the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 22 Feb 2021
| 22 Feb 2021
Introducing the extended volatility range proton-transfer-reaction mass spectrometer (EVR PTR-MS)
Felix Piel
Markus Müller
Klaus Winkler
Jenny Skytte af Sätra
Armin Wisthaler
Proton-transfer-reaction mass spectrometry (PTR-MS) is widely used in atmospheric sciences for measuring volatile organic compounds in real time. In the most widely used type of PTR-MS instruments, air is directly introduced into a chemical ionization reactor via an inlet capillary system. The reactor has a volumetric exchange time of ∼0.1 s, enabling PTR-MS analyzers to measure at a frequency of 10 Hz. The time response does, however, deteriorate if low-volatility analytes interact with surfaces in the inlet or in the instrument. Herein, we present the extended volatility range (EVR) PTR-MS instrument which mitigates this issue. In the EVR configuration, inlet capillaries are made of passivated stainless steel, and all wetted metal parts in the chemical ionization reactor are surface-passivated with a functionalized hydrogenated amorphous silicon coating. Heating the entire setup (up to 120 ∘C) further improves the time-response performance.
We carried out time-response performance tests on a set of 29 analytes having saturation mass concentrations C0 in the range between 10−3 and 105 µg m−3. The -signal decay times after instant removal of the analyte from the sampling flow were between 0.2 and 90 s for gaseous analytes. We also tested the EVR PTR-MS instrument in combination with the chemical analysis of aerosols online (CHARON) particle inlet, and -signal decay times were in the range between 5 and 35 s for particulate analytes. We show on a set of example compounds that the time-response performance of the EVR PTR-MS instrument is comparable to that of the fastest flow tube chemical ionization mass spectrometers that are currently in use. The fast time response can be used for rapid (∼1 min equilibration time) switching between gas and particle measurements. The CHARON EVR PTR-MS instrument can thus be used for real-time monitoring of both gaseous and particulate organics in the atmosphere. Finally, we show that the CHARON EVR PTR-MS instrument also rapidly detects highly oxygenated species (with up to eight oxygen atoms) in particles formed by limonene ozonolysis.
- Article
(1935 KB) - Full-text XML
-
Supplement
(505 KB) - BibTeX
- EndNote





The Earth's atmosphere contains a plethora of organic compounds, both in the gas and in the particulate phase (Goldstein and Galbally, 2007). Atmospheric organic compounds vary widely in their physicochemical properties (e.g., volatility, polarity, solubility), which makes their comprehensive measurement challenging (Heald and Kroll, 2020).
Proton-transfer-reaction mass spectrometry (PTR-MS) is widely used in atmospheric sciences for measuring volatile organic compounds (Hansel et al., 1995; de Gouw and Warneke, 2007; Yuan et al., 2017). In PTR-MS instruments, air is directly introduced into an ion–molecule reactor wherein organic molecules ionize in collisions with hydronium (H3O+) ions. An electric field is applied across the ion–molecule reactor, which is thus commonly referred to as the drift tube. Reagent and analyte ions are extracted from the drift tube and analyzed in a mass spectrometer.
One of the main advantages of PTR-MS is its rapidness. The drift tube has a volumetric exchange time of ∼0.1 s, enabling PTR-MS analyzers to measure at a frequency of 10 Hz (Müller et al., 2010). The time response does, however, deteriorate if low-volatility analytes interact with surfaces in the inlet or in the instrument. PTR-MS instrument users have mitigated this problem by (i) operating the drift tube at elevated temperatures (Mikoviny et al., 2010), (ii) increasing the flow through the drift tube (Breitenlechner et al., 2017; Krechmer et al., 2018) and (iii) minimizing the wall collisions of analyte molecules (Breitenlechner et al., 2017). The problem of analyte adsorption becomes even more pronounced when particles are analyzed with the chemical analysis of aerosols online (CHARON) inlet (Eichler et al., 2015; Müller et al., 2017). After particle vaporization, low-volatility gases adsorb onto surfaces in the vaporizer, in the transfer line from the vaporizer to the drift tube and in the drift tube itself. This slows down the instrumental response significantly (Piel et al., 2019).
A second major advantage of PTR-MS is that H3O+ ion chemistry detects a wide spectrum of organic analytes, from non-oxidized to highly oxidized species. It must, however, be ensured that the analyte molecules reach the ionization region and are not lost in the inlet line (e.g., Pagonis et al., 2017; Deming et al., 2019) or onto the drift tube walls.
Herein we will demonstrate how the use of heated inlet capillaries made of passivated stainless steel (SS) and of a heated drift tube with passivated metal surfaces significantly improves the time-response performance of PTR-MS analyzers. We will show that the heated and passivated instrument responds fast to low-volatility analytes, both for gas-phase and particle-phase measurements. The instrument setup described herein has thus been named the extended volatility range (EVR) configuration.
2.1 The EVR PTR-MS instrument
The PTR-MS instrument has been described in detail elsewhere (Yuan et al., 2017, and references therein). The data presented herein were obtained with two state-of-the-art CHARON PTR-MS analyzers (models PTR-TOF 4000X2 and PTR-TOF 6000X2) produced by IONICON Analytik (Innsbruck, Austria). In their conventional setup, these analyzers include inlet capillaries made of polyether ether ketone (PEEK) and a drift tube plus ion funnel consisting of electropolished SS drift rings and polytetrafluoroethylene (PTFE) spacers. PEEK and especially SS are known to adsorb certain analytes. In an effort to optimize the instrumental time response, we have eliminated all surfaces that are prone to analyte adsorption (Fig. 1). All PEEK capillaries were replaced by surface-treated SS capillaries. The surface treatment consisted in an application of functionalized hydrogenated amorphous silicon coating. The same surface passivation was applied to all wetted SS parts in the drift tube. In the CHARON inlet, the sampler and the vaporizer as well as the transfer tube from the vaporizer to the drift tube were surface-passivated. Figure 1 shows all surface-treated parts in blue. As for conventional PTR-MS analyzers, the drift tube and inlet lines were housed in a temperature-controlled enclosure that can be operated from room temperature to 120 ∘C. The enclosure temperature is referred to as the drift tube temperature (Tdrift).
