the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 31 Mar 2021
| 31 Mar 2021
Application of chemical derivatization techniques combined with chemical ionization mass spectrometry to detect stabilized Criegee intermediates and peroxy radicals in the gas phase
Alexander Zaytsev
Martin Breitenlechner
Anna Novelli
Hendrik Fuchs
Daniel A. Knopf
Jesse H. Kroll
Frank N. Keutsch
Short-lived highly reactive atmospheric species, such as organic peroxy radicals (RO2) and stabilized Criegee intermediates (SCIs), play an important role in controlling the oxidative removal and transformation of many natural and anthropogenic trace gases in the atmosphere. Direct speciated measurements of these components are extremely helpful for understanding their atmospheric fate and impact. We describe the development of an online method for measurements of SCIs and RO2 in laboratory experiments using chemical derivatization and spin trapping techniques combined with H3O+ and chemical ionization mass spectrometry (CIMS). Using chemical derivatization agents with low proton affinity, such as electron-poor carbonyls, we scavenge all SCIs produced from a wide range of alkenes without depleting CIMS reagent ions. Comparison between our measurements and results from numeric modeling, using a modified version of the Master Chemical Mechanism, shows that the method can be used for the quantification of SCIs in laboratory experiments with a detection limit of 1.4×107 molecule cm−3 for an integration time of 30 s with the instrumentation used in this study. We show that spin traps are highly reactive towards atmospheric radicals and form stable adducts with them by studying the gas-phase kinetics of the reaction of spin traps with the hydroxyl radical (OH). We also demonstrate that spin trap adducts with SCIs and RO2 can be simultaneously probed and quantified under laboratory conditions with a detection limit of 1.6×108 molecule cm−3 for an integration time of 30 s for RO2 species with the instrumentation used in this study. Spin trapping prevents radical secondary reactions and cycling, ensuring that measurements are not biased by chemical interferences, and it can be implemented for detecting RO2 species in laboratory studies and potentially in the ambient atmosphere.
- Article
(496 KB) - Full-text XML
-
Supplement
(700 KB) - BibTeX
- EndNote





Earth's atmosphere is an oxidizing environment. The initial oxidation step of volatile organic compounds (VOCs) involves the reaction of a parent hydrocarbon with an oxidant. The hydroxyl radical (OH) is the most important oxidant in the atmosphere, although oxidation can be also initiated by O3, NO3, and Cl or Br atoms. Generally, reactions of VOCs with OH, NO3, and Cl atoms occur via H abstraction or via addition to unsaturated carbon double bonds, leading to the formation of alkyl radicals. This reaction is quickly followed by O2 addition, resulting in the production of organic peroxy radicals (RO2). In an NO-rich environment, RO2 radicals predominantly react with NO, while reactions with the hydroperoxy radical (HO2) and potentially other RO2 become more important at lower NO concentrations, as do unimolecular reactions. The common tendency is to form closed-shell, more oxidized VOCs (OVOCs). OVOCs may have lower volatilities than the parent hydrocarbons and may partition to the particle phase, thereby contributing to secondary organic aerosol (SOA) formation. OH, HO2, and RO2 radicals can form a catalytic reaction cycle, which can lead to the production of tropospheric ozone as a consequence of the shift in the ratio to favor the formation of NO2. This cycle is terminated by the formation of organic hydroperoxides and nitrates, which can be viewed as reservoirs of the corresponding radicals. Overall, atmospheric radicals – especially their cycling – play an important role in the formation of SOA and tropospheric ozone, as well as in controlling atmospheric oxidation capacity.
Organic peroxy radicals can also be formed via ozonolysis of unsaturated organic compounds. Ozonolysis of alkenes results in the formation of primary ozonides that promptly decompose to a stable carbonyl and a vibrationally excited carbonyl oxide, also known as a Criegee intermediate (CI), some of which are thermally stabilized (SCI). SCIs primarily decompose or react with water vapor (Vereecken et al., 2017) but are also believed to play a role in the oxidation of SO2 to form H2SO4 in the tropical regions (Khan et al., 2018). syn-SCI can undergo a unimolecular reaction and form a vinyl hydroperoxide, which rapidly decomposes to an OH radical and a vinyl radical. This radical is in resonance with an acetonyl-type radical, which can combine with molecular oxygen to form an RO2 species (Johnson and Marston, 2008).
