particles) is applied to the mass spectrometers sampling line without positioning the filament inside it (Fig. 1a). This measurement accounts for the instrumental backgrounds of the CI-APi-TOF (ion source and detector) and is taken shortly before and after every evaporation. It is also used to correct the gas-phase measurements of the CI-APi-TOF.
-
Heating background. The filament is placed at its evaporation position and heated, but without an aerosol sample. As the evaporation takes place in the inlet line that is also used for the gas-phase measurements, the walls contain adsorbed material, which could re-enter into the gas phase due to the heating. The core sampling system, in which the filament is placed (see Fig. 2b), prevents most of the gas flow directly in contact with the walls from entering the ion source of the mass spectrometer. However, the inlet line downstream of the core sampling probe has been in contact with the gas phase. In the chemical settings tested, it has been observed that for filament temperatures lower than 400 ∘C, the signal from this process is negligible. This is the temperature range where most of the particle-phase sample desorbs. For higher temperatures, the background signals increase. When observing individual ion signals as a function of time during heating, the background contribution from the sampling line walls also shows a different shape compared to the particle-phase signal (Fig. 8). The signals from particles on the filament show distinct spikes with a fast increase and a rapid exponential decay, while the wall background results in slowly rising signals, which do not show a pronounced decay.
-
Gas-phase adsorption background. The gas-phase adsorption on the filament is corrected for by placing the filament in the sampling position inside the TD-DMA and performing the measurement in the same way as regular sampling, the only difference being that the filament is set to the same potential as the central electrode; thus, no particles are sampled. When the nitrogen sheath flow is used, as in the size-resolved measurements, the filament is only exposed to nitrogen and there is hardly any relevant gas-phase adsorption. However, for the integral mode, the sheath flow is turned off and the filament is thus exposed to the gas phase of the aerosol sample. When heating the filament, the signals show the same shape as for the particle measurements, as they are originating from the filament itself. This background measurement is taken for every major change in gas-phase chemistry. For most of our tested systems, this background was comparably low and mostly negligible.
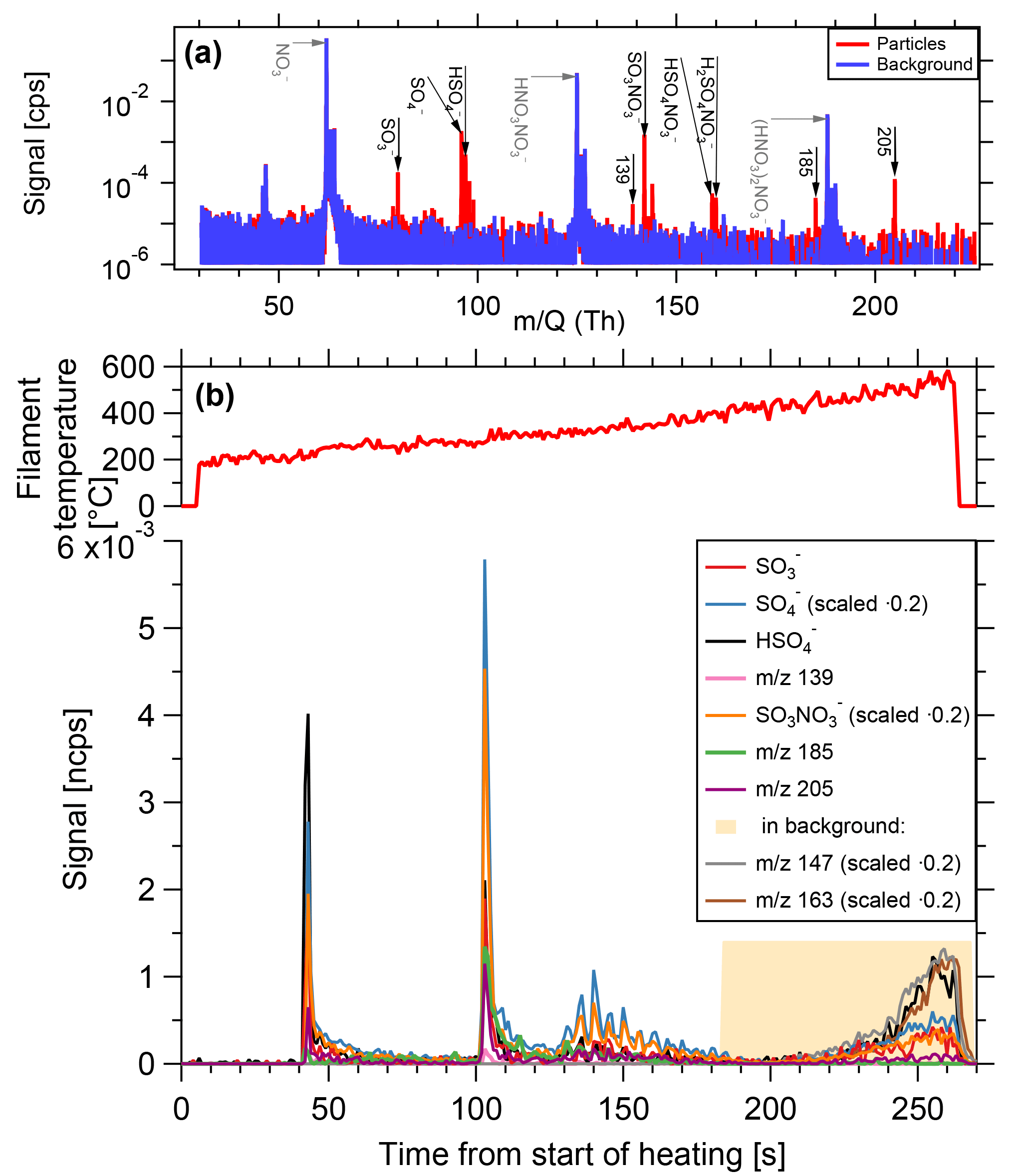
Figure 8Composition measurement of 15 nm particles generated from dimethylamine, sulfur dioxide and ozone in presence of UV light. (a) Spectrum of particle phase compared to the background. Prominent particle-phase peaks in black, primary ions in grey. (b) Time series of evaporation. As filament temperature increases, substances desorb from the filament and are depleted rapidly. At high temperatures, signals originating from the inlet line appear.
4.2 Example measurement
The instrument was tested during the CLOUD10T campaign at the Cosmics Leaving Outdoor Droplets (CLOUD) chamber at the European Organization for Nuclear Research (CERN). CLOUD is a 26 m3 stainless steel chamber used for investigating nucleation and growth of aerosol particles under atmospherically relevant and precisely controlled conditions (Kirkby et al., 2011). Figure 8 shows a measurement of size selected 15 nm particles generated from 119 pptv of dimethylamine, 4.3×106 cm−3 sulfuric acid, at a temperature of 4.8 ∘C and relative humidity of 40.2 %. The total particle number concentration (>2.5 nm) is approximately 1.7×105 cm−3 and the TD-DMA collected its analyte particles for a duration of 60 min. While the filament is gradually heated, substances desorb and are subsequently ionized by the negative nitrate primary ions and detected by the mass spectrometer. The example substances shown here desorb at different temperatures. The signal shape for a given ion signal as a function of time is the same as for the sample mass calibration (Sect. 3.4, Fig. 7); a sharp increase is followed by an exponential decrease, as the material on the filament is depleted. At filament temperatures beyond 350 to 400 ∘C, signals of different shapes appear (in this case, e.g., m∕z 147 and m∕z 163). These signals are also found in the heating background measurements and do not originate from the collected analyte particles. Instead, they are caused by desorption of substances from the inlet line due to the increased temperature of the carrier gas. The fact that the inlet line is a much larger reservoir than the filament explains why the shape of these background signals is different and why they do not decrease within a few seconds. The temperature range where valid measurements are taken is thus up to ∼350 ∘C. Nevertheless, the filament is heated up to 600 ∘C in order to ensure that all particulate material evaporates and memory effects are avoided.