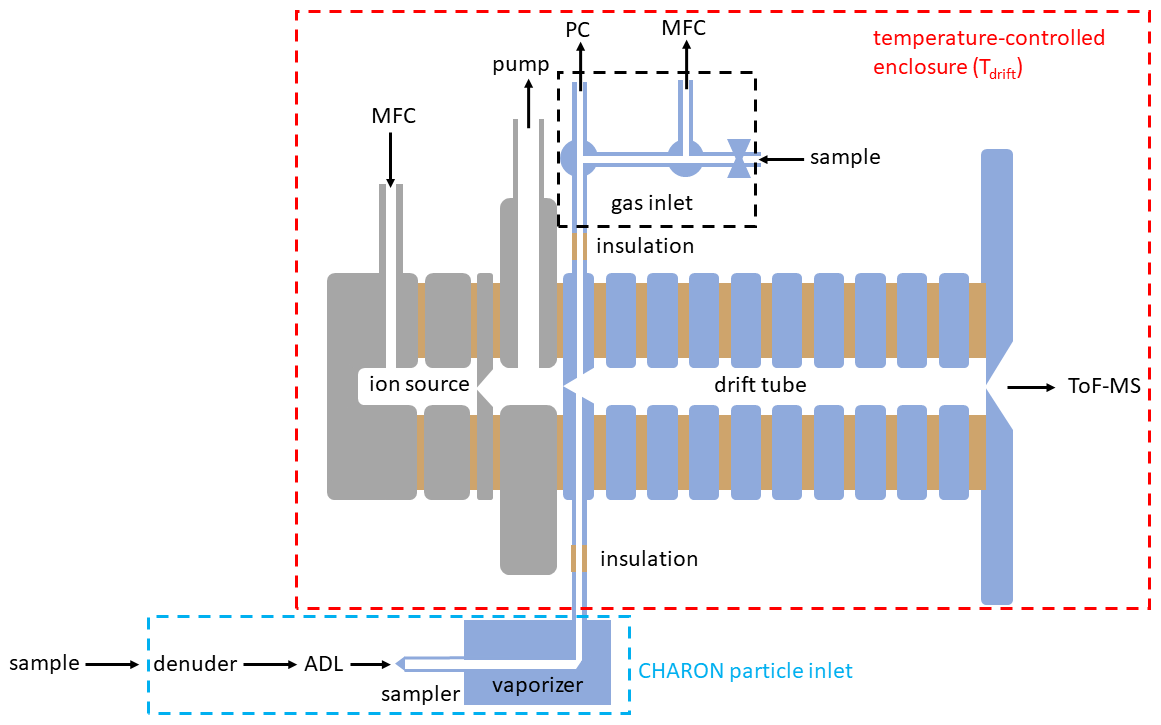
Figure 1Scheme of the EVR PTR-MS instrument including a gas inlet and the CHARON particle inlet. The parts in blue were passivated with a functionalized hydrogenated amorphous silicon coating. MFC: mass flow controller; PC: pressure controller, ADL: aerodynamic lens; ToF-MS: time-of-flight mass spectrometer.
2.2 Performance assessment of the EVR PTR-MS instrument
A laboratory study was carried out to measure signal decay times in the EVR PTR-MS analyzer for 29 analytes listed in the Supplement (Table S1). A single analyte was supplied in steady concentration to the analyzer and instantly removed from the inlet. is the time it took the analyte signal to decay to (36.8 %) of its initial value. τ90 is the time it took the analyte signal to decay to 10 % of its initial value. A stable gaseous analyte concentration, which is herein denoted with the subscript (g), was generated by placing a spatula tip of the solid sample into a 100 mL glass vial. The vial was heated and flushed with zero air (RH∼30 %). Heating temperatures ranged from 50 to 120 ∘C, depending upon the melting point of the analyte. The dynamic headspace of the vial was sampled through the gas inlet of the PTR-MS analyzer. The instrument and inlet were zeroed by overflowing the inlet with zero air (Fig. S1 in the Supplement). A stable particulate analyte concentration, which is herein denoted with the subscript (p), was generated by dissolving an aliquot of the solid sample in HPLC-grade water (Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany). The solution was then nebulized with a homebuilt nebulizer. The nebulizer outflow was dried with two homebuilt diffusion dryers, and gases were removed with an activated charcoal denuder (NovaCarb F, Mast Carbon International Ltd., Guildford, UK). The CHARON inlet and PTR-MS instrument were zeroed by diverting the sample flow through a high-efficiency particulate air (HEPA) filter (Fig. S1).
Example data from a field study were taken to show the signal response of the EVR PTR-MS analyzer when switching between the CHARON particle inlet and the gas inlet. The data were collected during a measurement campaign at the TROPOS Research Station Melpitz (Spindler et al., 2013) in Germany in February 2019.
A laboratory study was carried out for investigating the capability of the CHARON EVR PTR-MS analyzer to detect highly oxidized organic molecules in particles. For this purpose, we reacted ozone and limonene in a flow reactor to form secondary organic aerosol (SOA). The reactor outflow was passed through an activated charcoal denuder (NovaCarb F, Mast Carbon International Ltd., Guildford, UK) for removing gaseous organics and subsequently injected into a 210 L steel barrel (Wilai GmbH, Wiedemar, Germany). The CHARON EVR PTR-MS analyzer sampled from this reservoir. The instrument was operated at low reduced electric field strength ( Td; 1 Td V cm−2) and with as the reagent ion (Müller et al., 2020). With these instrumental settings, ionic fragmentation is largely suppressed, and highly oxidized organic molecules are detected in their ammonium adduct form (Zaytsev et al., 2019).
3.1 Effect of surface passivation on time response
Figure 2 shows data from two experiments wherein the signal decay of cis-pinonic acid(g) was measured with the same PTR-MS instrument before and after being modified to the EVR configuration. Tdrift was set to 100 ∘C in both experiments; all other experimental parameters were kept identical. In the conventional configuration, the analyte signal exhibited a long tailing while it rapidly dropped to near-zero levels in the EVR configuration. was 181 s for the conventional setup and 4 s in the EVR configuration. These data clearly demonstrate that surface passivation greatly improves the time response of PTR-MS analyzers.
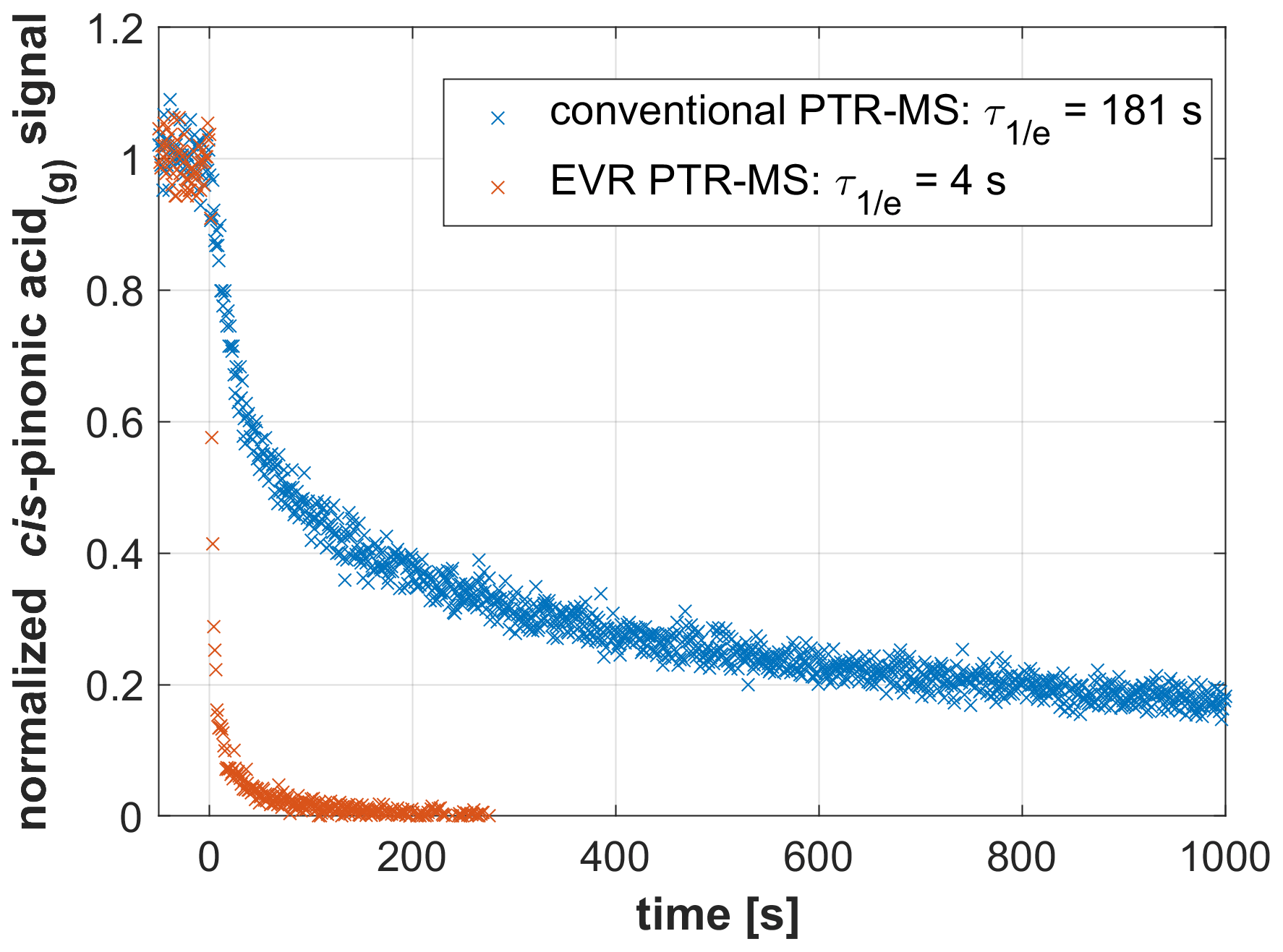
Figure 2Signal decay as observed in a conventional PTR-MS instrument (Tdrift=100 ∘C; in blue) and in an EVR PTR-MS analyzer (Tdrift=100 ∘C; in red) after a steady supply of gaseous cis-pinonic acid was instantly switched off at t=0 s.
