the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 26 Feb 2019
| 26 Feb 2019
Evaluation of cation exchange membrane performance under exposure to high Hg0 and HgBr2 concentrations
Matthieu B. Miller
Sarrah M. Dunham-Cheatham
Mae Sexauer Gustin
Grant C. Edwards
Reactive mercury (RM), the sum of both gaseous oxidized Hg and particulate bound Hg, is an important component of the global atmospheric mercury cycle, but measurement currently depends on uncalibrated operationally defined methods with large uncertainty and demonstrated interferences and artifacts. Cation exchange membranes (CEMs) provide a promising alternative methodology for quantification of RM, but method validation and improvements are ongoing. For the CEM material to be reliable, uptake of gaseous elemental mercury (GEM) must be negligible under all conditions and RM compounds must be captured and retained with high efficiency. In this study, the performance of CEM material under exposure to high concentrations of GEM (1.43×106 to 1.85×106 pg m−3) and reactive gaseous mercury bromide (HgBr2 ∼5000 pg m−3) was explored using a custom-built mercury vapor permeation system. Quantification of total permeated Hg was measured via pyrolysis at 600 ∘C and detection using a Tekran® 2537A. Permeation tests were conducted over 24 to 72 h in clean laboratory air, with absolute humidity levels ranging from 0.1 to 10 g m−3 water vapor. GEM uptake by the CEM material averaged no more than 0.004 % of total exposure for all test conditions, which equates to a non-detectable GEM artifact for typical ambient air sample concentrations. Recovery of HgBr2 on CEM filters was on average 127 % compared to calculated total permeated HgBr2 based on the downstream Tekran® 2537A data. The low HgBr2 breakthrough on the downstream CEMs (< 1 %) suggests that the elevated recoveries are more likely related to suboptimal pyrolyzer conditions or inefficient collection on the Tekran® 2537A gold traps.
- Article
(453 KB) - Full-text XML
-
Supplement
(464 KB) - BibTeX
- EndNote





Mercury (Hg) is a persistent environmental contaminant with a significant atmospheric lifetime, and the form and chemistry of Hg is an important determinant of its biogeochemical cycling. Mercury in the atmosphere is found in three forms: gaseous elemental mercury (GEM), gaseous oxidized mercury (GOM), and particulate bound mercury (PBM). PBM and GOM are often quantified together as reactive mercury (RM = GOM + PBM); while the term reactive mercury (RM) dilutes some specific information regarding the state of GOM in the atmosphere, it removes some uncertainty as to whether or not PBM contributes to the Hg collected by the cation exchange membranes (CEMs). Atmospheric GEM, at an average global background concentration of 1 to 2 ng m−3, can be reliably measured with calibrated analytical instruments (Gustin et al., 2015; Slemr et al., 2015). The measurement of GOM and PBM requires detection at part-per-quadrillion (pg m−3) concentrations and depends currently on uncalibrated operationally defined methods with demonstrated interferences and artifacts and concomitant large uncertainty (Marusczak et al., 2017; Jaffe et al., 2014; McClure et al., 2014; Gustin et al., 2013; Lyman et al., 2010). Recent reviews (Zhang et al., 2017; Gustin et al., 2015) detail the shortcomings, difficulties, developments, and ongoing improvements needed for atmospheric RM measurements.
One alternative methodology that may provide improved measurement of ambient RM involves use of CEMs. CEM materials have been used to selectively measure GOM concentrations in ambient air in previous studies (Huang et al., 2013, 2017; Marusczak et al., 2017; Pierce and Gustin, 2017; Huang and Gustin, 2015a; Sheu and Mason, 2001; Ebinghaus et al., 1999; Mason et al., 1997; Bloom et al., 1996). Use of CEM-type filters (then manufactured by Gelman Sciences and referred to as “ion exchange membranes”) for this purpose was first documented in the literature in a conference presentation (Bloom et al., 1996), though these had also been deployed in an earlier field-based international comparative study of RM measurement techniques in September 1995 (Ebinghaus et al., 1999). In the comparative study, one participating lab deployed a series of ion exchange membranes (for GOM) behind a quartz fiber filter (for PBM) at a sample flow rate of 9 to 10 L min−1 for 24 h measurements (filter pore sizes were not reported). Results for PBM and GOM were in similar ranges of 4.5 to 26 and 13 to 23 pg m−3, respectively (Ebinghaus et al., 1999).
The ion exchange membrane method was also applied in a 1995–1996 field campaign for determining the speciation of atmospheric Hg in the Chesapeake Bay area (Mason et al., 1997). This study used a five-stage Teflon filter pack system that included one up-front quartz fiber filter (0.8 µm pore size) to remove particles and four downstream Gelman ion exchange membranes (pore size not reported) to (1) capture GOM, (2) capture GOM breakthrough, (3) serve as deployment blanks, and (4) isolate the filter train on the downstream side (Mason et al., 1997). Concentrations of GOM were reported to be 5–10 pg m−3, essentially at or below the method detection limit, and it was speculated that even this small amount may have been an artifact from fine particulate Hg passing through the 0.8 µm quartz fiber filter (Mason et al., 1997). These low concentrations are likely due to GOM being degraded on the quartz fiber filter or inefficient uptake by the Gelman filter (see Supplement; Gustin et al., 2013). The third-in-series ion exchange membrane blanks were reported to be not significantly different in Hg concentration from unused membrane material, indicating that breakthrough was not a phenomenon that extended past the second ion exchange filter position.
The particulate Hg artifact problem was subsequently elaborated on in a further comparative study focusing exclusively on RM measurement techniques (Sheu and Mason, 2001). Specific concerns included physical particle breakthrough, re-evolution of gas-phase Hg2+ from PBM captured on the upstream particulate filters passing downstream to the ion exchange membranes, possible adsorption of GOM compounds to the particulate filters, or a GEM collection artifact on the ion exchange membranes. None of these concerns were proven or disproven conclusively.
Recent CEM-based sampling systems typically deploy a pair of CEM disk filters without a pre-particulate filter, in replicates of 2 to 3 at a flow rate of 1.0 L min−1 (Gustin et al., 2016). Each pair of filters constitutes one sample, the first filter serving as the primary RM collection surface and the second filter capturing breakthrough. Filters are deployed for 1 to 2 weeks and then collected for analysis (Huang et al., 2017). The CEM material consists of a negatively charged polyethersulfone-coated matrix (Pall Corporation) and at least one manufacturing evolution has occurred (Huang and Gustin, 2015b). Prior CEM material versions (I.C.E. 450) had a pore size of 0.45 µm, while the current CEM material (Mustang® S) has a manufacturer-reported pore size of 0.8 µm.