Some substances seem to desorb at two different temperatures. This can happen for two reasons: (1) the filament might be heated unevenly, but this would also result in less pronounced peaks instead of two sharp peaks. (2) Different chemical species that do not possess the same evaporation temperature, can result in the same product ions. In the example shown here, it is not exactly clear whether sulfuric acid belongs to dimethylamonium-bisulfate or dimethylamonium-sulfate, and what influence potential contaminants such as ammonia can have on the evaporation temperature (Lawler et al., 2016). (3) The signal appearing first originates from desorption of the molecule with this given mass to charge ratio, whereas the second appearance could be due to the fragmentation of a larger molecule. Some amount of the larger molecules seem to fragment and these fragments are then detected at higher temperatures than expected for a desorption (see also Sect. 6). In the chemical system of dimethylamine and sulfuric acid, one can also observe the selectivity of the ionization by nitrate ions. Sulfuric acid, for example, has a lower detection limit of 5×104 cm−3 (∼0.00185 pptv) (Kirkby et al., 2016), whereas for dimethylamine the lower detection limit of 1.7 pptv (Simon et al., 2016) is almost a factor of 103 higher. Therefore, the signals related to sulfuric acid are very strong whereas DMA related signals mostly do not reach the detection limit.
5.1 Collected particle mass during a measurement
The collected mass on the filament is calculated in order to quantify the measured signals. Additionally, it is estimated before the measurement from the present particle size distribution and the selected particle diameter so that the collected particle mass exceeds the detection limit.
As a DMA cannot provide an absolute monodisperse aerosol (see Sect. 3.1), the incoming number size distribution dn∕ddp of the sample in combination with the transmission efficiency τ(dp), collection efficiency η(dp) and charge fraction x(dp) is needed to calculate the collected mass precisely. The transmission and collection efficiency are retrieved from the TD-DMA characterization (Sect. 3, Figs. 4, 5, and 6) and the incoming size distribution can be provided by an SMPS or other instrument measuring the number size distribution of the analyte aerosol in the applicable size range. Thus, the collected mass on the filament can be calculated according to the following equation:
with Qaerosol the aerosol flow rate through the TD-DMA, ρ the particle density and the selected diameter. The amount of mass collected per time thus depends both on the selected diameter and on the size distribution of the aerosol analyte. In addition, as for any DMA, the effective diameter collected varies with the size distribution of the background aerosol. To get an impression of the amount of mass that can be collected under certain conditions, we look at the aerosol size distribution of the example event described in Sect. 4.2 (see Fig. 9). The lower panel displays the number size distribution and median diameter (black line), as well as the time period of collection (pink line). The upper panel shows the particle mass collected by the TD-DMA per unit time when the instrument is set to collect a certain diameter; it ranges between 10−5 and 10−3 ng s−1. Integration over time yields the total mass collected as described by Eq. (11).
The minimum mass concentration in the analyte aerosol where measurements with the TD-DMA are possible, depends not only on size distribution and selected size but also on sampling time and analyte aerosol composition. For example, the lower limit for sulfuric acid particles with a sampling time of 2 h is 27 pg m−3. Measuring in size-resolved mode from monodisperse 15 nm particles, 811 pg m−3 are needed. Assuming the number size distribution from the example nucleation event (Fig. 8) and measuring all sizes, the mass concentration should be higher than 385 pg m−3. The TD-DMA's sensitivity is thus comparable to other instruments operating in this size range (see Sect. 6.2).
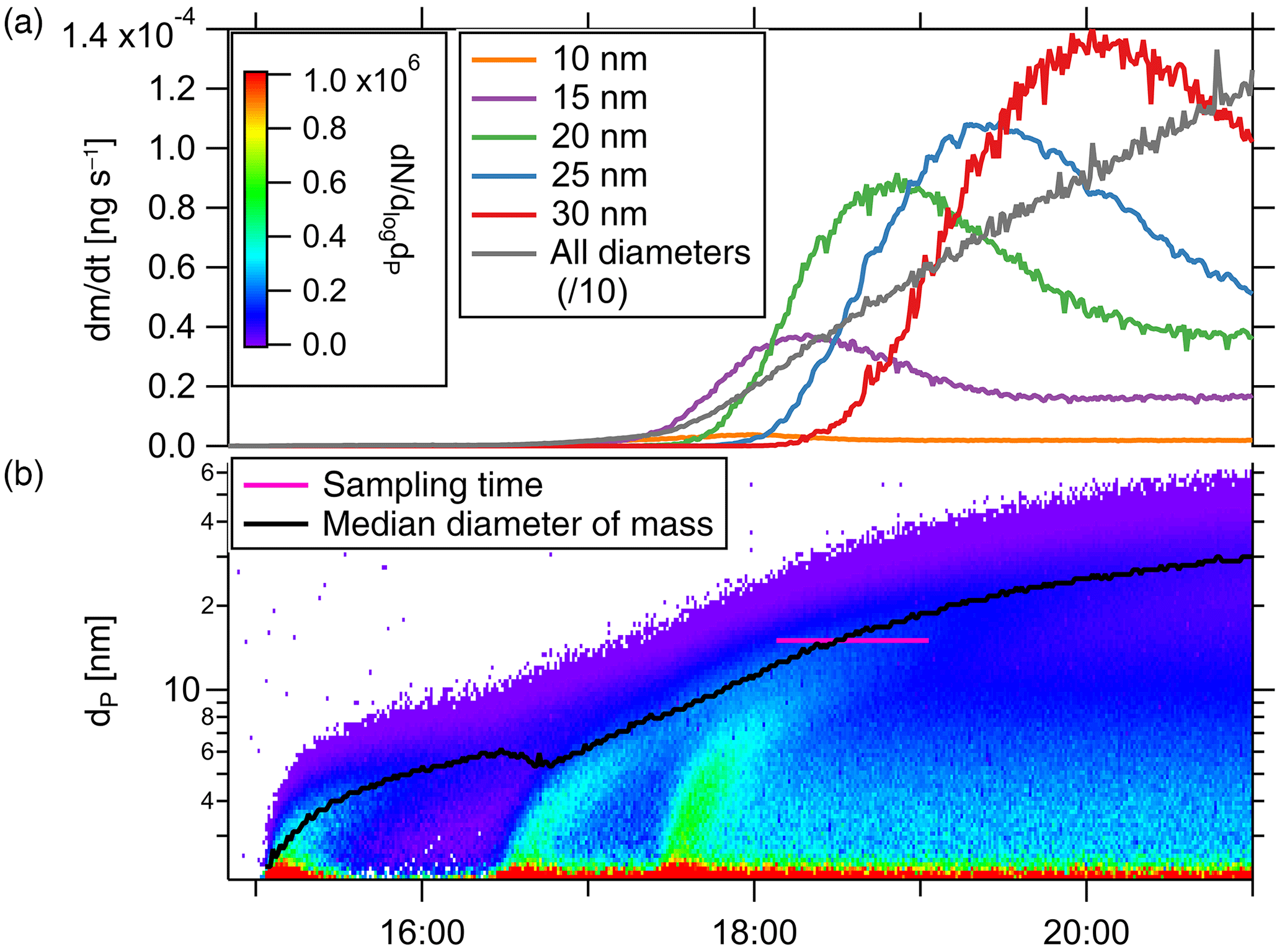
Figure 9Particle mass collected by the TD-DMA per unit time when the instrument is set to collect a certain diameter (a), for a given size distribution (b).