Figure 3 shows the response of the EVR PTR-MS analyzer (Tdrift=120 ∘C) to an example set of analytes, measured via the gas inlet (upper panel) and via the CHARON particle inlet (lower panel). 4-Nitrocatechol(g) (in dark yellow) exhibited a rather slow decay ( s). cis-Pinonic acid(g) (in red) decayed in a few seconds ( s). For 2-tridecanone (in blue), was 0.3 s, which is close to the volumetric exchange time of the drift tube (∼0.1 s). The three example compounds shown here cover the full 3 orders of magnitude span in (10−1 to 102 s) that was observed for gaseous analytes. For particulate analytes, ranged from a few seconds to a few tens of seconds. The lower panel of Fig. 3 shows example data obtained for 2,7-dihydroxynaphthalene(p) (in dark yellow; s), levoglucosan(p) (in red; s) and nitrate(p) (in blue; s). The nitrate(p) signal originated from ammonium nitrate particles.
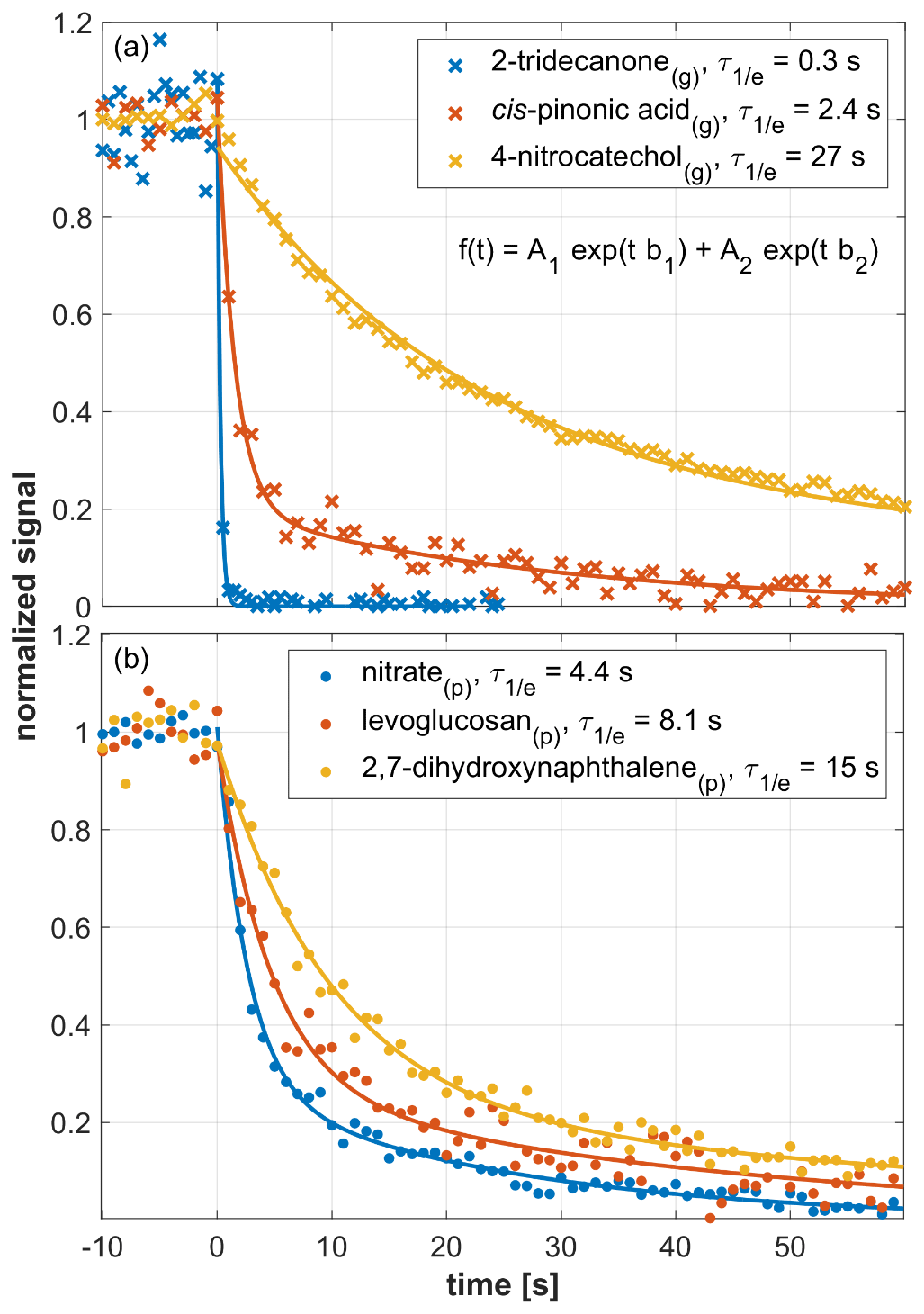
Figure 3Signal decay as observed in an EVR PTR-MS analyzer (Tdrift=120 ∘C) after a steady supply of analyte was instantly switched off at t=0 s. The decay of gaseous analytes is shown in the upper panel, while the lower panel refers to particulate analytes. Initial steady-state mixing ratios were as follows: 2-tridecanone(g), 1 ppbv; cis-pinonic acid(g), 2.2 ppbv; 4-nitrocatechol(g), 30 ppbv; nitrate, 3 ppbv; levoglucosan(p), 1.2 ppbv; 2,7-dihydroxynaphthalene(p), 0.3 ppbv. Signals were fitted using a double exponential decay function (see insert in a). All fitting parameters are listed in Table S2.