Measurements of atmospheric radicals and reactive intermediates such as RO2 and SCIs are challenging because of their high reactivity towards trace gases and surfaces and rapid cycling, which may lead to potential interferences. Highly sensitive detection systems are required to determine the minute concentrations of these species. Concentrations of the smallest organic peroxy radical, CH3O2, are typically on the order of 108 molecule cm−3, while concentrations of other RO2 species can be much lower (Fuchs et al., 2008). As for SCIs, their concentrations are expected to be less than 105 molecule cm−3 (Novelli et al., 2017). With respect to RO2 species, there are several field-deployable measurement techniques available for nonspeciated measurements of the sum of RO2. Matrix isolation electron spin resonance spectroscopy (MIESR) is an established, but rarely used, method for field measurements (Mihelcic et al., 1985). MIESR is an offline technique with a low time resolution (∼30 min); however, its main advantage is that it does not require instrument calibration. Besides MIESR, chemical amplification and conversion systems represent another class of instruments for field studies (Edwards et al., 2003; Hornbrook et al., 2011; Cantrell et al., 1984; Wood and Charest, 2014). In these systems, peroxy radicals are not measured directly but are rather converted to other radicals or closed-shell molecules (e.g., NO2 or H2SO4). A detection limit of 107 molecule cm−3 can be achieved at a temporal resolution of 15 s, but discrimination of different RO2 species is not possible (Edwards et al., 2003). In addition, secondary chemistry, i.e., additional sources of radical production and destruction, has to be considered, and care needs to be taken to ensure that measurements are not biased by any chemical interferences (Reiner et al., 1997). Finally, laser-induced fluorescence (LIF) was also applied for ambient measurements of RO2 radicals (Fuchs et al., 2008). This technique is characterized by an excellent detection limit of (2–7) ×107 molecule cm−3 for an integration time of 30 s. Similarly to chemical amplifier systems, however, LIF does not allow the differentiation of various RO2 species. Although it is indirect and converts RO2 to OH, it does not have an amplification chain. Recently, novel mass spectrometric techniques using different ionization schemes to directly detect individual RO2 species were developed (Hansel et al., 2018; Berndt et al., 2018, 2019; Nozière and Vereecken, 2019).
As for SCIs, indirect measurement techniques have been widely used. In these techniques, SCIs are chemically converted to other species (e.g., H2SO4 or hydroxymethyl hydroperoxide, HMHP) (Berndt et al., 2014; Sipilä et al., 2014; Neeb et al., 1997). In 2008, the simplest SCI, CH2OO, was directly detected for the first time (Taatjes et al., 2008). Later, synchrotron photoionization mass spectrometry was combined with the CI generation technique using diiodoalkane photolysis (Welz et al., 2012), which spurred several studies to examine the kinetics of bimolecular and unimolecular SCI reactions (Taatjes et al., 2012; Lewis et al., 2015; Chhantyal-Pun et al., 2016). Recently, two new techniques for direct measurements of SCIs using Fourier transform microwave spectroscopy and chemical ionization mass spectrometry (CIMS) were introduced (Womack et al., 2015; Berndt et al., 2017).
Despite the abundance of different analytical methods used for the detection of atmospheric radicals and reactive intermediates, there is still a need for an online, direct, field-deployable technique for measuring these short-lived highly reactive compounds in a speciated way. Free radicals have been conventionally detected by chemical derivatization (CD) techniques such as spin trapping in condensed-phase biological and chemical systems (Hawkins and Davies, 2014; Nosaka and Nosaka, 2017). Nonradical spin traps (e.g., nitrone spin traps) are known to react with free radicals to form stable radical adducts that can be detected with electron paramagnetic resonance spectroscopy (Roberts et al., 2016). In addition, radical spin traps (e.g., nitroxide radicals) are also highly reactive towards radical species such as C-centered radicals and form closed-shell adducts with them (Bagryanskaya and Marque, 2014). However, there are only a few studies in which these techniques were applied for probing atmospheric radicals and intermediates. Watanabe et al. (1982) presented an offline method to quantify hydroxyl radicals using the spin trap α-4-pyridyl-N-tert-butylnitrone α-1-oxide (4-POBN), where condensed-phase stable adducts were detected by electron spin resonance. Recently, Giorio et al. (2017) used the spin trap 5,5-dimethyl-1-pyrroline N-oxide (DMPO) to characterize SCIs by detecting gas-phase spin trap adducts with online mass spectrometry.