5.2 Mass fraction of individual substances from time integrated signals
The fraction of a substance in the aerosol sample shall be determined. Like this, the relative contribution of an individual substance or a group of substances (e.g., all organic compounds) to nucleation can be investigated, e.g., for the same size under different conditions or with increasing size under constant conditions.
The calculation requires the collected mass from Sect. 5.1 and the conversion factor from the sample mass calibration in Sect. 3.4. The fraction fi of an individual substance in the aerosol sample is thus
Note that the collected mass calculated from the SMPS data should be used instead of the sum of all identified components, . Using the latter would be based on the assumption that all compounds of the aerosol could be charged, detected, identified and quantified. As we know from gas-phase measurements, this is usually not the case for the chemically complex atmospheric aerosols. Chemical ionization, especially the negative nitrate ionization used here, is a selective technique. A substance needs to react with the primary ions to be charged and detected in the mass spectrometer. This can either be the transfer of a positively charged hydrogen atom from the substance to the primary ion, or clustering with the primary ion (Hyttinen et al., 2017). For the same reason, the signal of each substance also needs to be corrected with its ionization efficiency (see Sect. 3.4) and this property is not known for all substances. That circumstance can also be bypassed by comparing the contribution of compounds under different conditions rather than providing an absolute concentration.
6.1 Discussion on the newly developed TD-DMA
The detection limit and sensitivity are a limit to the parameters particle size, number concentration of the analyte aerosol, ionization efficiency, complexity of the spectrum and time resolution. A major challenge is the fact that small particles have an extremely low mass. For example, a particle of half the diameter contains less mass by a factor of 8. In addition, charging probability decreases with size. At 15 nm, the fraction of singly negatively charged particles is less than 0.07 (Fuchs, 1963; Tigges et al., 2015) for the soft X-ray charger used here. For an aerosol of multi component composition, it applies that the more particle mass is collected, the more substances overcome the lower detection limit and the more detailed the observed mass spectrum will be. The TD-DMA is thus designed for high transmission at a coarse resolution. In this way, a size selection is possible but still a sufficient amount of mass is collected. In case of very low particle concentrations or when a high time resolution is needed, it is also possible to sample all particle sizes in an integral mode by turning off the sheath flow. The low mass concentration of the particulate matter is also the reason why a discontinuous system with consecutive collection and evaporation is chosen. Conversely, the probability for multiple charging increases with particle diameter, so that particles larger than the target size can contaminate the sample. This can be corrected for when counting, but not in the chemical analysis. Within the size range of 10 to 30 nm that the TD-DMA is specialized for, multiple charging does not play a significant role. A typical particle size selected with the TD-DMA is 15 nm. Doubly charged particles of the same electrical mobility have a diameter of 21 nm (Stolzenburg and McMurry, 2008; Hinds, 1999). Considering the charging probabilities of the used soft X-ray charger (Tigges et al., 2015), a fraction of of the 15 nm particles carry one charge, whereas just a fraction of of the 21 nm particles carry two charges. With the particle mass being proportional to their cubic diameter, the contribution of 21 nm particles to a 15 nm sample would be . Thus, at the maximum only a fraction of 0.009 of the particle mass in the sample would originate from doubly charged larger particles. This assumes a uniform particle number size distribution. As for new particle formation events, the smallest particles have the highest number concentration, this estimate calculation gives an upper limit. In case of very large background particles in the accumulation mode size range, as, for example, in field use, an impactor should be used.
The soft X-ray charger was chosen for that reason and due to the fact that it does not change the chemistry much, compared to, e.g., a corona charger. Nevertheless, it would be beneficial to find a charger which offers a higher charging probability for small particles while still fulfilling the above criteria.
For optimizing the collection process, it would be beneficial to enhance the collection efficiency (see Sect. 3.2) also for larger sizes. Applying a larger collection voltage to the filament increases the collection efficiency, which is currently limited by leak currents occurring between filament and central electrode when the potential difference is too high. It is also a possibility to prepare the DMA for a higher flow of the analyte aerosol in order to collect more mass.
The exact position of the filament, in combination with the flow profiles at the place of evaporation, is crucial and optimization of these features has a high impact on the sensitivity. Also, the high temperature gradient when heating the filament releases the analyte in a very compact way, which benefits the lower detection limit. This method of direct heating ensures a homogeneous and precisely controlled temperature for the desorption of the substances. Currently we have a discrepancy between the TD-DMA's sample mass calibration and the gas-phase calibration of sulfuric acid (Kürten et al., 2012). This can be explained by losses in the transition from filament to mass spectrometer due to condensation on the mass spectrometer's inlet line. There have also been tests to evaporate the sample directly inside the ion source, but for the presently used ion source (Kürten et al., 2011) this was not expedient. Nevertheless, these losses are unsatisfactory and possibilities for further improvements are being investigated.
As discussed in Sect. 4.2, the filament material might be problematic in terms of influencing the analyte. Heating, especially from metallic surfaces, might lead to fragmentation or other chemical modification of the original molecules. We tested different filaments and saw that not only the material but also the way the wire is manufactured strongly influences this process and thus the suitability of the filament. Also, it was found that a careful heating procedure seems to reduce these effects. This was investigated by using the sample mass calibration method (see Sect. 3.4) with different substances and by careful examination of the spectra from nucleation events. Some fraction of the larger molecules (for example of the HOMs) seem to fragment and these fragments are then detected at higher temperatures than molecules that have the same elemental composition. A detailed analysis of this issue will be the subject of future studies. A complete degeneration of substances is hardly observed for the relevant temperature range, only very small peaks that might originate from such reactions are present in the spectra at higher temperatures. Nevertheless, it cannot be excluded that some material breaks up into substances that are not ionized by the negative nitrate CI-APi-TOF and are thus not visible in the spectra. By coating the filament with a thin layer of a chemically inert material, this could be reduced.