Figure 4 summarizes the values measured for 21 gaseous analytes (upper panel) and 15 particulate analytes (lower panel). The color coding and sizing are explained in the figure legend. Mixing ratios were quantified according to the procedure outlined in the Supplement of Müller et al. (2017) and were typically in the 0.1–10 ppbv range. For some compounds (vanillin(g), 2-tridecanone(g), 2,6-dimethoxyphenol(g), 4-nitroguaiacol(g), ammonia(g)) it was difficult to generate mixing ratios in the low-ppbv range, which is why we also included mixing ratios up to 100 ppbv in our analysis. We typically measured at three different mixing ratios for each compound. Three data points are thus typically shown for each analyte. Only small changes in the instrumental time response were observed because analyte concentrations typically only spanned a relatively narrow range. We will thus not discuss any concentration dependence of here. We observed an increase in with decreasing saturation mass concentrations (log C0) as a general trend, although with significant deviations for some compounds (Fig. S2). Glucose(g), for example, exhibited a much faster response than the sugar alcohols (xylitol(g), arabitol(g)) despite having a similar log C0. Structural effects may play a role here, since glucose is a cyclic molecule, while the sugar alcohols are both linear. It is also important to note that the SIMPOL.1 method has not been validated for saccharides and that the calculated log C0 may be inaccurate. 4-Nitrocatechol, with a relatively high log C0 of 4.2, was among the slowest responding gaseous analytes. This observation remains unexplained. With the CHARON inlet connected, was in the 5 to 20 s range for most analytes (Fig. 4, lower panel). Exceptions were diglycolic acid(p) and tartaric acid(p) with up to ∼35 s. Notably, no obvious dependence of on log C0 was observed. Levoglucosan, 2,7-dihydroxynaphthalene, stearic acid, azelaic acid, diglycolic acid and vanillic acid were studied in both phases. The instrumental response was typically ∼5 s slower in the particle measurements. The CHARON inlet has a larger surface area and a lower sample flow than the gas inlet. Stearic acid and azelaic acid responded faster with the CHARON inlet, which remains unexplained. The reader is cautioned that the values presented in Fig. 4 should not be taken as absolute and generally applicable values. The reported numbers should be seen as indicative figures for the time response of state-of-the-art IONICON EVR PTR-MS instruments. It is well known that analytes compete for surface adsorption with other matrix constituents such as water or other surface-affine compounds. All of our experiments were carried out with a single compound at one humidity level. We consider it beyond the scope of this work to investigate a matrix dependence of . Especially for low-volatility analytes, repeated sampling may passivate remaining active sites, which in turn improves the time response. This effect probably explains the relatively large variations in observed for the slow-responding analytes, even if all three experiments were carried out at similar mixing ratios. In previous work, we anecdotally observed that basic analytes exhibited a significantly slower time response when acidic samples had been sampled before. The sampling history was not considered in our study.
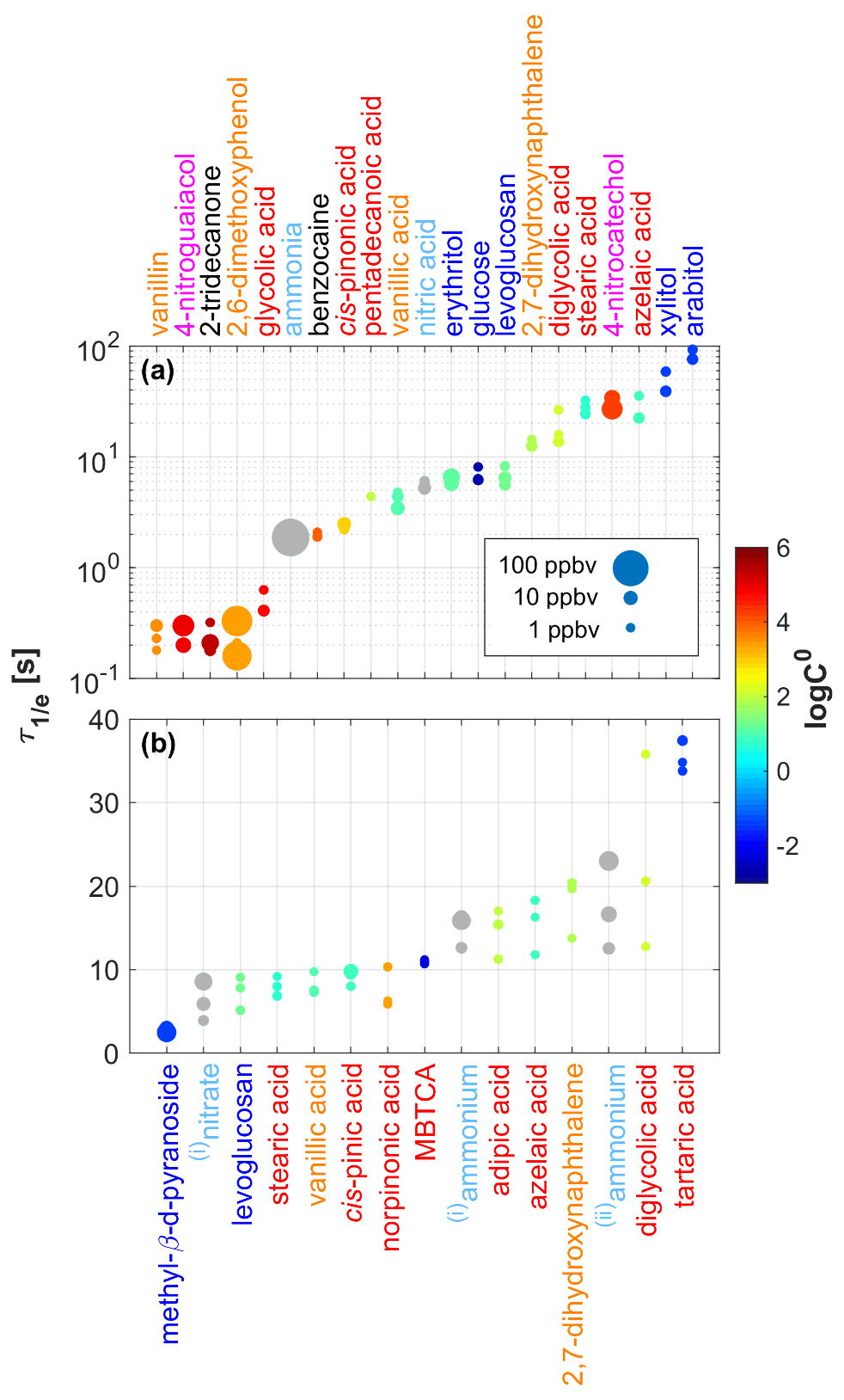
Figure 4Signal decay times () measured for 21 gaseous analytes (a) and 15 particulate analytes (b). Analytes were grouped and color-coded into six classes: saccharides (in dark blue), carboxylic acids (in red), substituted phenols (in orange), nitroaromatics (in magenta), small polar molecules (in light blue) and others (in black). The size of the dots indicates the initial steady-state mixing ratio (0.1–100 ppbv) used in the respective experiment. The color code of the data points indicates the saturation mass concentrations (log C0) of the analytes as calculated using the SIMPOL.1 method (Pankow and Asher, 2008). (i) Originating from ammonium nitrate; (ii) originating from ammonium sulfate.
3.2 Drift tube temperature effects
For long-chain ketones (Pagonis et al., 2017; Krechmer et al., 2018) and carboxylic acids (Fig. S2), exhibits the expected increase with decreasing log C0. Since log C0 depends upon temperature, changes in Tdrift should lead to predictable changes in . We thus measured for 2-tridecanone(g) and cis-pinonic acid(g) at variable Tdrift (Fig. 5). In the case of 2-tridecanone(g) (upper panel), an increase in Tdrift from room temperature to 60 ∘C decreased from 24 to 2 s. At Tdrift=100 ∘C, was 0.3 s, approaching the volumetric exchange time of the drift tube. In the case of cis-pinonic acid(g) (lower panel), we only investigated temperatures of 80 ∘C and above because the solid analyte sample was kept at 70 ∘C. When increasing Tdrift from 80 to 120 ∘C, dropped from 6.5 to 2.4 s. For the two compounds investigated, the decrease in can be explained by the increase in log C0 with temperature. According to Epstein et al. (2010), a 15 ∘C temperature rise increases log C0 by ∼1 in the 0 to 50 ∘C temperature range. The effect becomes less pronounced at higher temperatures. For cis-pinonic acid, the SIMPOL.1 method yields a log C0 increase from 5.2 to 6.2 in the 80 to 120 ∘C temperature range. At 45 ∘C, 2-tridecanone has roughly the same log C0 as cis-pinonic acid at 120 ∘C (Fig. S3). Consistently, we observed a similar time response for the two compounds at Tdrift=45 ∘C and Tdrift=120 ∘C, respectively. We conclude that increasing Tdrift to 120 ∘C is an effective way for reducing in EVR PTR-MS analyzers.