Here, we explore three types of CD agents, including two spin-trapping agents, and show how they can be used for the detection and quantification of various atmospheric radicals and reactive intermediates (Fig. 1). First, we implement the CD agent hexafluoroacetone (HFA) to characterize a wide range of gas-phase SCIs. HFA is selectively reactive towards SCIs (i.e., it is unreactive towards OH, HO2, and RO2), forms stable secondary ozonides with them, and has high vapor pressure and low proton affinity (Fig. 1). Next, we use the radical spin trap (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) to demonstrate that spin traps are highly reactive towards radicals in the gas phase – and can therefore effectively scavenge atmospheric radicals – by studying the kinetics of the TEMPO + OH reaction. Finally, we utilize the nonradical spin trap DMPO to simultaneously detect atmospheric gas-phase radicals and intermediates, including SCIs and RO2 species (Fig. 1). Spin trap adducts and secondary ozonides with CD agents are observed and quantified using H3O+ and CIMS, which allows for speciated online measurements of stabilized Criegee intermediates and speciated RO2 radicals formed via ozonolysis of a wide range of parent hydrocarbons. The analytical methods presented here can be used for the quantification of speciated SCIs and RO2 formed in laboratory experiments, as well as for field measurements.
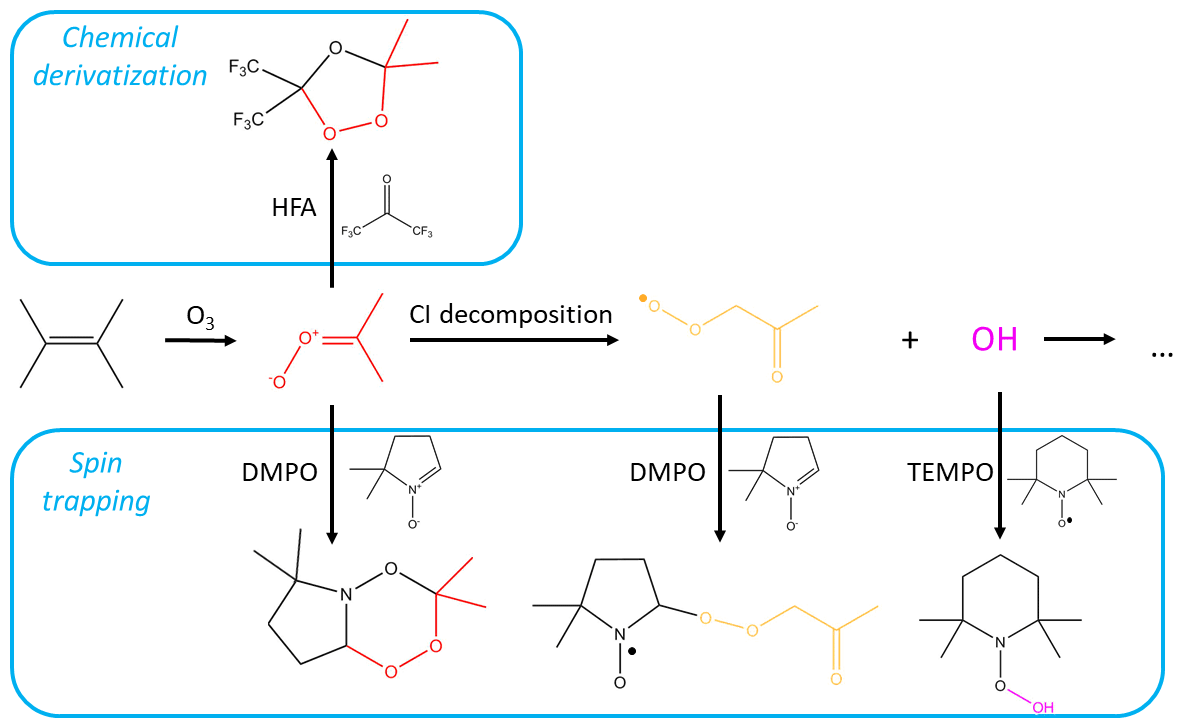
Figure 1Mechanism of tetramethylethylene (TME) ozonolysis. Stabilized Criegee intermediate (shown in red) can be scavenged by the chemical derivatization agent HFA or the spin trap DMPO, or can decompose to the peroxy radical (shown in yellow) and OH. RO2 and OH species can, in turn, react with spin traps. Reactions involving SCI are from MCM v3.3.1 (Jenkin et al., 1997) and Giorio et al. (2017).