The ionization with negative nitrate primary ions is a selective technique. To target more substances like, e.g., amines and ammonia, different primary ions can be chosen (Lawler et al., 2016). To distinguish the aerosol sample from potential gas-phase adsorption, a sheath flow of pure nitrogen surrounds the filament in the size selective mode. In the integral mode, the filament is also exposed to the gas phase of the analyte and a gas-phase background measurement is advised (see Sect. 4.1).
Up to now, the TD-DMA has been used in chamber experiments with timed nucleation events, but it is also intended for field use in the future. Here, the size selective mode is especially useful because the larger background particles would otherwise dominate the mass concentration. Also, background measurements, as suggested in Sect. 4.1 (3) are not straightforward when chemical conditions change over time. To look further into the chemistry of aerosol growth, it will be interesting to perform measurements at a flow tube, where a steady state production of the analyte aerosol is possible. When not being limited by the time resolution or small number concentrations, one can analyze particles starting at small sizes with a high size resolution, which is variable in this method, and thus gain a very detailed picture of the growth process.
6.2 Comparison of instruments capable of chemical analysis of sub-30 nm particles
The new instrument TD-DMA shall be placed in the context of existing techniques for the online chemical analysis of sub-30 nm aerosol particles. When comparing the different nanoparticle instruments, one has to focus on a selection of criteria as the instruments strongly differ from one another and have their individual functional principles and thus strengths and weaknesses.
To give an overview regarding the functional principle of the devices (see Table 1), we introduced the following criteria: (1) continuous vs. discontinuous, (2) size-selective vs. non-size-selective, (3) how the particulate material is evaporated and (4) whether the gas phase can be measured simultaneously.
One of the most important criteria is the sensitivity of the instruments, but the question arises of which is the appropriate parameter to describe this. Especially the fact that the cited studies do not all refer to the same parameter when discussing the sensitivities of their instruments, makes a comparison difficult.
One option for a meaningful number is the minimum mass concentration of the analyte aerosol (pg m−3) which is needed to perform measurements. However, this is not a constant number for each instrument but depends strongly on the number size distribution, target substances, chemical composition of the analyte aerosol, collection time and other factors. For example, an instrument specialized for size selective analysis of small particles will profit more from a mode of smaller particles, whereas an approach sampling all sizes will benefit more from a mode of larger particles as they contain more mass. Thus for small particles, the first instrument will have a higher sensitivity, and for larger particles, the second one. For the TD-DMA, we thus derived the sensitivity towards sulfuric acid for three different circumstances: measuring all particles (27 pg m−3), selecting 15 nm particles from a monodisperse mode (811 pg m−3) and a non-size-resolved measurement assuming the number size distribution of an example nucleation event (385 pg m−3, see also Sect. 5.1). Lopez-Hilfiker et al. (2014) compared their sensitivities between a chamber study and an ambient study with different background aerosol conditions, temperature, gas-phase concentrations and sample volume. Although their target molecules were all oxygenated organics, the resulting detection limits in pg m−3 span a range of two orders of magnitude. The minimum detectable mass (pg) would be another option, but this parameter is only meaningful in combination with the information how much mass can be sampled from the analyte aerosol. In addition, most techniques use chemical ionization to charge the sample after it is transferred to the gas phase. This is a selective technique and depending on the primary ion chosen, different target substances will result in different sensitivities. Horan et al. (2017) report a seemingly rather high lower detection limit of 105 pg m−3, but the instrument has nevertheless proven the ability to analyze particles as small as 13 nm. Therefore, a higher number in pg m−3 does not necessarily mean that an instrument is less suitable for analyzing small particles. However, the smallest detectable particle size certainly depends on parameters like the mass concentration of the analyte aerosol and collection time. Table 1 contains a selection of parameters considering the sensitivity. However, we emphasize that this table represents only an overview on the available techniques and care has to be taken for the exact interpretation due to the sensitivity's dependence on other parameters.
Apart from the sensitivity of a technique, other criteria are also relevant. An important example is in how far the aerosol components fragment when they are transferred to the gas phase, or in how far the instrument can detect semi stable reaction products from particle-phase reactions. These questions are not discussed in this short overview as it is beyond the scope of the present manuscript. Ultimately, it would be beneficial to have several instruments measure the same sub-30 nm aerosol particle population during an intercomparison campaign.
The TD-DMA is suitable for measuring nucleation and Aitken mode particles and is successfully analyzing aerosol mass concentrations in the pg m−3 range. The DMA unit is optimized for a high transmission at a coarse resolution to collect a sufficient amount of mass. The characterization of the DMA unit and the evaporation of a test substance show to be reproducible. The instruments ability to measure freshly nucleated particles was proven in chamber experiments.
The advantages of the instrument are as follows: (a) the TD-DMA allows for size selection, thus the chemical composition of different particle sizes can be compared in order to determine the relative importance of different vapors for particle growth as a function of size. (b) An integral, non-size-selective mode of operation is possible in order to maximize the mass of collected particles. (c) The TD-DMA is a modular and compact unit that can in principle be used with different mass spectrometers or other gas analyzers. (d) During the particle collection process, the gas phase can be analyzed. For this reason, the same mass spectrometer can be used for particle as well as gas-phase measurements and both phases can be directly compared. With this instrument, the processes affecting particle composition and growth can be investigated in detail.
The research data can be accessed from https://doi.org/10.5281/zenodo.1443176 (Wagner et al., 2018).
The instrument was mainly developed by ACW and AK, with major contributions from JC, AB, SB, CF, ME and JH. ACW, AB and SB carried out the measurements and analyzed the data. ACW wrote the manuscript. All authors contributed to the scientific discussion and commented on the manuscript.
The authors declare that they have no conflict of interest.
We thank the workshop team of our institute for manufacturing the components and for technical support. We appreciate the useful discussion with Jim Smith from University of California, Irvine, USA, and Alexander Vogel, Institute for Atmospheric and Environmental Science, Frankfurt, Germany. We also thank Jasmin Tröstl and Urs Baltensperger from Paul Scherrer Institute, Villigen, Switzerland, for providing a nSMPS and the whole CLOUD team for support during the CLOUD10T measurement campaign.
This research was supported by the German Federal Ministry of Education and
Research (project “CLOUD-12” 01LK1222A and “CLOUD-16” 01LK1601A) as well as
the EC Seventh Framework Programme (MC-ITN “CLOUD-TRAIN” 316662).
Edited by: Johannes Schneider
Reviewed by: two anonymous referees
Arnold, F., Curtius, J., Spreng, S., and Deshler, T.: Stratospheric aerosol sulfuric acid: First direct in situ measurements using a novel balloon-based mass spectrometer apparatus, J. Atmos. Chem., 30, 3=-10, https://doi.org/10.1023/A:1006067511568, 1998.
Breitenlechner, M., Fischer, L., Hainer, M., Heinritzi, M., Curtius, J., and Hansel, A.: PTR3: An Instrument for Studying the Lifecycle of Reactive Organic Carbon in the Atmosphere, Anal. Chem., 89, 5824–5831, https://doi.org/10.1021/acs.analchem.6b05110, 2017.
Bzdek, B. R., Pennington, M. R., and Johnston, M. V.: Single particle chemical analysis of ambient ultrafine aerosol: A review, J. Aerosol Sci., 52, 109–120, https://doi.org/10.1016/j.jaerosci.2012.05.001, 2012.