Previous work with the I.C.E. 450 material indicated it does not adsorb significant quantities of GEM in passive exposures but does selectively uptake gas-phase Hg2+ species (Lyman et al., 2007). The CEM material was subsequently adapted for use in active sample flow systems, with the presumption of continued inertness to GEM and selectivity for GOM (Huang and Gustin, 2015a; Huang et al., 2013). These studies and others (Lyman et al., 2016) have shown better GOM recovery on CEM material compared to potassium chloride (KCl)-coated denuder methods.
Despite these tests, the transparency of the CEM material to GEM uptake has not been conclusively demonstrated for active sampling flow rates or for high GEM concentrations, though limited data using low-concentration manual Hg0 injections through CEM filters suggested little or no GEM uptake (Lyman et al., 2016). However, even small rates of GEM uptake by the CEM material could result in a significant measurement artifact (e.g., a modest 1 % to 2 % GEM uptake could easily overwhelm detection of typical ambient GOM concentrations). It is therefore important that a GEM artifact be ruled out if the CEM material is to be successfully deployed for ambient RM measurements.
Additionally, previous studies observed significant amounts of “breakthrough” GOM on the secondary filter. The amount of breakthrough is not consistent as a constant mass, with total Hg ranging from zero to as high as 400 pg (Huang et al., 2017), or as a percentage of Hg collected on the primary filter, ranging from 0 % to 40 % (Pierce and Gustin, 2017). Similar variable breakthrough issues were observed in the earliest field-based CEM measurements as well (Mason et al., 1997). In contrast to ambient measurements, previous laboratory experiments have reported only minor (0 % to 16 %) or no breakthrough (Huang and Gustin, 2015a; Huang et al., 2013). Limited experimental work with flow rates of 1.0 and 16.7 L min−1 in ambient air could not provide an explanation for differing breakthrough rates (Pierce and Gustin, 2017).
In this research we investigated the potential for GEM uptake on CEM material using a custom-built permeation system. Tests were done to investigate the ability of a pyrolyzer to convert GEM to GOM. In addition, the ability of the CEM material to capture and retain a representative GOM compound (mercury(II) bromide, HgBr2) was explored, and the collection efficiency for this compound was estimated. We attempted to explain or rule out possible mechanisms of RM breakthrough for both dry and humid conditions.
2.1 System for sampling configuration
A Tekran® 2537A ambient mercury analyzer was integrated with a custom-built permeation system designed to enable controlled exposures of GEM and GOM to CEM filters (Fig. 1). The 2537A analyzer was calibrated at the beginning and periodically throughout the study (Fig. S1 in the Supplement) and checked for accuracy by manual Hg0 injections (mean recovery of 101.1 %±4.3, n=10). The entire system was checked for Hg contamination in clean air prior to permeation tests and periodically during sampling (Fig. S2); see the Supplement for additional information on Tekran quality control. All tubing and connections used in the permeation system were polytetrafluoroethylene (PTFE), except for the quartz glass pyrolyzer tube and perfluoroalkoxy (PFA) filter holders. Given its reactive nature, some GOM inevitably adsorbs to internal line surfaces, but the capacity of these materials to sorb and retain GOM is not infinite and a steady state of adsorption and desorption is expected after 5–6 h of exposure to a stable concentration (Xiao et al., 1997; Gustin et al., 2013).
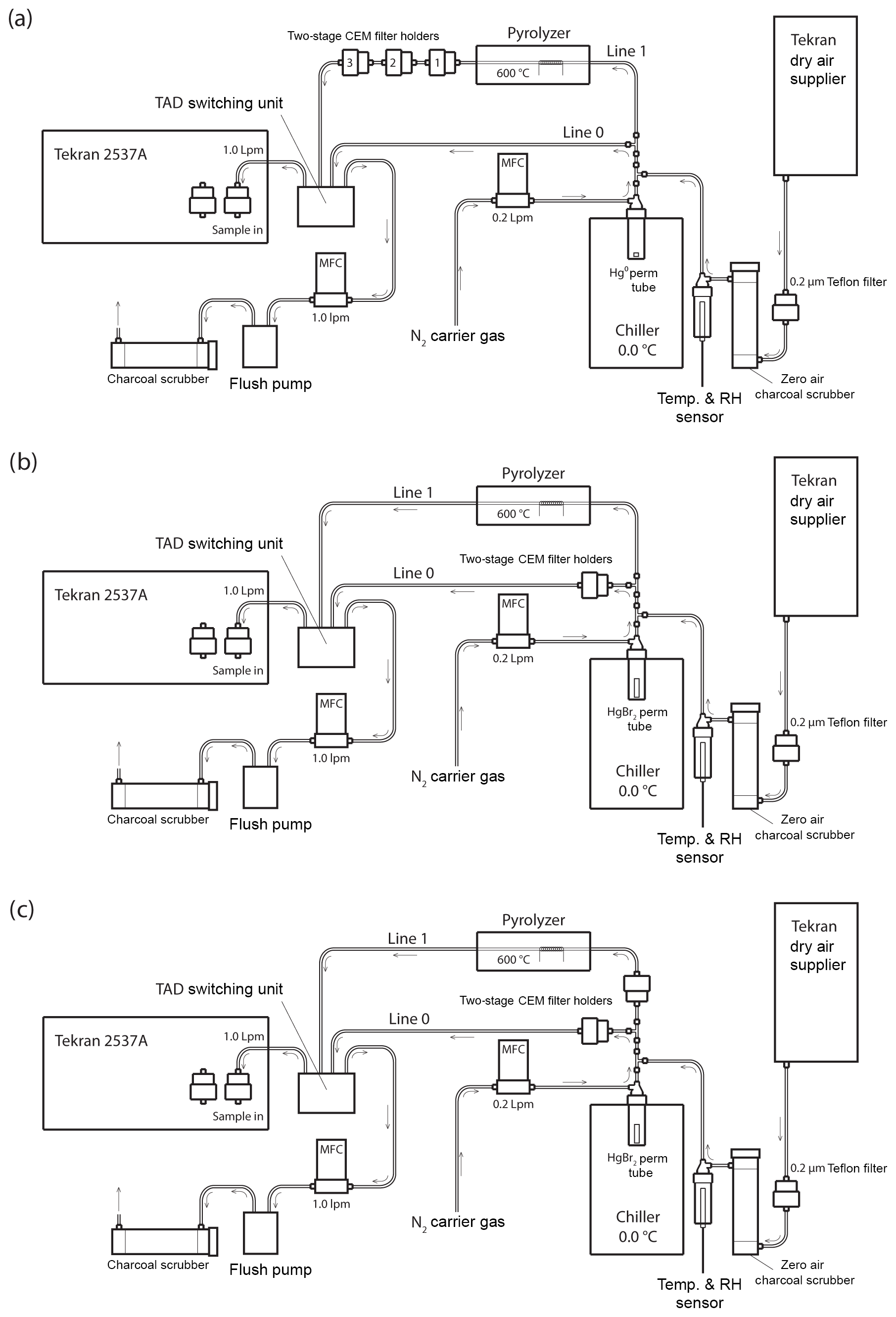
Figure 1Schematic of the Hg vapor permeation system configurations for (a) GEM permeations, (b) HgBr2 permeations, and (c) simultaneous HgBr2 loading on two sample lines. Note that the dry air supplier is disconnected for ambient and elevated humidity HgBr2 permeations, with the sample path starting at a 0.2 µm Teflon particulate filter and water bath inserted immediately in front of the charcoal scrubber. All tubing is PTFE, except for the quartz glass pyrolyzer tube and PFA filter holders.