Figure 5Signal decay as observed in an EVR PTR-MS analyzer for 2-tridecanone(g) (a) and cis-pinonic acid(g) (b), respectively, at different drift tube temperatures. Initial steady-state mixing ratios were as follows: 2-tridecanone(g), 1–1.3 ppbv; cis-pinonic acid, 2.2 ppbv. Signals were fitted using a double exponential decay function (see insert in b). All fitting parameters are listed in Table S3.
Exposing the sample gas to heated surfaces in an analyzer may thermally degrade some analytes. It is important to note that none of the acids studied in this work decarboxylated at Tdrift=120 ∘C. It may, however, be necessary to use a lower Tdrift when more thermally labile analytes are targeted.
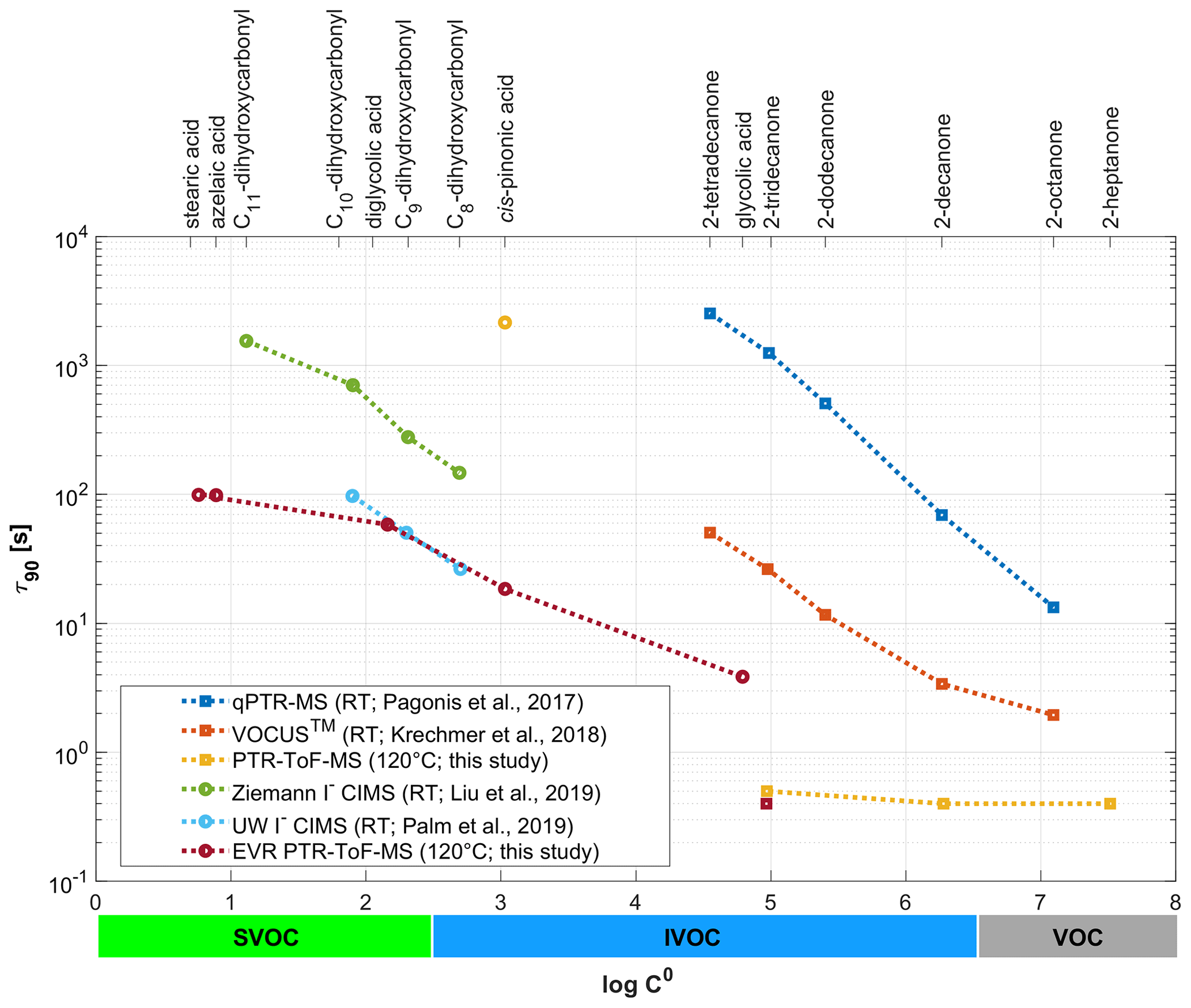
Figure 6Signal decay times (here reported as τ90) as observed in different online chemical ionization mass spectrometry (CIMS) instruments for a set of ketones, carboxylic acids and dihydroxycarbonyls. τ90 is plotted as a function of the SIMPOL.1-derived saturation mass concentration (log C0). Note that the figure legend shows the different operating temperatures of the instruments used (RT: room temperature).
3.3 Response times of different online CIMS instruments
Figure 6 compares the signal decay times (here reported as τ90 and not as ) as observed in different online chemical ionization mass spectrometry (CIMS) instruments. The plot includes data from this study, which were obtained with two state-of-the-art IONICON PTR-MS analyzers. The data points in yellow were obtained with a conventional instrument, while the data points in dark red were collected with an EVR-type analyzer. Both instruments were operated at Tdrift=120 ∘C. The figure also includes literature data obtained with an old quadrupole PTR-MS instrument (qPTR-MS; in blue: Pagonis et al., 2017), with a PTR-ToF-MS analyzer from a different manufacturer (VOCUS™; in orange: Krechmer et al., 2018) and with two iodide (I−) CIMS instruments (in green: Liu et al., 2019; in light blue: Palm et al., 2019). The latter four instruments were all operated at room temperature (RT). τ90 is plotted against log C0 of the respective analyte, which was calculated using the SIMPOL.1 method (Pankow and Asher, 2008). The upper horizontal axis lists the compound names; the lower horizontal axis shows in which log C0 range molecules are classified as volatile organic compounds (VOCs), intermediate-volatility organic compounds (IVOCs) and semi-volatile organic compounds (SVOCs). Since the instruments did not measure the same SVOCs, we only use our carboxylic acid data for the log C0-based comparison. For the acids, log C0 appears to be the main determinant of τ90, whereas for the saccharides and substituted phenols other properties affect τ90 (Fig. S2). For VOCs and the more volatile IVOCs (log C0≥5), τ90 is close to the volumetric exchange time of the drift tube for both the EVR and the conventional PTR-ToF-MS instrument, if the drift tube and inlet are heated to 120 ∘C. Conventional unheated PTR-MS instruments have a much slower time response. For the less volatile IVOCs (log C0≤3; cis-pinonic acid) surface passivation reduces τ90 by 2 orders of magnitude, even if the drift tube is heated to 120 ∘C in both the conventional and the EVR PTR-ToF-MS analyzer. For the SVOCs, the heated EVR PTR-ToF-MS has a similar time response as an optimized I− CIMS instrument with reduced instrument wall interactions (Palm et al., 2019).