2.1 Ozonolysis experiments with the chemical derivatization agent HFA
The ozonolysis experiments with multiple hydrocarbons including tetramethylethylene (TME), isoprene, pentene, hexene, α-pinene, and limonene were conducted in a flow tube reactor at ambient pressure and temperature (∼290 K) and under dry conditions (relative humidity <2 %). The experimental setup consisted of a flow reactor system with a residence time of ∼10 s. The parent hydrocarbon was mixed with ozone and the chemical derivatization agent HFA (C3F6O) in the flow reactor, leading to SCI formation and scavenging as SCI • HFA adducts. SCIs are known to be highly reactive towards ketones, especially electron-poor ones such as HFA (Horie et al., 1999; Taatjes et al., 2012). HFA has been previously used to effectively scavenge SCIs and prevent their secondary chemistry, thus allowing SCI formation to be probed directly (Drozd et al., 2011; Drozd and Donahue, 2011). The other advantage of employing this chemical derivatization agent is its relatively low proton affinity (PA 670.4 kJ mol−1; Hunter and Lias, 1998). Since the PA of HFA is lower than that of water, HFA cannot be protonated in H3O+ CIMS. Hence, one can introduce a significant amount of HFA into the system to make sure that all SCIs are scavenged very rapidly without any concern that the H3O+ reagent ions will be depleted. The parent hydrocarbon was vaporized from a flask filled with the pure substance by passing a constant flow of zero air regulated via a 0.1–10 cm3 min−1 mass flow controller (Bronkhorst). The HFA flow was regulated by another mass flow controller (Bronkhorst). Ozone was produced by passing zero air through an ozone generator using a low-pressure mercury ultraviolet lamp. The ozone concentration was measured using an ozone monitor (2B Technologies) (Table S2 in the Supplement).
A proton-transfer-reaction mass spectrometer (PTR-8000, IONICON Analytik) was used to observe the resulting SCI • HFA adducts as well as the parent hydrocarbons and their oxidation products. This instrument was operated using H3O+ reagent ions (H3O+ CIMS) and was directly calibrated to 10 VOCs with different functional groups (Isaacman-Van Wertz et al., 2017; Isaacman-Van Wertz et al., 2018).
2.2 Experiments with spin traps
2.2.1 Kinetics experiments with the spin trap TEMPO
Highly reactive spin traps are needed for the effective derivatization of radicals and reactive intermediates in the gas phase. A set of experiments in which the reaction rate coefficient between the spin trap TEMPO (C9H18NO) and OH was measured were conducted in a flow-tube experimental setup at Forschungszentrum Jülich. TEMPO is commonly used to detect carbon-centered radicals in chemical and biological systems (Bagryanskaya and Marque, 2014) and is known to be highly reactive towards OH in the aqueous phase (Samuni et al., 2002). TEMPO was introduced in the flow-tube setup using a liquid calibration unit (LCU, IONICON Analytik). The LCU quantitatively evaporates aqueous standards into the gas stream. The TEMPO standard was prepared gravimetrically with an aqueous volume mixing ratio of 485 parts per million (ppm). A known amount (up to 10 µL min−1) of this solution was then evaporated into a humidified gas stream of synthetic air (31 SLPM), resulting in a gas-phase TEMPO concentration of up to 4.5×1011 molecule cm−3. Some of the setup outflow was drawn to a laser photolysis–laser-induced fluorescence (LP-LIF) instrument (Lou et al., 2010), with which the OH reactivity of TEMPO was measured. Laser flash photolysis of ozone was used to produce OH in the experimental setup, while LIF was applied to monitor the time-dependent OH decay. Another part of the outflow was drawn to a CIMS instrument (PTR3, IONICON Analytik) to monitor concentrations of TEMPO and its oxidation products. This instrument was operated in two ionization modes: using H3O+•(H2O)n, n=0–1 (as H3O+ CIMS; Breitenlechner et al., 2017) and n, n=0–2 (as CIMS; Zaytsev et al., 2019) reagent ions. The PTR3 is designed to minimize inlet losses of sampled compounds. It was directly calibrated to 10 VOCs with different functional groups using LCU. Collision-dissociation methods were used to constrain the sensitivities of CIMS to compounds that cannot be calibrated directly (Zaytsev et al., 2019). Sensitivities were calculated in normalized duty-cycle-corrected counts per second per part per billion by volume (ndcps ppb−1; the duty-cycle correction was done to the reference 100; ion signals were normalized to the primary ion signal of 106 dcps).
2.2.2 Ozonolysis experiments with the spin trap DMPO
An additional set of ozonolysis experiments with several hydrocarbons including TME and α-pinene were conducted in a double flow reactor setup (Fig. 2). The goal of these experiments was to examine how spin traps can be used for the simultaneous detection of stabilized Criegee intermediates and peroxy radicals. The experimental setup consisted of two identical ∼2.1 L flow reactors. The parent hydrocarbon was mixed with ozone in the first flow-tube reactor with a residence time of ∼28 s. Similar to the previous ozonolysis experiments described in Sect. 2.1, the parent hydrocarbon was vaporized from a flask filled with the pure substance by passing zero air regulated by a mass flow controller, and ozone was generated using a low-pressure mercury ultraviolet lamp. We used an LCU to introduce the spin trap DMPO (C6H11NO) in the second flow reactor with a residence time of ∼23 s. A known amount (up to 10 µL min−1) of the DMPO solution was evaporated into a humidified gas stream of synthetic air (5.4–7 SLPM), resulting in a gas-phase DMPO concentration of up to 1.1×1013 molecule cm−3. The second flow reactor was used to derivatize SCIs and RO2 species with DMPO while the parent hydrocarbon was still reacting with ozone. Hence, we conducted integrated production measurements of the SCIs and RO2 species formed in both flow reactors. DMPO represents a class of nonradical spin traps and is widely used to detect oxygen-centered radicals such as OH, HO2, and RO2 in chemical and biological systems (Roberts et al., 2016; Van Der Zee at al., 1996). Recently, DMPO was also employed to detect SCIs in the gas phase (Giorio et al., 2017). The PTR3 was used to detect SCI • DMPO and RO2 • DMPO adducts, while ozone levels were observed using an ozone monitor (2B Technologies) (Table S3 in the Supplement).