Sample flow through the system was alternated between two PTFE sample lines (designated Line 0 and Line 1) using a Tekran® automated dual switching (TADS) unit. Sample air was constantly pulled through each line at 1.0 L min−1 by the internal pump and mass flow controller (MFC) in the 2537A, or by an external flush pump (KNF Laboport® N86 KNP) and MFC (Sierra Smart-Trak® 2). Laboratory air was pulled through a single inlet at the combined rate of 2.0 L min−1, passing through a 0.2 µm PTFE particulate filter and an activated charcoal scrubber (granular activated carbon 6–12 mesh, FisherChemical®) to produce clean sample air. Additionally, for dry air permeations sample air was pulled through a Tekran® 1102 Air Dryer installed upstream of the particulate filter, and for elevated humidity permeations sample air was pulled through the headspace of a distilled water bath (DIW, < 0.2 ng L−1 total Hg) that was located upstream from the charcoal scrubber to eliminate the DIW being a potential Hg source to the system. Temperature and RH were measured in line (Campbell Scientific CS215) and used for calculation of absolute humidity.
Pure liquid Hg0 and crystalline HgBr2 (purity > 99.998 % Sigma-Aldrich®) were used as Hg vapor sources. The elemental Hg0 bead was contained in a PTFE vial. Solid HgBr2 crystals were packed in thin-walled PTFE heat-shrink tubing (O.D. 0.635 cm) with solid Teflon plugs in both ends to create a permeation tube with an active permeation length of 2 mm (Huang et al., 2013). The HgBr2 permeation tube was also placed in the bottom of a PTFE vial, and the permeation vials were submerged in a temperature-controlled laboratory chiller (0.06±0.13 ∘C, Cole-Parmer Polystat®). A low source temperature was favored, because higher temperatures would have produced unacceptably high concentrations, and there is evidence that at higher temperatures a small amount of Hg0 can be evolved from Hg2+ compounds (Xiao et al., 1997).
An ultra-high-purity nitrogen (N2) carrier gas was passed through the permeation vials at 0.2 L min−1 to carry the target Hg vapor into the main sample line through a PTFE T-junction. The main sample line was split into Line 0 and Line 1 immediately downstream from the permeation flow junction, with flow on each line controlled by MFC. Line 0 proceeded directly to the 2537A without modification during GEM permeations (Fig. 1a) but housed CEM filters during the HgBr2 permeations (Fig. 1b, c). Line 1 contained an in-line pyrolyzer unit. The goal of the pyrolyzer was to convert all Hg to GEM for detection on the Tekran® 2537A.
2.2 Pyrolyzer
The pyrolyzer used in the study (Fig. S3) consisted of a 25.4 cm long quartz glass tube of 0.625 cm diameter (custom, URG Corporation). A loosely packed 3 cm section of quartz wool was lodged in the midsection of the tube and this 3 cm section was wrapped with 22 gauge nichrome wire (18 loops). The quartz tube was closely contained within 2.5 cm thick quartz fiber insulation within a 1.6 mm aluminum casing, except for an enclosed air space around the heated nichrome coil section. The coil wire was connected to 16 American wire gauge stranded copper wire with all metal disconnects that were buried within the quartz fiber insulation to reduce thermal fatigue on the connections. The copper wire insulation was stripped and replaced with higher temperature heat-shrink insulation where the wiring passed through the pyrolyzer case to the external power supply. The tip of a 150 mm long K-type thermocouple (Auber WRNK-191) was inserted through the insulation into the heated air space next to the coil to provide a temperature feedback for a proportional–integral–derivative (PID) controller (Auber SYL-1512A). Power to the nichrome coil was supplied by a 12 V DC transformer through a solid-state relay (Auber MGR-1D4825) switched by the PID controller. It was found that the position of the feedback thermocouple in the airspace outside of the heating coil caused a large discrepancy between nominal temperature set point and actual temperature inside the heated section of pyrolyzer tube. In general, much higher temperatures are achieved inside the coil than outside. To compensate for this, actual temperature at the heated coil section was verified as 600 ∘C by an external infrared sensor and internal thermocouple probe.
To test if higher pyrolyzer temperatures converted more GOM to GEM for detection by the Tekran 2537, the pyrolyzer temperature was increased to 650, 800, and 1000 ∘C (Fig. S4). Pyrolyzer temperatures were measured by placing a thermocouple inside the pyrolyzer. GOM concentrations measured as GEM by the Tekran 2537A increased at 600 and 800 ∘C relative to 375 ∘C. There was no significant difference between the amount of mercury concentrations in the downstream Tekran 2537A when the pyrolyzer was at 600 and 800 ∘C (t test, p=0.08), indicating that the increased pyrolyzer temperature did not convert more GOM to GEM. However, when the pyrolyzer temperature was increased to 1000 ∘C, significantly more mercury was measured by the downstream Tekran 2537A relative to when the pyrolyzer was at 650 ∘C (t test, p=0.00), indicating that the higher temperature was more efficient at converting GOM to GEM; however, the pyrolyzer design could not sustain the 1000 ∘C temperature and was deemed unsafe to use in the experimental permeation system. Thus, all experiments were performed with a pyrolyzer temperature of 600 ∘C.