Chen, D. R., Pui, D. Y. H., Hummes, D., Fissan, H., Quant, F. R., and Sem, G. J.: Design and evaluation of a nanometer aerosol differential mobility analyzer (Nano-DMA), J. Aerosol Sci., 29, 497–509, https://doi.org/10.1016/S0021-8502(97)10018-0, 1998.
Core Writing Team, Pachauri, R. K., and Reisinger, A. (Eds.): Climate Change 2007: Synthesis Report. Contribution of Working Groups I, II and III to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change, IPCC, Geneva, Switzerland, 2007.
Curtius, J. and Arnold, F.: Measurement of aerosol sulfuric acid: 1. Experimental setup, characterization, and calibration of a novel mass spectrometric system, J. Geophys. Res.-Atmos., 106, 31965–31974, https://doi.org/10.1029/2001JD000604, 2001.
Curtius, J., Sierau, B., Arnold, F., Baumann, R., Busen, R., Schulte, P., and Schumann, U.: First direct sulfuric acid detection in the exhaust plume of a jet aircraft in flight, Geophys. Res. Lett., 25, 923–926, https://doi.org/10.1029/98GL00512, 1998.
Donaldson, K. and Borm, P.: Particle Toxicology, CRC Press, Boca Raton, Florida, USA, ISBN: 1420003143, 2006.
Dunne, E. M., Gordon, H., Kürten, A., Almeida, J., Duplissy, J., Williamson, C., Ortega, I. K., Pringle, K. J., Adamov, A., Baltensperger, U., Barmet, P., Benduhn, F., Bianchi, F., Breitenlechner, M., Clarke, A., Curtius, J., Dommen, J., Donahue, N. M., Ehrhart, S., Flagan, R. C., Franchin, A., Guida, R., Hakala, J., Hansel, A., Heinritzi, M., Jokinen, T., Kangasluoma, J., Kirkby, J., Kulmala, M., Kupc, A., Lawler, M. J., Lehtipalo, K., Makhmutov, V., Mann, G., Mathot, S., Merikanto, J., Miettinen, P., Nenes, A., Onnela, A., Rap, A., Reddington, C. L. S., Riccobono, F., Richards, N. A. D., Rissanen, M. P., Rondo, L., Sarnela, N., Schobesberger, S., Sengupta, K., Simon, M., Sipilä, M., Smith, J. N., Stozkhov, Y., Tomé, A., Tröstl, J., Wagner, P. E., Wimmer, D., Winkler, P. M., Worsnop, D. R., and Carslaw, K. S.: Global atmospheric particle formation from CERN CLOUD measurements, Science, 354, 1119, https://doi.org/10.1126/science.aaf2649, 2016.
Ehn, M., Thornton, J. A., Kleist, E., Sipilä, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I.-H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurtén, T., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., Canagaratna, M., Maso, M. D., Berndt, T., Petäjä, T., Wahner, A., Kerminen, V.-M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476, https://doi.org/10.1038/nature13032, 2014.
Eisele, F. L. and Tanner, D. J.: Measurement of the gas phase concentration of H2SO4 and methane sulfonic acid and estimates of H2SO4 production and loss in the atmosphere, J. Geophys. Res.-Atmos., 98, 9001–9010, https://doi.org/10.1029/93JD00031, 1993.
Flagan, R. C.: Differential Mobility Analysis of Aerosols: A Tutorial, KONA Powder Part. J., 26, 254–268, https://doi.org/10.14356/kona.2008023, 2008.
Franchin, A., Downard, A., Kangasluoma, J., Nieminen, T., Lehtipalo, K., Steiner, G., Manninen, H. E., Petäjä, T., Flagan, R. C., and Kulmala, M.: A new high-transmission inlet for the Caltech nano-RDMA for size distribution measurements of sub-3 nm ions at ambient concentrations, Atmos. Meas. Tech., 9, 2709–2720, https://doi.org/10.5194/amt-9-2709-2016, 2016.
Fuchs, N. A.: On the stationary charge distribution on aerosol particles in a bipolar ionic atmosphere, Geofisica pura e applicata, 56, 185–193, https://doi.org/10.1007/BF01993343, 1963.
Fuzzi, S., Baltensperger, U., Carslaw, K., Decesari, S., Denier van der Gon, H., Facchini, M. C., Fowler, D., Koren, I., Langford, B., Lohmann, U., Nemitz, E., Pandis, S., Riipinen, I., Rudich, Y., Schaap, M., Slowik, J. G., Spracklen, D. V., Vignati, E., Wild, M., Williams, M., and Gilardoni, S.: Particulate matter, air quality and climate: lessons learned and future needs, Atmos. Chem. Phys., 15, 8217–8299, https://doi.org/10.5194/acp-15-8217-2015, 2015.
Gonser, S. G. and Held, A.: A chemical analyzer for charged ultrafine particles, Atmos. Meas. Tech., 6, 2339–2348, https://doi.org/10.5194/amt-6-2339-2013, 2013.
Graus, M., Müller, M., and Hansel, A.: High Resolution PTR-TOF: Quantification and Formula Confirmation of VOC in Real Time, J. Am. Soc. Mass Spectr., 21, 1037–1044, https://doi.org/10.1016/j.jasms.2010.02.006, 2010.
Hallquist, M., Wenger, J. C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N. M., George, C., Goldstein, A. H., Hamilton, J. F., Herrmann, H., Hoffmann, T., Iinuma, Y., Jang, M., Jenkin, M. E., Jimenez, J. L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, Th. F., Monod, A., Prévôt, A. S. H., Seinfeld, J. H., Surratt, J. D., Szmigielski, R., and Wildt, J.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009.
Hansel, A., Jordan, A., Holzinger, R., Prazeller, P., Vogel, W., and Lindinger, W.: Proton transfer reaction mass spectrometry: on-line trace gas analysis at the ppb level, Int. J. Mass Spectrom., 149–150, 609–619, https://doi.org/10.1016/0168-1176(95)04294-U, 1995.
He, S., Li, L., Duan, H., Naqwi, A., and Hogan, C. J.: Aerosol Analysis via Electrostatic Precipitation-Electrospray Ionization Mass Spectrometry, Anal. Chem., 87, 6752–6760, https://doi.org/10.1021/acs.analchem.5b01183, 2015.
Heinritzi, M., Simon, M., Steiner, G., Wagner, A. C., Kürten, A., Hansel, A., and Curtius, J.: Characterization of the mass-dependent transmission efficiency of a CIMS, Atmos. Meas. Tech., 9, 1449–1460, https://doi.org/10.5194/amt-9-1449-2016, 2016.
Hermann, M., Wehner, B., Bischof, O., Han, H. S., Krinke, T., Liu, W., Zerrath, A., and Wiedensohler, A.: Particle counting efficiencies of new TSI condensation particle counters, J. Aerosol Sci., 38, 674–682, https://doi.org/10.1016/j.jaerosci.2007.05.001, 2007.
Hinds, W. C.: Aerosol Technology, 2nd ed., Wiley Interscience, ISBN: 978-0-471-19410-1, 1999.