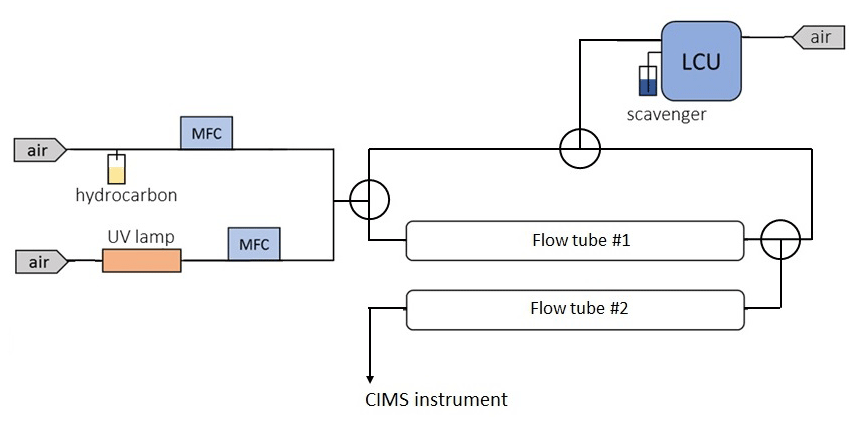
Figure 2Schematic of the experimental setup used to detect SCIs and RO2 with the spin trap DMPO. DMPO was introduced in the experimental setup using a liquid calibration unit (LCU, IONICON Analytik).
2.3 Kinetic model and quantum-chemical calculations
The Framework for 0-D Atmospheric Modeling v3.1 (F0AM; Wolfe et al., 2016) containing reactions from the Master Chemical Mechanism (MCM v3.3.1) (Jenkin et al., 1997; Saunders et al., 2003) was used to simulate the photooxidation of studied alkenes in the flow reactor system and to compare the modeled concentrations of the products with the measurements. Model calculations were constrained using physical parameters of the experimental setup (pressure and temperature) as well as to observed concentrations of the parent hydrocarbon, ozone, and the chemical derivatization agent.
In order to estimate the proton affinities of the SCI • HFA adducts, we performed geometry optimization and proton affinity calculations with the Gaussian 09 package (Frisch et al., 2009) using the B3LYP functional (Stephens et al., 1994) and TZVP basis sets.
3.1 Detection of speciated stabilized Criegee intermediates using chemical derivatization techniques
The primary goal of the first set of experiments was the detection of speciated stabilized Criegee intermediates as adducts with the chemical derivatization agent HFA to prevent secondary reactions within the experimental setup. Starting with (CH3)2COO, an SCI produced via the ozonolysis of TME, we tested the formation of SCI • HFA adducts under different experimental conditions (Fig. 3). (CH3)2COO • HFA (C6H6O3F6•H+, 241.03) can be easily identified in the mass spectrum due to its unique mass defect associated with six F atoms (Fig. S2 in the Supplement). SCI • HFA adducts were observed when TME, ozone, and HFA were present in the experimental setup. Ozonolysis of TME also results in the formation of acetone (C3H6O•H+, 59.05), which was detected in the presence of TME and ozone and was not affected by HFA addition (Fig. 3).