The residence time in the pyrolyzer tube was approximately 1.5 s. Quartz wool was added to increase the amount of surface area available to facilitate reactions and maximize the amount of GOM converted to GEM in the pyrolyzer. Based on supplemental experiments, the downstream Tekran® 2537A Hg measurements when the pyrolyzer was at 650 ∘C were 75 % compared to the measurements when the pyrolyzer was at 1000 ∘C (Fig. S4), indicating a higher GOM to GEM conversion efficiency with higher pyrolyzer temperatures. Though this conversion efficiency value does not describe the exact inefficiency of the experimental system in this study, it provides an estimate for the efficiency of the pyrolyzer design in this study. Having an efficient pyrolyzer provides us with a means of constraining permeation tube permeation rates.
2.3 Sample deployment
CEM filters were deployed in two-stage, 47 mm disk PFA filter holders (Savillex©). The primary “A” filter in the two-stage holder is the first to be exposed to the permeated Hg, with the secondary “B” filter mounted immediately behind the A filter (A to B distance: ∼3 mm) to measure potential breakthrough. For GEM permeations, three two-stage filter holders were placed in series on Line 1 behind the pyrolyzer unit (Fig. 1a), while total Hg coming through the system was measured on Line 0 with no filters in place. This allowed simultaneous exposure of six CEM filters in one GEM sample exposure. The first in-line CEM filter served to scrub any small residual RM passing through the system and pyrolyzer, and these first in-line filters were removed for the calculations of mean GEM uptake rate (Sect. S5 in the Supplement and discussion). A controlled experiment was also performed to ensure that both Lines 0 and 1 were conducting comparable concentrations of mercury under the experimental conditions. Two-stage filter packs were deployed with CEM filters in each line at equal distances from the permeation tube. The membranes were deployed for the same amount of time in triplicate and analyzed to quantify the amount of total mercury sorbed to the membranes. The average percentage deviation between lines was 2.9 %, with a maximum deviation of 5.4 %. These results indicated that though there may be some difference in the amount of mercury passing through Lines 0 and 1, the difference was relatively small.
For determining the potential for GOM breakthrough, two system configurations were used. In the first configuration (Fig. 1b), the total Hg concentration of air that passed through the pyrolyzer on Line 1 was measured without any filters, while Line 0 held one two-stage CEM filter pair for HgBr2 loading. This configuration allowed for 10 min interval quantification of the HgBr2 permeation concentration through Line 1 using the 2537A and comparison with total Hg loading on the CEM filters on Line 0.
In the second configuration, replicate filters were concurrently loaded with HgBr2 by placing two-stage CEM filter holders on both Line 0 and Line 1 (upstream of the pyrolyzer, Fig. 1c). In all HgBr2 exposures, the filter holders were placed as close to the permeation vial as possible, with a total distance from vial to filter surface of approximately 20 cm. Mercury bromide permeation was conducted in dry air and elevated humidity air. The difference between one line being fully open to the HgBr2 permeation flow (configuration Fig. 1b) and then closed by deployment of the CEM filters (configuration Fig. 1c) enabled a rough determination of the amount of HgBr2 line loss within the system.
2.4 Analyses of cation exchange membranes
After permeation, CEM filters were collected into clean, sterile polypropylene vials and analyzed for total Hg by digestion in an oxidizing acid solution, reduction to Hg0, gold amalgamation, and final quantification by cold vapor atomic fluorescence spectrometry (CVAFS, EPA Method 1631, Rev. E) using a Tekran® 2600 system. The system background Hg signal was determined for every analytical run by analyzing pure reagent solution in the same vials and at the same volume as used for actual filter samples. Total Hg standards (5 to 100 ppb) were analyzed before and after each batch of 10 filter samples to check precision and recovery, and the mean recovery for all Hg standards was 97.2±5.0 % (n=37). Analysis for total Hg on the CEM filters provided for comparison of total Hg filter loading and verification of in-line results. A to B filter breakthrough was calculated by comparison of total Hg recoveries on the primary and secondary CEM filters using Eq. (1):
Blank CEM filters were collected and analyzed in the same manner with every set of sample filters deployed on the permeation system, and the overall mean filter blank value was subtracted from all total Hg values to calculate the final blank-corrected Hg values used for data analyses. All data were analyzed in Microsoft® Excel (version 16.12) and RStudio® (version 3.2.2).
3.1 Elemental mercury uptake on CEM filters
Elemental Hg uptake on CEM material was negligible for permeated Hg0 vapor concentrations ranging from 1.43×106 to 1.85×106 pg m−3 (Fig. 2). High GEM concentrations were employed in this study under the logic that if no GEM uptake was observed at high concentrations, a similar lack of GEM uptake can be expected for lower concentrations.
The mean Hg mass on blank CEM filters was 50±20 pg (n=28). For permeations into dry sample air of 0.5±0.1 g m−3 water vapor (WV), total mean Hg0 permeation exposures of 2.7×106 pg (24 h) and 7.3×106 pg (72 h) resulted in total (blank-corrected) Hg recoveries on the CEM filters of 100±40 pg (n=10) and 280±110 pg (n=5), respectively. These quantities of total recovered Hg equate to a mean GEM uptake rate on the CEM filters of 0.004±0.002 % (0.006±0.006 % including the first in-line filter). For GEM permeations into ambient humidity sample air (2 to 4 g m−3 WV), at a slightly lower total mean permeated Hg0 24 h exposure of 2.1×106 pg, total (blank-corrected) Hg recoveries on the CEM filters were 55±30 pg (n=10), equating to a GEM uptake rate of 0.003±0.001 % (0.005±0.005 % including the first in-line filter).