3.4 Rapid switching between the gas inlet and the CHARON particle inlet
For certain applications, it is desirable to periodically switch between gas and particle measurements. Figure 7 shows example data collected by a CHARON EVR PTR-MS instrument during ambient air measurements at a rural background station in Germany.
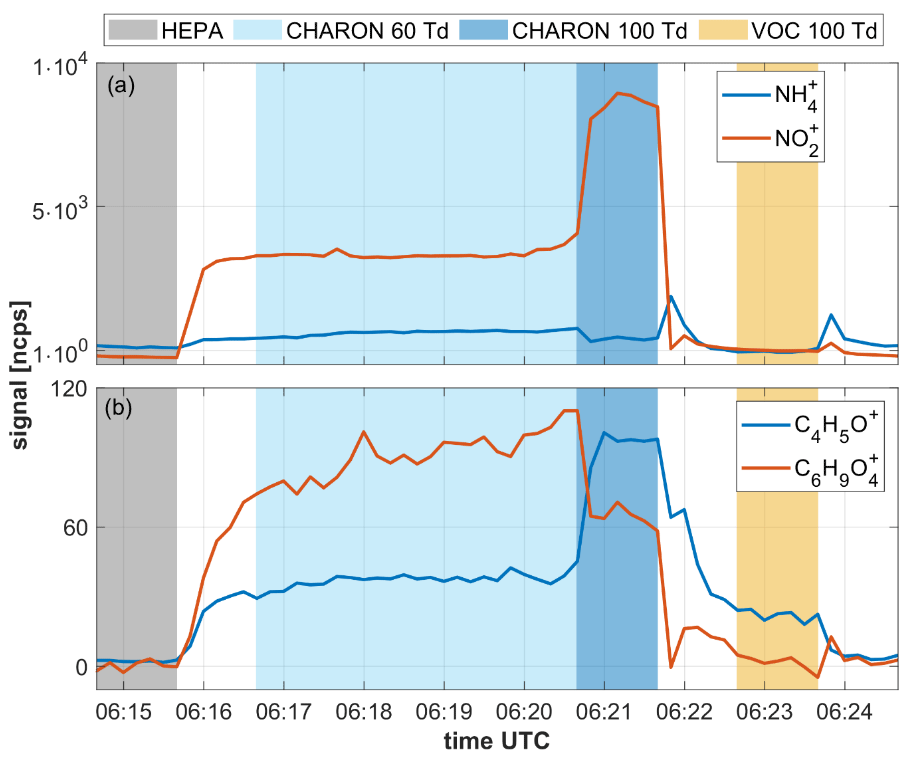
Figure 7Time series showing various analyte signals as recorded during a 10 min measurement cycle of ambient air. The 10 min measurement cycle included (i) 1 min of instrumental background measurements with the CHARON inlet including a HEPA filter, (ii) 4 min of particulate measurements at an of 60 Td, (iii) 1 min of particulate measurements at an of 100 Td and (iv) 1 min of gas measurements at an of 100 Td. The benefit of measuring particles at 60 and 100 Td is explained in Leglise et al. (2019) and is not discussed here. The CHARON inlet enriched the particle concentration by a factor of ∼20, which explains the higher signal intensities in the particle measurement (in blue) as compared to the gas measurement (in yellow).
The reader should focus on the transition from background to ambient particle measurements at 06:15:40 UTC and the transition from particle to gas measurements at 06:21:40 UTC, which is when the slow signal response becomes most evident. The upper panel shows the time evolution of the ( 18.034) and ( 45.996) signals. In the CHARON inlet, ammonium nitrate particles evaporate to yield gaseous ammonia (NH3) and nitric acid (HNO3). Ammonia is detected in its protonated form, while nitric acid dehydrates upon protonation to yield the nitronium ion (). Both ammonia and nitric acid are particularly prone to adsorptive losses on stainless steel (Neuman et al., 1999; Nowak et al., 2007). In the EVR-type instrument, wetted surfaces do not include any untreated stainless steel, which results in a fast instrumental response to both compounds. Both the and the signal equilibrated within 1 min when switching from HEPA to ambient CHARON measurements and from particle to gas measurements, respectively. The lower panel shows the evolution of the C4H5O+ ( 69.033) and ( 145.049) signals within one measurement cycle. is the main ionic fragment from levoglucosan (Leglise et al., 2019). C4H5O+ is believed to be a fragment of larger furanoid compounds in particles and protonated furan in the gas measurement. The instrumental response to these analytes was somewhat slower than to ammonium nitrate but equilibration still occurred within 1 min. The instrumental response to levoglucosan is similar to what we observed in single compound measurements in the laboratory, suggesting that the presence of a complex matrix does not negatively affect instrumental response times.
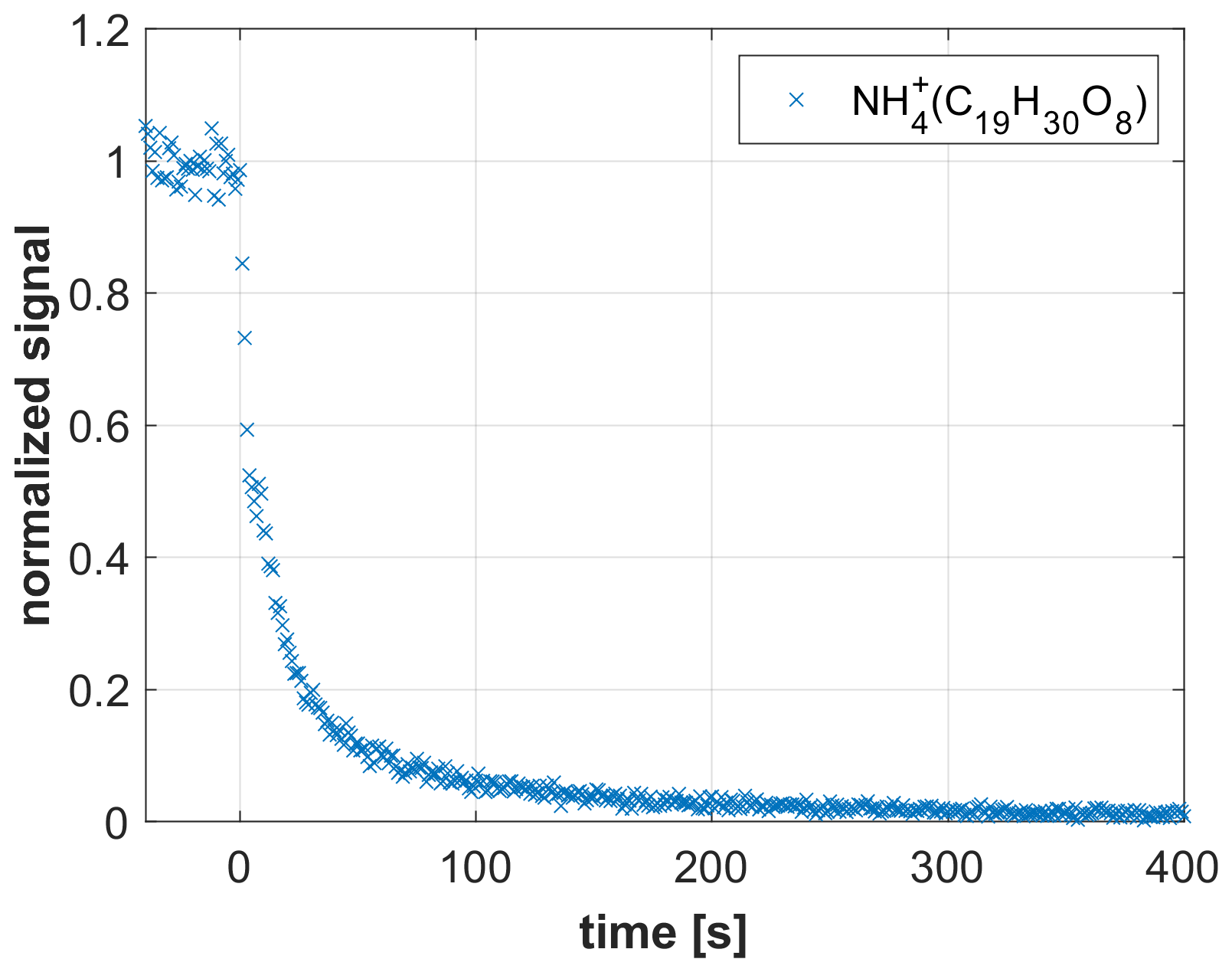
Figure 8Decay of the signal as recorded by a CHARON EVR PTR-MS analyzer when SOA was sampled and the instrument was switched from the CHARON particle inlet to the gas inlet at t=0.