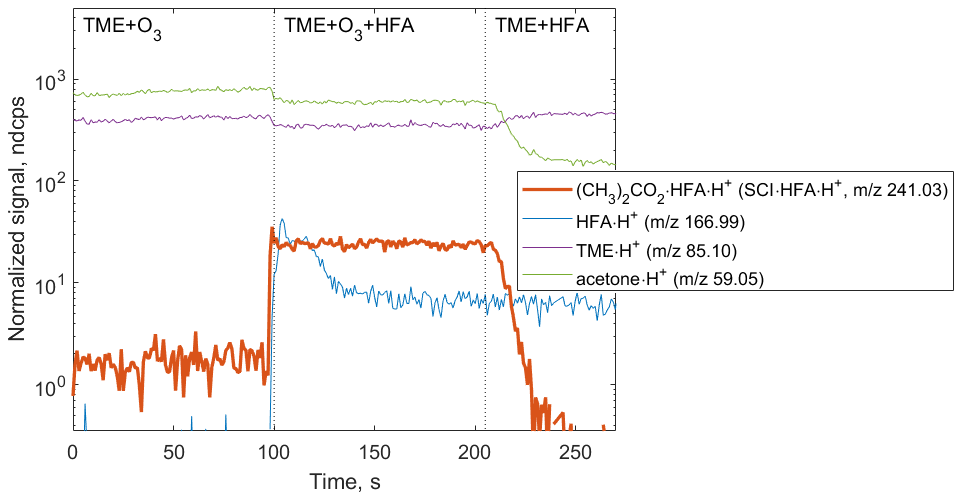
Figure 3Ion tracers observed in a TME ozonolysis experiment as a function of the reactant conditions. Reactant concentrations are [TME] ; ; [HFA] molecule cm−3. (CH3)2COO • HFA • H+ ion (red tracer, 241.03) is observed when TME, HFA, and O3 are present in the system.
We measured the (CH3)2COO•HFA adduct signal as a function of different reactant conditions: initial TME concentrations were in the range of (1.48–1.85) ×1011 molecule cm−3; ozone, (6.77–108.2) ×1012 molecule cm−3; HFA, (1.17–6.13) ×1015 molecule cm−3. The measurements are compared to the predictions of the kinetic model in Fig. 4. Concentrations of (CH3)2COO species were calculated using the MCM with updated kinetics data from the literature (Newland et al., 2015; Chhantyal-Pun et al., 2016; Long et al., 2018). For more details see the Supplement.
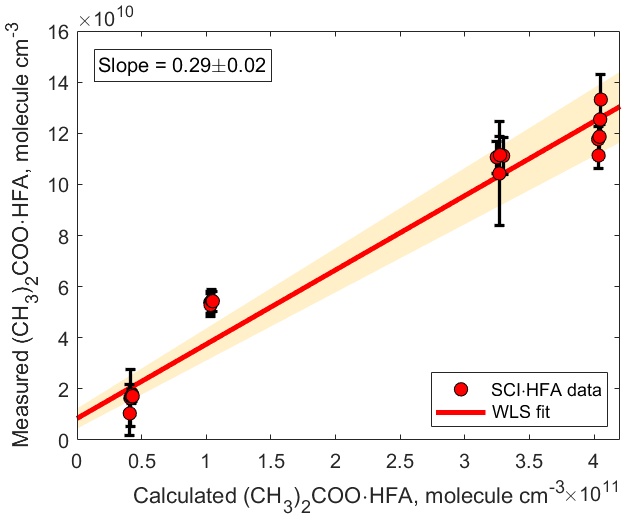
Figure 4Correlation plot comparing measured and calculated concentrations of (CH3)2COO•HFA. The adducts were detected using H3O+ CIMS as (CH3)2COO•HFA•H+ ( 241.03). The slope was calculated using weighted least squares (WLS). The 95 % confidence interval was estimated via a Monte Carlo simulation (N=5000) and is shown using red shading.
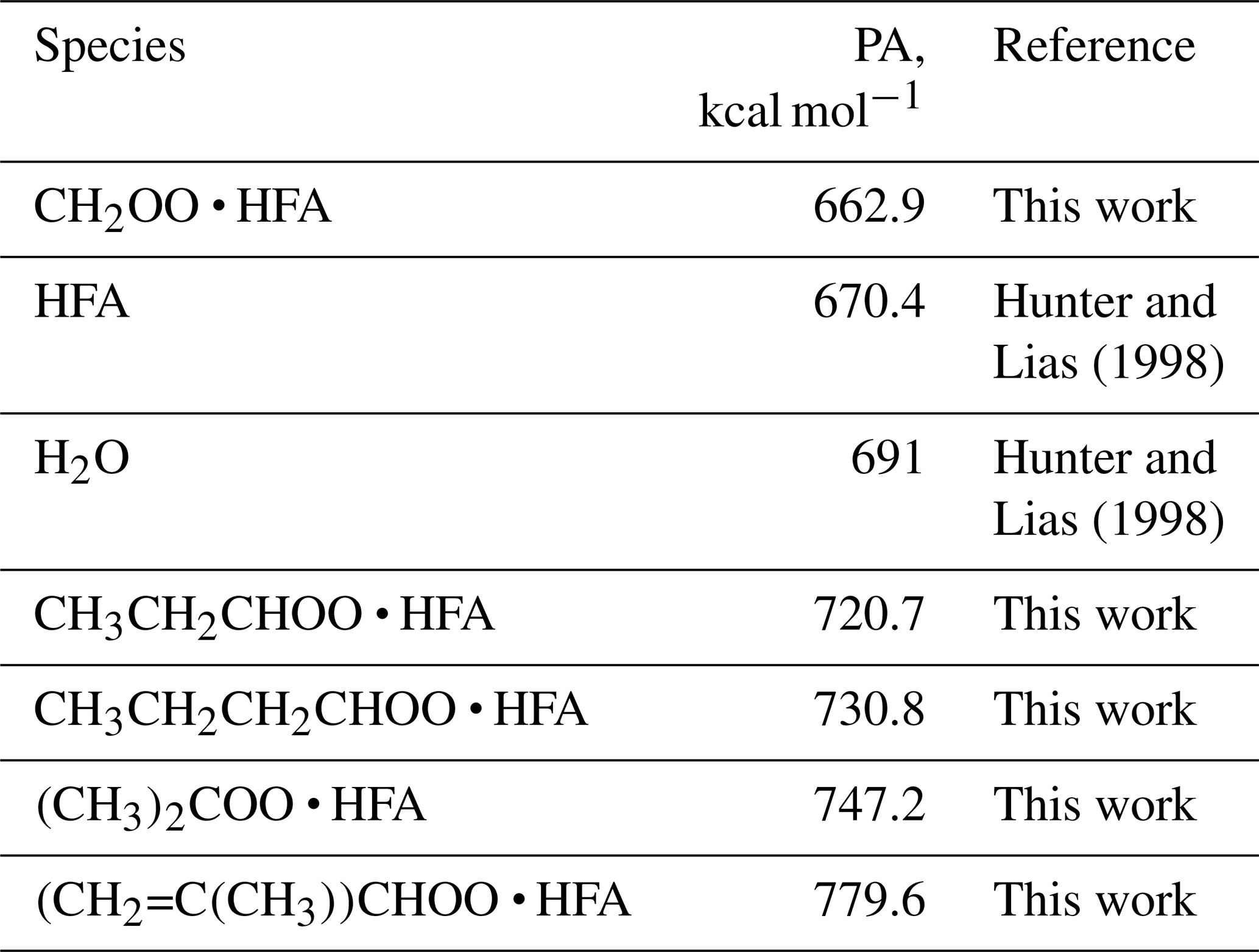
In the presence of HFA, SCIs can react with HFA and form stable adducts:
The reaction rate coefficient k1 was not measured experimentally, and we used the k value for the CH2OO + HFA reaction: cm3 molecule−1 s−1 (Taatjes et al., 2012). It has been suggested that the reaction between HFA and acetone oxide may be slower compared to the CH2OO one (Murray et al., 1965; Taatjes et al., 2012), while molecule cm−3 s−1 was used in previous studies (Drozd et al., 2011). However, the concentration of HFA was 2 orders of magnitude higher than the concentrations of other chemical compounds, so, even at lower k values, the reaction with HFA remains the main chemical loss pathway for (CH3)2COO (Fig. S3 in the Supplement).