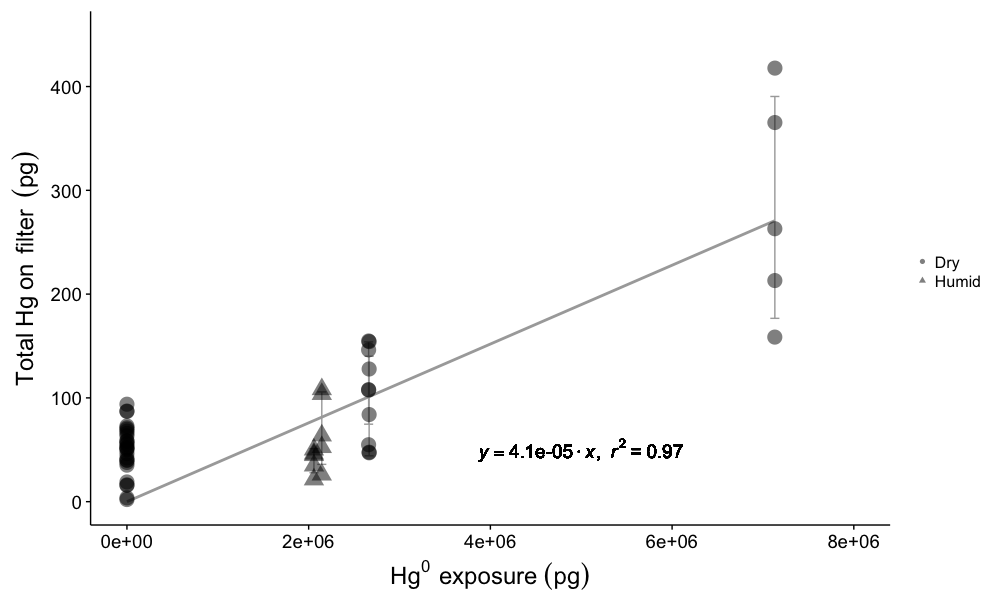
Figure 2Total Hg recovered on CEM material for blank filters (Hg exposure =0 pg) and different Hg0 vapor permeations in dry (0.5±0.1 g m−3 WV) and humid air (2–4 g m−3 WV). Circles represent dry air permeations, triangles represent humid air exposures, and all permeation exposures were blank-corrected. The regression line shows the relationship between total Hg0 exposure and blank-corrected mean total Hg recovered on CEM filters (error bars ±one standard deviation), with a slope of indicating a linear uptake rate of 0.004 %.
The first in-line CEM filter during the GEM permeations always showed more total Hg than the following five downstream filters that were not significantly different from each other (Fig. S5). It is unlikely that the Hg observed on the first CEM filters resulted from GEM uptake. Even at the highest GEM permeation rate, the first filter captured only ∼1700 pg of Hg, out of a total permeated amount of over 7.3 million pg (a 0.02 % uptake rate). This means that the downstream CEM filters were still exposed to about 7.2985 million pg of GEM but captured less total Hg. As we cannot entirely rule out the possibility of some small rate of in situ oxidation of GEM in the system, at the surface of the Hg0 bead or in the vapor phase, the first in-line filters were not included in the calculation of GEM uptake rates because of suspicion that some component of the Hg captured on the first filter was GOM. The overall GEM uptake rate was linear (r2=0.69, p=0.00; Fig. S5) for the range of concentrations used in this study, indicating a similar low uptake rate can be expected down to lower GEM concentrations. GEM uptake by the first in-line filters was also linear (r2=0.92, p=0.00), though these results are based on five datapoints. Exponential models poorly fit the experimental data for both the overall GEM uptake and uptake by the first in-line filters (Fig. S5).
Table 1Summary of CEM filter loading and breakthrough during HgBr2 permeations. Samples denoted P indicate approximate permeation rate check through Line 1 via pyrolyzer and Tekran 2537A, italicized text indicates filter deployments on Line 1, and * indicates high values due to a leak around the first filter seal. NA – not available.

3.2 Mercury bromide uptake on CEM filters
Breakthrough of HgBr2 vapor from the primary (A) to secondary (B) CEM filters was low for all conditions tested in this study (Table 1). These conditions included HgBr2 permeated into clean dry laboratory air with < 0.5 g m−3 WV, clean air at ambient room humidity (4 to 5 g m−3 WV), and clean air at elevated humidity (10 to 11 g m−3 WV) at line temperatures between 17 to 19 ∘C. Overall, the mean A to B filter breakthrough ranged from 0 to 0.5 % and averaged 0.2±0.2 % (n=17), with no statistical difference observed in mean breakthrough rates for the three levels of humidity (ANOVA, p=0.124).
The first HgBr2 permeation in clean dry (< 0.5 g m−3 WV) laboratory air was over a 96 h period, using the system configuration in Fig. 1b to establish an approximate permeation rate (Fig. 3). Total Hg reaching the 2537A through the pyrolyzer on Line 1 (red line, Fig. 3) indicated an average HgBr2 exposure concentration of 4540 pg m−3, or about 4.5 pg min−1 from the permeation tube. This permeated concentration of HgBr2 was deliberately much higher than ambient concentrations in order to test retention and break through at high levels. It should be noted that these concentrations are 50–1000 times above background ambient concentrations and the performance of the CEM filters at low concentrations could be slightly different. After this permeation, total blank-corrected HgBr2 loading on the primary CEM filter on Line 0 was 49 400 pg but only 50 pg on the secondary CEM filter, indicating a breakthrough rate of approximately 0.1 %. Total Hg reaching the 2537A through the CEM filters on Line 0 (black line, Fig. 3) over this time period was 15 pg, mostly at the beginning of the deployment when some ambient Hg entered the opened system. The low concentrations of Hg measured downstream in Line 0 on the 2537A corroborate that breakthrough of HgBr2 was low. These data also demonstrate that the CEM material did not saturate with a HgBr2 loading of ∼50 000 pg, a loading far higher than could be expected in ambient conditions.

Figure 3HgBr2 permeations in clean dry laboratory air using the configuration in Fig. 1b (red line) and Fig. 1c (blue line). The red line indicates total Hg released from permeation tube and passing through the pyrolyzer on Line 1 before being measured by Tekran 2537A. The black line indicates Hg reaching 2537A through CEM filters on Line 0. Vertical grey lines indicate open system during filter deployments.