3.5 Detection of highly oxidized organic compounds
The CHARON PTR-MS instrument has been successfully used for measuring the chemical composition of SOA (e.g., Gkatzelis et al., 2018). The detection of highly oxidized species has, however, been hampered by the adsorption of low-volatility species onto instrumental surfaces. In an attempt to demonstrate the improved detection capability of the CHARON EVR PTR-MS instrument, we sampled SOA generated from limonene ozonolysis. Up to eight oxygen atoms were detected in the analyte ions (Fig. S4). Figure 8 shows the rapid decay ( s) of an O8 compound (C19H30O8, detected as ammonium adduct) when switching from the CHARON particle inlet to the gas inlet. The rapidness of the decay suggests that surface adsorption effects play a minor role even for such highly oxidized species. Hammes et al. (2019) claimed that the C19H30O8 molecule formed from limonene ozonolysis is a multifunctional compound containing three keto groups, one carboxyl group, one hydroperoxy group and one ester group. A calculation using SIMPOL.1 (Pankow and Asher, 2008) yields a log C0 of −5.9 for this molecule, suggesting that even extremely low volatility organic compounds (ELVOCs) can be monitored in real time by CHARON EVR PTR-MS. The fact that the detected C19H30O8 molecule contains a hydroperoxy group is notable, because hydroperoxides are known to efficiently decompose on metal surfaces in conventional PTR-MS instruments (Rivera-Rios et al., 2014). An additional benefit of surface passivation may thus be that the metal-catalyzed decomposition of peroxides and hydroperoxides is suppressed. Preliminary laboratory tests with cumene hydroperoxide and dicumylperoxide (i.e., the only peroxides that are commercially available, although with significant impurities) indicate that these compounds do indeed not decompose in EVR-type instruments. More work is needed to confirm this preliminary finding.
We have described and characterized the novel EVR PTR-MS instrument, which exhibits a significantly improved time-response performance as compared to conventional IONICON PTR-MS analyzers. The time response of this optimized instrument is comparable to that of the fastest flow tube CIMS instruments that are currently in use. This allows us to rapidly switch between gas and particle measurements, making the CHARON EVR PTR-MS instrument the only direct sample introduction CIMS instrument that can monitor gaseous and particulate organics in the atmosphere in real time. Besides being faster, the EVR PTR-MS instrument also allows us to target new analyte classes such as highly oxygenated organic molecules and potentially also hydroperoxides. We believe that the CHARON EVR PTR-MS instrument will be a valuable tool for overcoming current challenges in the measurement of atmospheric organic carbon (Heald and Kroll, 2020).
The data used in this paper are available from the corresponding author upon request.
The supplement related to this article is available online at: https://doi.org/10.5194/amt-14-1355-2021-supplement.
KW developed the EVR system and performed initial tests. MM, FP and AW designed the experimental studies. MM, FP and JSaS carried out the experiments. FP performed the data analysis, with support from JSaS and MM. FP and MM drafted the manuscript. AW wrote the final manuscript. All authors commented and accepted the final version of the manuscript.
Markus Müller, Felix Piel and Klaus Winkler work for IONICON Analytik. IONICON Analytik commercializes PTR-MS instruments (both in the conventional and in the EVR configuration) and the CHARON particle inlet. Markus Müller and Armin Wisthaler profit from a license agreement (CHARON inlet) between the University of Innsbruck and IONICON Analytik.
We would like to thank Laurent Poulain and Gerald Spindler from TROPOS for their support during the measurements in Melpitz. Special thanks go to Magda Claeys for providing many of the substances studied in this work.
Felix Piel has been supported by the Horizon 2020 Framework Programme (H2020) Marie Skłodowska-Curie Actions (IMPACT (grant no. 674911)). Jenny Skytte af Sätra has received support from the Erasmus+ program of the European Union. Measurements in Melpitz have been supported by the European Union's Horizon 2020 research and innovation program (ACTRIS-2 (grant no. 654109)).
This paper was edited by Andreas Hofzumahaus and reviewed by two anonymous referees.
Breitenlechner, M., Fischer, L., Hainer, M., Heinritzi, M., Curtius, J., and Hansel, A.: PTR3: An Instrument for Studying the Lifecycle of Reactive Organic Carbon in the Atmosphere, Anal. Chem., 89, 5824–5831, https://doi.org/10.1021/acs.analchem.6b05110, 2017.
de Gouw, J. and Warneke, C.: Measurements of volatile organic compounds in the earth's atmosphere using proton-transfer-reaction mass spectrometry, Mass Spectrom. Rev., 26, 223–257, https://doi.org/10.1002/mas.20119, 2007.
Deming, B. L., Pagonis, D., Liu, X., Day, D. A., Talukdar, R., Krechmer, J. E., de Gouw, J. A., Jimenez, J. L., and Ziemann, P. J.: Measurements of delays of gas-phase compounds in a wide variety of tubing materials due to gas–wall interactions, Atmos. Meas. Tech., 12, 3453–3461, https://doi.org/10.5194/amt-12-3453-2019, 2019.
Eichler, P., Müller, M., D'Anna, B., and Wisthaler, A.: A novel inlet system for online chemical analysis of semi-volatile submicron particulate matter, Atmos. Meas. Tech., 8, 1353–1360, https://doi.org/10.5194/amt-8-1353-2015, 2015.
Epstein, S. A., Riipinen, I., and Donahue, N. M.: A Semiempirical Correlation between Enthalpy of Vaporization and Saturation Concentration for Organic Aerosol, Environ. Sci. Technol., 44, 743–748, https://doi.org/10.1021/es902497z, 2010.
Gkatzelis, G. I., Tillmann, R., Hohaus, T., Müller, M., Eichler, P., Xu, K.-M., Schlag, P., Schmitt, S. H., Wegener, R., Kaminski, M., Holzinger, R., Wisthaler, A., and Kiendler-Scharr, A.: Comparison of three aerosol chemical characterization techniques utilizing PTR-ToF-MS: a study on freshly formed and aged biogenic SOA, Atmos. Meas. Tech., 11, 1481–1500, https://doi.org/10.5194/amt-11-1481-2018, 2018.
Goldstein, A. H. and Galbally, I. E.: Known and Unexplored Organic Constituents in the Earth's Atmosphere, Environ. Sci. Technol., 41, 1514–1521, https://doi.org/10.1021/es072476p, 2007.