Observed concentrations of (CH3)2COO • HFA agree to within a factor of 3 with concentrations predicted by the kinetic model (Fig. 4). This discrepancy can be explained by a combination of the following factors. First, a fraction of the (CH3)2COO • HFA adducts might be irreversibly deposited on the surfaces inside the experimental setup and the PTR 8000 instrument (Pagonis et al., 2017). In addition, the sensitivity of the observed SCI • HFA adducts depends on the reaction rate constant of the adduct with H3O+ ion and the degree of fragmentation of the protonated product ions SCI • HFA • H+ (Yuan et al., 2017). Since the reaction rate constant of SCI • HFA with H3O+ ions is unknown, we assumed that all SCI • HFA adducts were ionized via proton transfer from hydronium ions and therefore used the sensitivity we obtained from acetone calibration to quantify the detected SCI • HFA species. In addition, we did not take into account possible fragmentation of SCI • HFA • H+ ions, which may impede their detection, although a first bond cleavage would likely only break the ozonide ring structure without any loss of mass. Finally, the uncertainty in the kinetic model output is determined by the uncertainty in the SCI yield and the unimolecular and bimolecular reaction rate coefficients. The detection limit for (CH3)2COO • HFA adducts was 1.4×107 molecule cm−3 and was calculated for an integration time of 30 s as 3 standard deviations of the measured background divided by the derived sensitivity.