Subsequent replicate 24 h HgBr2 permeations in clean dry air resulted in consistent total Hg loading on CEM filters placed on both lines concurrently (8560±320 pg, n=6, Samples 2–7 Table 1), and mean total Hg on the secondary CEM filters was 20±10 pg (average breakthrough of 0.3 %). On Line 0 (black line, Fig. 3), which was never open to HgBr2 vapor downstream from the CEM filters at any point in the study, Hg measured at the 2537A was zero for all three 24 h permeations, indicating no breakthrough (Samples 2, 4, and 6, Table 1). However, when Line 1 had been exposed to the full HgBr2 vapor concentration of 4540 pg m−3 over the duration of the 96 h permeation test, 1155 pg of Hg was measured downstream in the first 24 h sample (Sample 3, Table 1). The amount of downstream Hg dropped to 10 pg in the second 24 h and 6 pg in the third 24 h (Samples 5 and 7, Table 1). This downstream Hg in Line 1 (compared to the zero Hg simultaneously observed on Line 0) is attributed to volatilization of HgBr2 that had adsorbed to the line material during the open permeation flow. At the moment CEM filters were deployed on Line 1 (red-to-blue transition, Fig. 3), a rapid asymptotic decline in the Hg signal began. This decay curve supports drawdown and depletion of a Hg reservoir on the interior line surfaces behind the CEM filters and not a continuous source such as breakthrough from the permeation tube that was still supplying HgBr2 to both sample lines. The total mass of Hg volatized from the interior line surfaces (1155 pg) represents 4 % to 5 % of the total HgBr2 that had passed through Line 1 (∼25 000 pg based on 2537A measurement). Eventually, Hg reaching the 2537A through Line 1 decreased to zero during the same 24 h filter deployment, indicating the majority of HgBr2 line contamination in a high-concentration permeation system can be expected to flush out within ∼12 h. However, we caution that materials used in high-concentration permeation systems, despite being flushed out, should not be used for background ambient air work without at least a very thorough acid cleaning.
Additional HgBr2 permeations were made at two levels of in-line humidity. At ambient room humidity (4 to 5 g m−3 WV), mean total Hg measured on the CEM filters was 7910±520 pg (n=4; Samples H2-5, Table 1), with an average breakthrough to the secondary filters of 0.3 %. When normalized for sample volume, the mean HgBr2 loading on CEM filters during ambient humidity (5968±125 pg) and dry air (5995±188 pg) permeations was not statistically significantly different (t test p=0.790). HgBr2 breakthrough rates were also the same (0.3 %) as during the dry air permeations, indicating that the permeation system was operating similarly at the two humidity levels and suggesting that absolute humidity concentrations of 4 to 5 g m−3 WV had insignificant effects on collection of HgBr2 in clean laboratory air by the CEM material.
An increase in humidity resulted in an initial large increase in Hg measured at the 2537A downstream of the CEM filters on Line 0 (Sample H1, Table 1), concurrently with an open HgBr2 permeation flow through Line 1 while both lines were subjected to increased RH. This downstream Hg on Line 0 dropped substantially to zero in ∼10 h in the first 24 h deployment (Sample H2, Table 1) and was zero for the duration of the second 24 h deployment (Sample H4, Table 1). Hg rapidly declined to zero, due to off-gassing from the tubing induced by the increased humidity, which facilitated a heterogeneous surface reduction of HgBr2 to GEM in the short section of line between the permeation source and CEM filters. This phenomenon was also observed during the Reno Atmospheric Mercury Intercomparison eXperiment (RAMIX; Gustin et al., 2013). Reduced HgBr2 then passed through to the 2537A as GEM. As the breakthrough rate and the mean HgBr2 loading on the CEM filters did not change between the dry air and ambient humidity permeations, the downstream Hg observed at the 2537A during the ambient humidity permeations cannot be attributed to a loss of Hg from the CEM filters and is more likely due to a process in the sample lines.
As a further test of possible humidity effects, two replicate 24 h CEM filter deployments were conducted in elevated humidity conditions (10 to 11 g m−3 WV) created by an in-line water bath. Mean total Hg loading on the primary CEM filters was higher compared to the previous permeations (11 700±720 pg, n=4, Samples H9-12, Table 1), indicating an increase in the effective HgBr2 permeation rate, possibly due to the perturbation caused by a poor filter seal and small leak in the preceding deployment (Sample H7-8, Table 1). However, mean total Hg on the secondary CEM filters was 20±20 pg, indicating an average breakthrough of 0.1 %, less than the breakthrough observed for the lower humidity permeations.
GEM uptake on the CEM material was negligible under the laboratory conditions and high GEM loading rates (3 orders of magnitude above ambient conditions) tested in this study, with an overall linear uptake rate of 0.004 % (Fig. S5). This uptake rate would be insignificant at typical ambient atmospheric Hg concentrations (1 to 2 ng m−3). As a hypothetical example, a CEM filter sampling ambient air at an average GEM concentration of 2 ng m−3 for a typical 2-week sample period would have a total Hg0 exposure of ∼40 000 pg. At the calculated uptake rate of 0.004 %, a maximum 1.6 pg of Hg observed on the sample filter could be attributed to GEM artifact. Although further work is required to more definitively determine detection and quantification limits of the various CEM methodologies, based on the mean total Hg mass of 50±20 pg observed in this study, the artifact of GEM uptake by the CEMs would be below the detection limit observed here. This corroborates the lack of GEM uptake seen by Lyman et al. (2016) for manual Hg0 injections on CEM filters at lower total mass loadings of 300 to 6000 pg.
Mean HgBr2 breakthrough from primary to secondary CEM filters averaged 0.2±0.2 % over all test conditions. A to B filter breakthrough was derived from a comparison between the large amount of HgBr2 permeated onto the primary CEM filters, to the small amount of HgBr2 that collected on the secondary CEM filters, 3 mm immediately downstream. The measurement of thousands of picogram of Hg on the primary filter, and only tens of picograms on the secondary filter, leads to the conclusion that the primary filter removed the majority of HgBr2 from the sample air stream under the laboratory conditions applied in this study. In addition, low breakthrough was corroborated by downstream measurement of the air stream passing through the CEM filters, using the Tekran® 2537A. The average breakthrough to the 2537A was 0 pg for 24 h permeations in dry air and 0 to 40 pg in humid air for filter deployments at steady state (> 24 h without large perturbations).