Horan, A. J., Apsokardu, M. J., and Johnston, M. V.: Droplet Assisted Inlet Ionization for Online Analysis of Airborne Nanoparticles, Anal. Chem., 89, 1059–1062, https://doi.org/10.1021/acs.analchem.6b04718, 2017.
Hyttinen, N., Otkjær, R. V., Iyer, S., Kjaergaard, H. G., Rissanen, M. P., Wennberg, P. O., and Kurtén, T.: Computational Comparison of Different Reagent Ions in the Chemical Ionization of Oxidized Multifunctional Compounds, J. Phys. Chem. A, 122, 269–279, https://doi.org/10.1021/acs.jpca.7b10015, 2017.
International Electrotechnical Commission: IEC 60751 – Industrial platinum resistance thermometers and platinum temperature sensors, Geneva, Switzerland, ISBN: 2-8318-9849-8, 2008.
Jayne, J. T., Leard, D. C., Zhang, X., Davidovits, P., Smith, K. A., Kolb, C. E., and Worsnop, D. R.: Development of an Aerosol Mass Spectrometer for Size and Composition Analysis of Submicron Particles, Aerosol Sci. Tech., 33, 49–70, https://doi.org/10.1080/027868200410840, 2000.
Jiang, J., Attoui, M., Heim, M., Brunelli, N. A., McMurry, P. H., Kasper, G., Flagan, R. C., Giapis, K., and Mouret, G.: Transfer Functions and Penetrations of Five Differential Mobility Analyzers for Sub-2 nm Particle Classification, Aerosol Sci. Tech., 45, 480–492, https://doi.org/10.1080/02786826.2010.546819, 2011.
Jokinen, T., Sipilä, M., Junninen, H., Ehn, M., Lönn, G., Hakala, J., Petäjä, T., Mauldin III, R. L., Kulmala, M., and Worsnop, D. R.: Atmospheric sulphuric acid and neutral cluster measurements using CI-APi-TOF, Atmos. Chem. Phys., 12, 4117–4125, https://doi.org/10.5194/acp-12-4117-2012, 2012.
Kirkby, J., Curtius, J., Almeida, J., Dunne, E., Duplissy, J., Ehrhart, S., Franchin, A., Gagne, S., Ickes, L., Kurten, A., Kupc, A., Metzger, A., Riccobono, F., Rondo, L., Schobesberger, S., Tsagkogeorgas, G., Wimmer, D., Amorim, A., Bianchi, F., Breitenlechner, M., David, A., Dommen, J., Downard, A., Ehn, M., Flagan, R. C., Haider, S., Hansel, A., Hauser, D., Jud, W., Junninen, H., Kreissl, F., Kvashin, A., Laaksonen, A., Lehtipalo, K., Lima, J., Lovejoy, E. R., Makhmutov, V., Mathot, S., Mikkila, J., Minginette, P., Mogo, S., Nieminen, T., Onnela, A., Pereira, P., Petaja, T., Schnitzhofer, R., Seinfeld, J. H., Sipila, M., Stozhkov, Y., Stratmann, F., Tome, A., Vanhanen, J., Viisanen, Y., Vrtala, A., Wagner, P. E., Walther, H., Weingartner, E., Wex, H., Winkler, P. M., Carslaw, K. S., Worsnop, D. R., Baltensperger, U., and Kulmala, M.: Role of sulphuric acid, ammonia and galactic cosmic rays in atmospheric aerosol nucleation, Nature, 476, 429–433, https://doi.org/10.1038/nature10343, 2011.
Kirkby, J., Duplissy, J., Sengupta, K., Frege, C., Gordon, H., Williamson, C., Heinritzi, M., Simon, M., Yan, C., Almeida, J., Tröstl, J., Nieminen, T., Ortega, I. K., Wagner, R., Adamov, A., Amorim, A., Bernhammer, A.-K., Bianchi, F., Breitenlechner, M., Brilke, S., Chen, X., Craven, J., Dias, A., Ehrhart, S., Flagan, R. C., Franchin, A., Fuchs, C., Guida, R., Hakala, J., Hoyle, C. R., Jokinen, T., Junninen, H., Kangasluoma, J., Kim, J., Krapf, M., Kürten, A., Laaksonen, A., Lehtipalo, K., Makhmutov, V., Mathot, S., Molteni, U., Onnela, A., Peräkylä, O., Piel, F., Petäjä, T., Praplan, A. P., Pringle, K., Rap, A., Richards, N. A. D., Riipinen, I., Rissanen, M. P., Rondo, L., Sarnela, N., Schobesberger, S., Scott, C. E., Seinfeld, J. H., Sipilä, M., Steiner, G., Stozhkov, Y., Stratmann, F., Tomé, A., Virtanen, A., Vogel, A. L., Wagner, A. C., Wagner, P. E., Weingartner, E., Wimmer, D., Winkler, P. M., Ye, P., Zhang, X., Hansel, A., Dommen, J., Donahue, N. M., Worsnop, D. R., Baltensperger, U., Kulmala, M., Carslaw, K. S., and Curtius, J.: Ion-induced nucleation of pure biogenic particles, Nature, 533, 521, https://doi.org/10.1038/nature17953, 2016.
Kreyling, W. G., Möller, W., Semmler-Behnke, M., Oberdörster, G., Donaldson, K., and Borm, P.: Particle dosimetry: deposition and clearance from the respiratory tract and translocation towards extrapulmonary sites, in: Particle toxicology, edited by: Donaldson, K. and Borm, P., CRC Press, Boca Raton, FL, USA, 46–69, 2006.
Kulmala, M., Petäjä, T., Ehn, M., Thornton, J., Sipilä, M., Worsnop, D. R., and Kerminen, V.-M.: Chemistry of Atmospheric Nucleation: On the Recent Advances on Precursor Characterization and Atmospheric Cluster Composition in Connection with Atmospheric New Particle Formation, Annu. Rev. Phys. Chem., 65, 21–37, https://doi.org/10.1146/annurev-physchem-040412-110014, 2014.
Kürten, A., Rondo, L., Ehrhart, S., and Curtius, J.: Performance of a corona ion source for measurement of sulfuric acid by chemical ionization mass spectrometry, Atmos. Meas. Tech., 4, 437–443, https://doi.org/10.5194/amt-4-437-2011, 2011.
Kürten, A., Rondo, L., Ehrhart, S., and Curtius, J.: Calibration of a Chemical Ionization Mass Spectrometer for the Measurement of Gaseous Sulfuric Acid, J. Phys. Chem. A, 116, 6375–6386, https://doi.org/10.1021/jp212123n, 2012.