Hammes, J., Lutz, A., Mentel, T., Faxon, C., and Hallquist, M.: Carboxylic acids from limonene oxidation by ozone and hydroxyl radicals: insights into mechanisms derived using a FIGAERO-CIMS, Atmos. Chem. Phys., 19, 13037–13052, https://doi.org/10.5194/acp-19-13037-2019, 2019.
Hansel, A., Jordan, A., Holzinger, R., Prazeller, P., Vogel, W., and Lindinger, W.: Proton transfer reaction mass spectrometry: on-line trace gas analysis at the ppb level, Int. J. Mass. Spectrom., 149–150, 609–619, https://doi.org/10.1016/0168-1176(95)04294-U, 1995.
Heald, C. L. and Kroll, J. H.: The fuel of atmospheric chemistry: Toward a complete description of reactive organic carbon, Sci. Adv., 6, eaay8967, https://doi.org/10.1126/sciadv.aay8967 2020.
Krechmer, J., Lopez-Hilfiker, F., Koss, A., Hutterli, M., Stoermer, C., Deming, B., Kimmel, J., Warneke, C., Holzinger, R., Jayne, J., Worsnop, D., Fuhrer, K., Gonin, M., and de Gouw, J.: Evaluation of a New Reagent-Ion Source and Focusing Ion–Molecule Reactor for Use in Proton-Transfer-Reaction Mass Spectrometry, Anal. Chem., 90, 12011–12018, https://doi.org/10.1021/acs.analchem.8b02641, 2018.
Leglise, J., Müller, M., Piel, F., Otto, T., and Wisthaler, A.: Bulk organic aerosol analysis by PTR-MS: an improved methodology for the determination of total organic mass, O:C and H:C elemental ratios and the average molecular formula, Anal. Chem., 91, 12619–12624, https://doi.org/10.1021/acs.analchem.9b02949, 2019.
Liu, X., Deming, B., Pagonis, D., Day, D. A., Palm, B. B., Talukdar, R., Roberts, J. M., Veres, P. R., Krechmer, J. E., Thornton, J. A., de Gouw, J. A., Ziemann, P. J., and Jimenez, J. L.: Effects of gas–wall interactions on measurements of semivolatile compounds and small polar molecules, Atmos. Meas. Tech., 12, 3137–3149, https://doi.org/10.5194/amt-12-3137-2019, 2019.
Mikoviny, T., Kaser, L., and Wisthaler, A.: Development and characterization of a High-Temperature Proton-Transfer-Reaction Mass Spectrometer (HT-PTR-MS), Atmos. Meas. Tech., 3, 537–544, https://doi.org/10.5194/amt-3-537-2010, 2010.
Müller, M., Graus, M., Ruuskanen, T. M., Schnitzhofer, R., Bamberger, I., Kaser, L., Titzmann, T., Hörtnagl, L., Wohlfahrt, G., Karl, T., and Hansel, A.: First eddy covariance flux measurements by PTR-TOF, Atmos. Meas. Tech., 3, 387–395, https://doi.org/10.5194/amt-3-387-2010, 2010.
Müller, M., Eichler, P., D'Anna, B., Tan, W., and Wisthaler, A.: Direct Sampling and Analysis of Atmospheric Particulate Organic Matter by Proton-Transfer-Reaction Mass Spectrometry, Anal. Chem., 89, 10889–10897, https://doi.org/10.1021/acs.analchem.7b02582, 2017.
Müller, M., Piel, F., Gutmann, R., Sulzer, P., Hartungen, E., and Wisthaler, A.: A novel method for producing NH4+ reagent ions in the hollow cathode glow discharge ion source of PTR-MS instruments, Int. J. Mass. Spectrom., 447, 116254, https://doi.org/10.1016/j.ijms.2019.116254, 2020.
Neuman, J. A., Huey, L. G., Ryerson, T. B., and Fahey, D. W.: Study of Inlet Materials for Sampling Atmospheric Nitric Acid, Environ. Sci. Technol., 33, 1133–1136, https://doi.org/10.1021/es980767f, 1999.
Nowak, J. B., Neuman, J. A., Kozai, K., Huey, L. G., Tanner, D. J., Holloway, J. S., Ryerson, T. B., Frost, G. J., McKeen, S. A., and Fehsenfeld, F. C.: A chemical ionization mass spectrometry technique for airborne measurements of ammonia, J. Geophys. Res., 112, D10S02, https://doi.org/10.1029/2006JD007589, 2007.
Pagonis, D., Krechmer, J. E., de Gouw, J., Jimenez, J. L., and Ziemann, P. J.: Effects of gas–wall partitioning in Teflon tubing and instrumentation on time-resolved measurements of gas-phase organic compounds, Atmos. Meas. Tech., 10, 4687–4696, https://doi.org/10.5194/amt-10-4687-2017, 2017.
Palm, B. B., Liu, X., Jimenez, J. L., and Thornton, J. A.: Performance of a new coaxial ion–molecule reaction region for low-pressure chemical ionization mass spectrometry with reduced instrument wall interactions, Atmos. Meas. Tech., 12, 5829–5844, https://doi.org/10.5194/amt-12-5829-2019, 2019.
Pankow, J. F. and Asher, W. E.: SIMPOL.1: a simple group contribution method for predicting vapor pressures and enthalpies of vaporization of multifunctional organic compounds, Atmos. Chem. Phys., 8, 2773–2796, https://doi.org/10.5194/acp-8-2773-2008, 2008.
Piel, F., Müller, M., Mikoviny, T., Pusede, S. E., and Wisthaler, A.: Airborne measurements of particulate organic matter by proton-transfer-reaction mass spectrometry (PTR-MS): a pilot study, Atmos. Meas. Tech., 12, 5947–5958, https://doi.org/10.5194/amt-12-5947-2019, 2019.
Rivera-Rios, J. C., Nguyen, T. B., Crounse, J. D., Jud, W., St. Clair, J. M., Mikoviny, T., Gilman, J. B., Lerner, B. M., Kaiser, J. B., Gouw, J., Wisthaler, A., Hansel, A., Wennberg, P. O., Seinfeld, J. H., and Keutsch, F. N.: Conversion of hydroperoxides to carbonyls in field and laboratory instrumentation: Observational bias in diagnosing pristine versus anthropogenically controlled atmospheric chemistry, Geophys. Res. Lett., 41, 8645–8651, https://doi.org/10.1002/2014GL061919, 2014.
Spindler, G., Grüner, A., Müller, K., Schlimper, S., and Herrmann, H.: Long-term size-segregated particle (PM10, PM2.5, PM1) characterization study at Melpitz – influence of air mass inflow, weather conditions and season, J. Atmos. Chem., 70, 165–195, https://doi.org/10.1007/s10874-013-9263-8, 2013.
Yuan, B., Koss, A. R., Warneke, C., Coggon, M., Sekimoto, K., and de Gouw, J. A.: Proton-Transfer-Reaction Mass Spectrometry: Applications in Atmospheric Sciences, Chem. Rev., 117, 13187–13229, https://doi.org/10.1021/acs.chemrev.7b00325, 2017.
Zaytsev, A., Breitenlechner, M., Koss, A. R., Lim, C. Y., Rowe, J. C., Kroll, J. H., and Keutsch, F. N.: Using collision-induced dissociation to constrain sensitivity of ammonia chemical ionization mass spectrometry ( CIMS) to oxygenated volatile organic compounds, Atmos. Meas. Tech., 12, 1861–1870, https://doi.org/10.5194/amt-12-1861-2019, 2019.
stickonto and slowly come off surfaces in the PTR-MS analyzer, which makes it impossible to measure rapid changes in the atmosphere. Herein, we present a new type of PTR-MS instrument with a specially treated surface that mitigates this problem.