While the permeation system was not specifically optimized for a quantitative mass balance between permeated HgBr2 and HgBr2 recovered on the CEM filters, a rough estimation of the CEM collection efficiency is possible. Using the HgBr2 permeations conducted in clean dry air (mean loading 8560 pg) and comparing this to the mean Hg concentration measured at the 2537A analyzer during the last 24 h of the 96 h permeation measurement (4680 pg m−3 or 6739 pg per 24 h), HgBr2 recovery on the CEM filters averaged 127 %. Adjusting the expected permeated HgBr2 mass for our estimated line loss (∼4 %–5 %) changed the recoveries to ∼123 %. Still, HgBr2 loading on the CEM filters was ∼23 % higher than expected based on the pyrolyzed total measurement on the 2537A, indicating not all HgBr2 was converted to GEM. This can be explained by the pyrolyzer design used in this study not being 100 % efficient at thermally reducing HgBr2 to Hg0, based on the higher total Hg recoveries on the CEM filters versus total Hg measured through the pyrolyzer on the Tekran 2537.
The technique of gold amalgamation in general, and specifically including the Tekran® 2537A analyzer, is widely considered to provide a quantitative total gaseous Hg measurement, at or very near 100 % collection efficiency for Hg0 and Hg compounds (Temme et al., 2003; Landis et al., 2002; Schroeder et al, 1995; Dumarey et al., 1985; Schroeder and Jackson, 1985). However, to our knowledge collection and desorption efficiencies on gold traps have not been demonstrated for HgBr2. The stated desorption temperature of the Tekran® 2537A gold traps is 500 ∘C, but temperatures as low as 375 ∘C have been reported (Gustin et al., 2013). This would cause reduced thermal decomposition efficiency for all captured GOM compounds, including HgBr2. We speculate that a combination of incomplete thermal decomposition to Hg0 at both the 600 ∘C pyrolyzer and during the best-case 500 ∘C desorption of the 2537A gold traps contributed to the ∼20 % non-detection of total permeated HgBr2 as it passed through the CVAFS optical path. Evidence for this conclusion can be seen in the 134 % increase in Hg collection by the Tekran® 2537A when the pyrolyzer was at 1000 ∘C, as compared to at 650 ∘C (Fig. S4), in supplementary experiments.
While our results validated some basic performance metrics for the CEM material, they did not provide data that could fully explain the higher levels of breakthrough observed for CEM filters deployed in ambient air over the 1- to 2-week sample periods in previous studies. Increasing humidity by itself did not affect observed HgBr2 breakthrough. A HgBr2 loading of ∼50 000 pg also did not lead to increased breakthrough, indicating there is no saturation effect on CEM filter capacity at a GOM loading far greater than expected from ambient concentrations. It remains unclear, though, whether breakthrough results from different collection efficiencies for GOM compounds other than HgBr2 or whether breakthrough results from a degradation of GOM retention capacity in the CEM material when exposed to ambient air chemistries not simulated in this study. Also, our experiments were conducted in particulate-free air, which leaves open the possibility that breakthrough is related to capture (or lack thereof) of PBM by the CEM material.
Further testing and refinements are necessary, beginning with optimization of the pyrolyzer parameters (e.g., temperature, volume) to allow for a more accurate quantitative comparison between the CEM and Tekran® 2537A results. Permeation rates of HgBr2 were variable and need to be more precisely controlled, with a standardized and stable GOM permeation system being needed in general. This study was undertaken using controlled laboratory conditions, but CEM performance needs to be further validated in ambient air. Specifically, the reasons for RM breaking through CEM filters deployed in ambient air still need to be determined.
Data are available from the corresponding author.
The supplement related to this article is available online at: https://doi.org/10.5194/amt-12-1207-2019-supplement.
MM and SDC performed the experiments and constructed the manuscript. MSG and GE guided the experiments and helped write the manuscript.
The authors declare that they have no conflict of interest.
The authors would like to acknowledge funding from Macquarie University
iMQRES 2015148 and NSF grant 629679. Valuable input and assistance were
received from Ashley Pierce, Seth Lyman, and the students of
Mae Sexauer Gustin's laboratory. We bid an untimely farewell to Grant C. Edwards,
who was ever a cheerful friend, mentor, and colleague. Grant C. Edwards passed
away unexpectedly on 10 September 2018. We thank the diligent reviewers for
their valuable and constructive comments.
Edited by: Jonathan Abbatt
Reviewed by: three anonymous referees
Bloom, N., Prestbo, E., and VonderGeest, E.: Determination of atmospheric gaseous Hg(II) at the pg/m 3 level by collection onto cation exchange membranes, followed by dual amalgamation/cold vapor atomic fluorescence spectrometry, 4th International Conference on Mercury as a Global Pollutant, Hamburg, 1996.
Dumarey, R., Dams, R., and Hoste, J.: Comparison of the collection and desorption efficiency of activated charcoal, silver, and gold for the determination of vapor phase atmospheric mercury, Anal. Chem., 57, 2638–2643, https://doi.org/10.1021/ac00290a047, 1985.
Ebinghaus, R., Jennings, S. G., Schroeder, W. H., Berg, T., Donaghy, T., Guentzel, J., Kenny, C., Kock, H. H., Kvietkus, K., Landing, W., Muhleck, T., Munthe, J., Prestbo, E. M., Schneeberger, D., Slemr, F., Sommar, J., Urba, A., Wallschlager, D., and Xiao, Z.: International field intercomparison measurements of atmospheric mercury species at Mace Head, Ireland, Atmos. Environ., 33, 3063–3073, 1999.
Gustin, M. S., Huang, J., Miller, M. B., Peterson, C., Jaffe, D. A., Ambrose, J., Finley, B. D., Lyman, S. N., Call, K., Talbot, R., Feddersen, D., Mao, H., and Lindberg, S. E.: Do We Understand What the Mercury Speciation Instruments Are Actually Measuring? Results of RAMIX, Environ. Sci. Technol., 47, 7295–7306, https://doi.org/10.1021/es3039104, 2013.
Gustin, M. S., Amos, H. M., Huang, J., Miller, M. B., and Heidecorn, K.: Measuring and modeling mercury in the atmosphere: a critical review, Atmos. Chem. Phys., 15, 5697–5713, https://doi.org/10.5194/acp-15-5697-2015, 2015.
Gustin, M. S., Pierce, A. M., Huang, J., Miller, M. B., Holmes, H. A., and Loria-Salazar, S. M.: Evidence for Different Reactive Hg Sources and Chemical Compounds at Adjacent Valley and High Elevation Locations, Environ. Sci. Technol., 50, 12225–12231, https://doi.org/10.1021/acs.est.6b03339, 2016.
Huang, J. and Gustin, M. S.: Uncertainties of Gaseous Oxidized Mercury Measurements Using KCl-Coated Denuders, Cation-Exchange Membranes, and Nylon Membranes: Humidity Influences, Environ. Sci. Technol., 49, 6102–6108, https://doi.org/10.1021/acs.est.5b00098, 2015a.