Kürten, A., Jokinen, T., Simon, M., Sipilä, M., Sarnela, N., Junninen, H., Adamov, A., Almeida, J., Amorim, A., Bianchi, F., Breitenlechner, M., Dommen, J., Donahue, N. M., Duplissy, J., Ehrhart, S., Flagan, R. C., Franchin, A., Hakala, J., Hansel, A., Heinritzi, M., Hutterli, M., Kangasluoma, J., Kirkby, J., Laaksonen, A., Lehtipalo, K., Leiminger, M., Makhmutov, V., Mathot, S., Onnela, A., Petäjä, T., Praplan, A. P., Riccobono, F., Rissanen, M. P., Rondo, L., Schobesberger, S., Seinfeld, J. H., Steiner, G., Tomé, A., Tröstl, J., Winkler, P. M., Williamson, C., Wimmer, D., Ye, P., Baltensperger, U., Carslaw, K. S., Kulmala, M., Worsnop, D. R., and Curtius, J.: Neutral molecular cluster formation of sulfuric acid–dimethylamine observed in real time under atmospheric conditions, P. Natl. Acad. Sci. USA, 111, 15019–15024, https://doi.org/10.1073/pnas.1404853111, 2014.
Laitinen, T., Hartonen, K., Kulmala, M., and Riekkola, M.-L.: Aerosol time-of-flight mass spectrometer for measuring ultrafine aerosol particles, Boreal Environ. Res., 14, 539–549, 2009.
Laitinen, T., Ehn, M., Junninen, H., Ruiz-Jimenez, J., Parshintsev, J., Hartonen, K., Riekkola, M.-L., Worsnop, D. R., and Kulmala, M.: Characterization of organic compounds in 10- to 50-nm aerosol particles in boreal forest with laser desorption-ionization aerosol mass spectrometer and comparison with other techniques, Atmos. Environ., 45, 3711–3719, https://doi.org/10.1016/j.atmosenv.2011.04.023, 2011.
Lawler, M. J., Whitehead, J., O'Dowd, C., Monahan, C., McFiggans, G., and Smith, J. N.: Composition of 15–85 nm particles in marine air, Atmos. Chem. Phys., 14, 11557–11569, https://doi.org/10.5194/acp-14-11557-2014, 2014.
Lawler, M. J., Winkler, P. M., Kim, J., Ahlm, L., Tröstl, J., Praplan, A. P., Schobesberger, S., Kürten, A., Kirkby, J., Bianchi, F., Duplissy, J., Hansel, A., Jokinen, T., Keskinen, H., Lehtipalo, K., Leiminger, M., Petäjä, T., Rissanen, M., Rondo, L., Simon, M., Sipilä, M., Williamson, C., Wimmer, D., Riipinen, I., Virtanen, A., and Smith, J. N.: Unexpectedly acidic nanoparticles formed in dimethylamine–ammonia–sulfuric–acid nucleation experiments at CLOUD, Atmos. Chem. Phys., 16, 13601–13618, https://doi.org/10.5194/acp-16-13601-2016, 2016.
Lehtipalo, K., Rondo, L., Kontkanen, J., Schobesberger, S., Jokinen, T., Sarnela, N., Kürten, A., Ehrhart, S., Franchin, A., Nieminen, T., Riccobono, F., Sipilä, M., Yli-Juuti, T., Duplissy, J., Adamov, A., Ahlm, L., Almeida, J., Amorim, A., Bianchi, F., Breitenlechner, M., Dommen, J., Downard, A. J., Dunne, E. M., Flagan, R. C., Guida, R., Hakala, J., Hansel, A., Jud, W., Kangasluoma, J., Kerminen, V.-M., Keskinen, H., Kim, J., Kirkby, J., Kupc, A., Kupiainen-Määttä, O., Laaksonen, A., Lawler, M. J., Leiminger, M., Mathot, S., Olenius, T., Ortega, I. K., Onnela, A., Petäjä, T., Praplan, A., Rissanen, M. P., Ruuskanen, T., Santos, F. D., Schallhart, S., Schnitzhofer, R., Simon, M., Smith, J. N., Tröstl, J., Tsagkogeorgas, G., Tomé, A., Vaattovaara, P., Vehkamäki, H., Vrtala, A. E., Wagner, P. E., Williamson, C., Wimmer, D., Winkler, P. M., Virtanen, A., Donahue, N. M., Carslaw, K. S., Baltensperger, U., Riipinen, I., Curtius, J., Worsnop, D. R., and Kulmala, M.: The effect of acid–base clustering and ions on the growth of atmospheric nano-particles, Nat. Commun., 7, 11594, https://doi.org/10.1038/ncomms11594, 2016.
Lelieveld, J., Evans, J. S., Fnais, M., Giannadaki, D., and Pozzer, A.: The contribution of outdoor air pollution sources to premature mortality on a global scale, Nature, 525, 367–371, https://doi.org/10.1038/nature15371, 2015.
Lopez-Hilfiker, F. D., Mohr, C., Ehn, M., Rubach, F., Kleist, E., Wildt, J., Mentel, Th. F., Lutz, A., Hallquist, M., Worsnop, D., and Thornton, J. A.: A novel method for online analysis of gas and particle composition: description and evaluation of a Filter Inlet for Gases and AEROsols (FIGAERO), Atmos. Meas. Tech., 7, 983–1001, https://doi.org/10.5194/amt-7-983-2014, 2014.
Lopez-Hilfiker, F. D., Mohr, C., Ehn, M., Rubach, F., Kleist, E., Wildt, J., Mentel, Th. F., Carrasquillo, A. J., Daumit, K. E., Hunter, J. F., Kroll, J. H., Worsnop, D. R., and Thornton, J. A.: Phase partitioning and volatility of secondary organic aerosol components formed from a-pinene ozonolysis and OH oxidation: the importance of accretion products and other low volatility compounds, Atmos. Chem. Phys., 15, 7765–7776, https://doi.org/10.5194/acp-15-7765-2015, 2015.
McMurry, P. H., Ghimire, A., Ahn, H.-K., Sakurai, H., Moore, K., Stolzenburg, M., and Smith, J. N.: Sampling Nanoparticles for Chemical Analysis by Low Resolution Electrical Mobility Classification, Environ. Sci. Technol., 43, 4653–4658, https://doi.org/10.1021/es8029335, 2009.
Merikanto, J., Spracklen, D. V., Mann, G. W., Pickering, S. J., and Carslaw, K. S.: Impact of nucleation on global CCN, Atmos. Chem. Phys., 9, 8601–8616, https://doi.org/10.5194/acp-9-8601-2009, 2009.
Nel, A.: Air pollution-related illness: effects of particles, Science, 308, 804–806, https://doi.org/10.1126/science.1108752, 2005.
Noble, C. A. and Prather, K. A.: Real-time single particle mass spectrometry: A historical review of a quarter century of the chemical analysis of aerosols, Mass Spectrom. Rev., 19, 248–274, https://doi.org/10.1002/1098-2787(200007)19:4<248::AID-MAS3>3.0.CO;2-I, 2000.
Phares, D. J. and Collier, S.: Direct Collection of Aerosols by Electrostatic Classification for Size-Resolved Chemical Analysis, Aerosol Sci. Tech., 44, 173–181, https://doi.org/10.1080/02786820903482914, 2010.
Pope III, C. A. and Dockery, D. W.: Health effects of fine particulate air pollution: lines that connect, J. Air Waste Manage., 56, 709–742, https://doi.org/10.1080/10473289.2006.10464485, 2006.
Reischl, G. P., Mäkelä, J. M., and Necid, J.: Performance of Vienna Type Differential Mobility Analyzer at 1.2–20 Nanometer, Aerosol Sci. Tech., 27, 651–672, https://doi.org/10.1080/02786829708965503, 1997.
Riccobono, F., Rondo, L., Sipilä, M., Barmet, P., Curtius, J., Dommen, J., Ehn, M., Ehrhart, S., Kulmala, M., Kürten, A., Mikkilä, J., Paasonen, P., Petäjä, T., Weingartner, E., and Baltensperger, U.: Contribution of sulfuric acid and oxidized organic compounds to particle formation and growth, Atmos. Chem. Phys., 12, 9427–9439, https://doi.org/10.5194/acp-12-9427-2012, 2012.
Simon, M., Heinritzi, M., Herzog, S., Leiminger, M., Bianchi, F., Praplan, A., Dommen, J., Curtius, J., and Kürten, A.: Detection of dimethylamine in the low pptv range using nitrate chemical ionization atmospheric pressure interface time-of-flight (CI-APi-TOF) mass spectrometry, Atmos. Meas. Tech., 9, 2135–2145, https://doi.org/10.5194/amt-9-2135-2016, 2016.
Smith, J. N., Moore, K. F., McMurry, P. H., and Eisele, F. L.: Atmospheric Measurements of Sub-20 nm Diameter Particle Chemical Composition by Thermal Desorption Chemical Ionization Mass Spectrometry, Aerosol Sci. Tech., 38, 100–110, https://doi.org/10.1080/02786820490249036, 2004.
Smith, J. N., Barsanti, K. C., Friedli, H. R., Ehn, M., Kulmala, M., Collins, D. R., Scheckman, J. H., Williams, B. J., and McMurry, P. H.: Observations of aminium salts in atmospheric nanoparticles and possible climatic implications, P. Natl. Acad. Sci. USA, 107, 6634–6639, https://doi.org/10.1073/pnas.0912127107, 2010.
Stolzenburg, M. R. and McMurry, P. H.: Equations Governing Single and Tandem DMA Configurations and a New Lognormal Approximation to the Transfer Function, Aerosol Sci. Tech., 42, 421–432, https://doi.org/10.1080/02786820802157823, 2008.
Tigges, L., Wiedensohler, A., Weinhold, K., Gandhi, J., and Schmid, H. J.: Bipolar charge distribution of a soft X-ray diffusion charger, J. Aerosol Sci., 90, 77–86, https://doi.org/10.1016/j.jaerosci.2015.07.002, 2015.
Tröstl, J., Chuang, W. K., Gordon, H., Heinritzi, M., Yan, C., Molteni, U., Ahlm, L., Frege, C., Bianchi, F., Wagner, R., Simon, M., Lehtipalo, K., Williamson, C., Craven, J. S., Duplissy, J., Adamov, A., Almeida, J., Bernhammer, A.-K., Breitenlechner, M., Brilke, S., Dias, A., Ehrhart, S., Flagan, R. C., Franchin, A., Fuchs, C., Guida, R., Gysel, M., Hansel, A., Hoyle, C. R., Jokinen, T., Junninen, H., Kangasluoma, J., Keskinen, H., Kim, J., Krapf, M., Kürten, A., Laaksonen, A., Lawler, M., Leiminger, M., Mathot, S., Möhler, O., Nieminen, T., Onnela, A., Petäjä, T., Piel, F. M., Miettinen, P., Rissanen, M. P., Rondo, L., Sarnela, N., Schobesberger, S., Sengupta, K., Sipilä, M., Smith, J. N., Steiner, G., Tomè, A., Virtanen, A., Wagner, A. C., Weingartner, E., Wimmer, D., Winkler, P. M., Ye, P., Carslaw, K. S., Curtius, J., Dommen, J., Kirkby, J., Kulmala, M., Riipinen, I., Worsnop, D. R., Donahue, N. M., and Baltensperger, U.: The role of low-volatility organic compounds in initial particle growth in the atmosphere, Nature, 533, 527, https://doi.org/10.1038/nature18271, 2016.
TSI Incorporated: Ultrafine Condensation Particle Counter Model 3776, Instruction Manual, Shoreview, MN, USA, 2007.
Vehkamäki, H. and Riipinen, I.: Thermodynamics and kinetics of atmospheric aerosol particle formation and growth, Chem. Soc. Rev., 41, 5160–5173, https://doi.org/10.1039/C2CS00002D, 2012.
Voisin, D., Smith, J. N., Sakurai, H., McMurry, P. H., and Eisele, F. L.: Thermal Desorption Chemical Ionization Mass Spectrometer for Ultrafine Particle Chemical Composition, Aerosol Sci. Tech., 37, 471–475, https://doi.org/10.1080/02786820300959, 2003.
Wagner, A. C., Bergen, A., Brilke, S., Fuchs, C., Ernst, M., Hoker, J., Heinritzi, M., Simon, M., Bühner, B., Curtius, J., and Kürten, A.: Dataset for 10.5194/amt-11-1-2018, https://doi.org/10.5281/zenodo.1443176, 2018.
Wang, S., Zordan, C. A., and Johnston, M. V.: Chemical Characterization of Individual, Airborne Sub-10-nm Particles and Molecules, Anal. Chem., 78, 1750–1754, https://doi.org/10.1021/ac052243l, 2006.
Wang, S. C. and Flagan, R. C.: Scanning Electrical Mobility Spectrometer, Aerosol Sci. Tech., 13, 230–240, https://doi.org/10.1080/02786829008959441, 1990.
Wellenius, G. A., Burger, M. R., Coull, B. A., Schwartz, J., Suh, H. H., Koutrakis, P., Schlaug, G., Gold, D. R., and Mittleman, M. A.: Ambient air pollution and the risk of acute ischemic stroke, Arch. Intern. Med., 172, 229–234, https://doi.org/10.1001/archinternmed.2011.732, 2012.
Wimmer, D., Lehtipalo, K., Nieminen, T., Duplissy, J., Ehrhart, S., Almeida, J., Rondo, L., Franchin, A., Kreissl, F., Bianchi, F., Manninen, H. E., Kulmala, M., Curtius, J., and Petäjä, T.: Technical Note: Using DEG-CPCs at upper tropospheric temperatures, Atmos. Chem. Phys., 15, 7547–7555, https://doi.org/10.5194/acp-15-7547-2015, 2015.
Zhang, Q., Jimenez, J. L., Canagaratna, M. R., Ulbrich, I. M., Ng, N. L., Worsnop, D. R., and Sun, Y.: Understanding atmospheric organic aerosols via factor analysis of aerosol mass spectrometry: a review, Anal. Bioanal. Chem., 401, 3045–3067, https://doi.org/10.1007/s00216-011-5355-y, 2011.
Zhang, R., Khalizov, A., Wang, L., Hu, M., and Xu, W.: Nucleation and Growth of Nanoparticles in the Atmosphere, Chem. Rev., 112, 1957–2011, https://doi.org/10.1021/cr2001756, 2012.
Zhang, S.-H. and Flagan, R. C.: Resolution of the radial differential mobility analyzer for ultrafine particles, J. Aerosol Sci., 27, 1179–1200, https://doi.org/10.1016/0021-8502(96)00036-5, 1996.