Huang, J. and Gustin, M. S.: Use of Passive Sampling Methods and Models to Understand Sources of Mercury Deposition to High Elevation Sites in the Western United States, Environ. Sci. Technol., 49, 432–441, https://doi.org/10.1021/es502836w, 2015b.
Huang, J., Miller, M. B., Weiss-Penzias, P., and Gustin, M. S.: Comparison of Gaseous Oxidized Hg Measured by KCl-Coated Denuders, and Nylon and Cation Exchange Membranes, Environ. Sci. Technol., 47, 7307–7316, https://doi.org/10.1021/es4012349, 2013.
Huang, J., Miller, M. B., Edgerton, E., and Sexauer Gustin, M.: Deciphering potential chemical compounds of gaseous oxidized mercury in Florida, USA, Atmos. Chem. Phys., 17, 1689–1698, https://doi.org/10.5194/acp-17-1689-2017, 2017.
Jaffe, D. A., Lyman, S., Amos, H. M., Gustin, M. S., Huang, J., Selin, N. E., Levin, L., ter Schure, A., Mason, R. P., Talbot, R., Rutter, A., Finley, B., Jaeglé, L., Shah, V., McClure, C., Ambrose, J., Gratz, L., Lindberg, S., Weiss-Penzias, P., Sheu, G.-R., Feddersen, D., Horvat, M., Dastoor, A., Hynes, A. J., Mao, H., Sonke, J. E., Slemr, F., Fisher, J. A., Ebinghaus, R., Zhang, Y., and Edwards, G.: Progress on Understanding Atmospheric Mercury Hampered by Uncertain Measurements, Environ. Sci. Technol., 48, 7204–7206, https://doi.org/10.1021/es5026432, 2014.
Landis, M. S., Stevens, R. K., Schaedlich, F., and Prestbo, E. M.: Development and characterization of an annular denuder methodology for the measurement of divalent inorganic reactive gaseous mercury in ambient air, Environ. Sci. Technol., 36, 3000–3009, https://doi.org/10.1021/es015887t, 2002.
Lyman, S., Jones, C., O'Neil, T., Allen, T., Miller, M., Gustin, M. S., Pierce, A. M., Luke, W., Ren, X., and Kelley, P.: Automated Calibration of Atmospheric Oxidized Mercury Measurements, Environ. Sci. Technol., 50, 12921–12927, https://doi.org/10.1021/acs.est.6b04211, 2016.
Lyman, S. N., Gustin, M. S., Prestbo, E. M., and Marsik, F. J.: Estimation of Dry Deposition of Atmospheric Mercury in Nevada by Direct and Indirect Methods, Environ. Sci. Technol., 41, 1970–1976, https://doi.org/10.1021/es062323m, 2007.
Lyman, S. N., Jaffe, D. A., and Gustin, M. S.: Release of mercury halides from KCl denuders in the presence of ozone, Atmos. Chem. Phys., 10, 8197–8204, https://doi.org/10.5194/acp-10-8197-2010, 2010.
Marusczak, N., Sonke, J. E., Fu, X., and Jiskra, M.: Tropospheric GOM at the Pic du Midi Observatory – Correcting Bias in Denuder Based Observations, Environ. Sci. Technol., 51, 863–869, https://doi.org/10.1021/acs.est.6b04999, 2017.
Mason, R., Lawson, N., and Sullivan, K.: The concentration, speciation and sources of mercury in Chesapeake Bay precipitation, Atmos. Environ., 31, 3541–3550, https://doi.org/10.1016/S1352-2310(97)00206-9, 1997.
McClure, C. D., Jaffe, D. A., and Edgerton, E. S.: Evaluation of the KCI Denuder Method for Gaseous Oxidized Mercury using HgBr2 at an In-Service AMNet Site, Environ. Sci. Technol., 48, 11437–11444, https://doi.org/10.1021/es502545k, 2014.
Pierce, A. M. and Gustin, M. S.: Development of a Particulate Mass Measurement System for Quantification of Ambient Reactive Mercury, Environ. Sci. Technol., 51, 436–445, https://doi.org/10.1021/acs.est.6b04707, 2017.
Schroeder, W. and Jackson, R.: An instrumental analytical technique for speciation of atmospheric mercury, Int. J. Environ. An. Ch., 22, 1–18, https://doi.org/10.1080/03067318508076405, 1985.
Schroeder, W., Keeler, G., Kock, H., Roussel, P., Schneeberger, D., and Schaedlich, F.: International field intercomparison of atmospheric mercury measurement methods, Water Air Soil Pollut., 80, 611–620, https://doi.org/10.1007/BF01189713, 1995.
Sheu, G. R. and Mason, R. P.: An examination of methods for the measurements of reactive gaseous mercury in the atmosphere, Environ. Sci. Technol., 35, 1209–1216, https://doi.org/10.1021/es001183s, 2001.
Slemr, F., Angot, H., Dommergue, A., Magand, O., Barret, M., Weigelt, A., Ebinghaus, R., Brunke, E.-G., Pfaffhuber, K. A., Edwards, G., Howard, D., Powell, J., Keywood, M., and Wang, F.: Comparison of mercury concentrations measured at several sites in the Southern Hemisphere, Atmos. Chem. Phys., 15, 3125–3133, https://doi.org/10.5194/acp-15-3125-2015, 2015.
Temme, C., Einax, J. W., Ebinghaus, R., and Schroeder, W. H.: Measurements of Atmospheric Mercury Species at a Coastal Site in the Antarctic and over the South Atlantic Ocean during Polar Summer, Environ. Sci. Technol., 37, 22–31, https://doi.org/10.1021/es025884w, 2003.
Xiao, Z., Sommar, J., Wei, S., and Lindqvist, O.: Sampling and determination of gas phase divalent mercury in the air using a KCl coated denuder, Fresen. J. Anal. Chem., 358, 386–391, 1997.
Zhang, L., Lyman, S., Mao, H., Lin, C.-J., Gay, D. A., Wang, S., Sexauer Gustin, M., Feng, X., and Wania, F.: A synthesis of research needs for improving the understanding of atmospheric mercury cycling, Atmos. Chem. Phys., 17, 9133–9144, https://doi.org/10.5194/acp-17-9133-2017, 2017.