Besides TME, we also observed the formation of SCI • HFA for a series of precursors including isoprene, pentene, and hexene (Figs. S4–S6 in the Supplement). Proton affinities (PAs) of different CI • HFA adducts were calculated using density-functional theory (DFT) methods (Table 1). A variety of these adducts can be detected using H3O+ CIMS since their PAs are significantly higher than that of water, which is in agreement with experimental data (Figs. S4–S6). CH2OO • HFA cannot be detected because of its low PA (Table 1). We also did not observe SCI • HFA adducts for larger C10 SCIs produced via ozonolysis of α-pinene and limonene. This can be explained by the lower reactivity of larger SCIs with HFA, potential instability of these secondary ozonides in the gas phase, or their gas-wall partitioning in tubing and inside the PTR-8000 instrument.
Table 1Proton affinities (PAs) of HFA, water, and secondary ozonides produced in reactions of SCIs with HFA. Species with PAs higher than that of water can be detected in H3O+ CIMS.
3.2 Reactivity of spin traps with OH
Spin traps have been shown to be highly reactive towards free radicals and to efficiently form adducts with them in the aqueous phase. However, their reactivity with atmospheric radicals and the stability of the formed adducts in the gas phase remain largely unknown. In order to address these questions, we conducted a set of experiments to estimate the reaction rate between the spin trap TEMPO and the hydroxyl radical by measuring its OH reactivity.
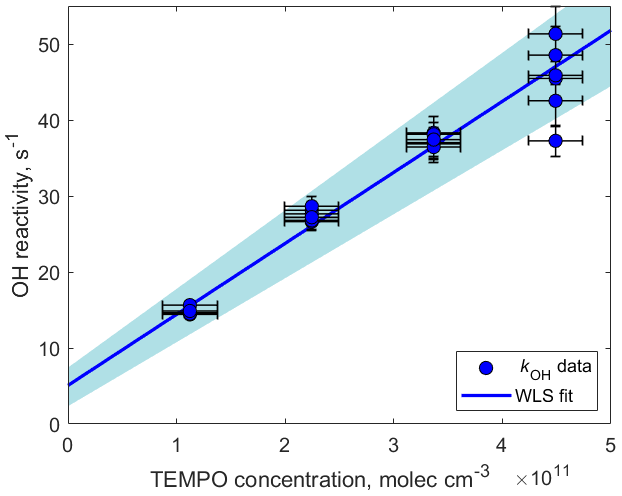
Figure 5OH reactivity measured as a function of TEMPO concentration. The slope determining the reaction rate coefficient cm3 molecule−1 s−1 was calculated using weighted least squares (WLS). The 95 % confidence interval was estimated via a Monte Carlo simulation (N=5000) and is shown using blue shading. The intercept (5.1±24) s−1 can be explained by the presence of other OH reactants such as O3, NO, NO2, and CO.
The OH reactivity of a specific reactant can be calculated as a product of the reactant concentration and its reaction rate with OH (Fuchs et al., 2017):
kOH was measured as a function of TEMPO concentration by varying the amount of TEMPO introduced in the experimental setup using the LCU (Fig. 5). The slope of the fitted line in Fig. 5 determines the reaction rate coefficient cm3 molecule−1 s−1. This rate constant is close to the collisional limit of typical radical–molecule reactions in the atmosphere and is 1 order of magnitude greater than the rate constant for the same reaction in the aqueous phase ( cm3 molecule−1 s−1; Samuni et al., 2002). This demonstrates that TEMPO is highly reactive towards OH in the gas phase, emphasizing the applicability of spin trapping for atmospheric measurements. Furthermore, the TEMPO+OH reaction leads to the formation of stable TEMPO•OH adducts that can be detected by H3O+ CIMS (C9H18NO•H+, 174.149) and therefore could be used for the quantification of hydroxyl radicals in the atmosphere (Fig. S7 in the Supplement). Further tests are needed to compare the measurement capability of this method (e.g., sensitivity, wall losses, and potential interferences) with that of a well-established technique such as LIF.
3.3 Simultaneous detection of SCIs and RO2 species from the ozonolysis of alkenes using spin-trapping techniques
Next, we implemented spin trapping for the detection of speciated SCIs and RO2 species formed via the ozonolysis of alkenes, starting with TME. Decomposition of the TME primary ozonide leads to the formation of acetone oxide, (CH3)2COO. This SCI can further undergo a unimolecular reaction followed by O2 addition to form the peroxy radical CH3C(=O)CH2OO• and OH (Fig. 1). In order to detect SCIs and RO2 species produced via the ozonolysis of TME, we used a measurement method based on the stabilization of these species using the spin trap DMPO followed by detection via and H3O+ CIMS. DMPO was shown to form stable secondary ozonides with SCIs in the gas phase (Giorio et al., 2017):
DMPO has been shown to be highly reactive towards SCIs ( cm3 molecule−1 s−1; see the Supplement for more details).
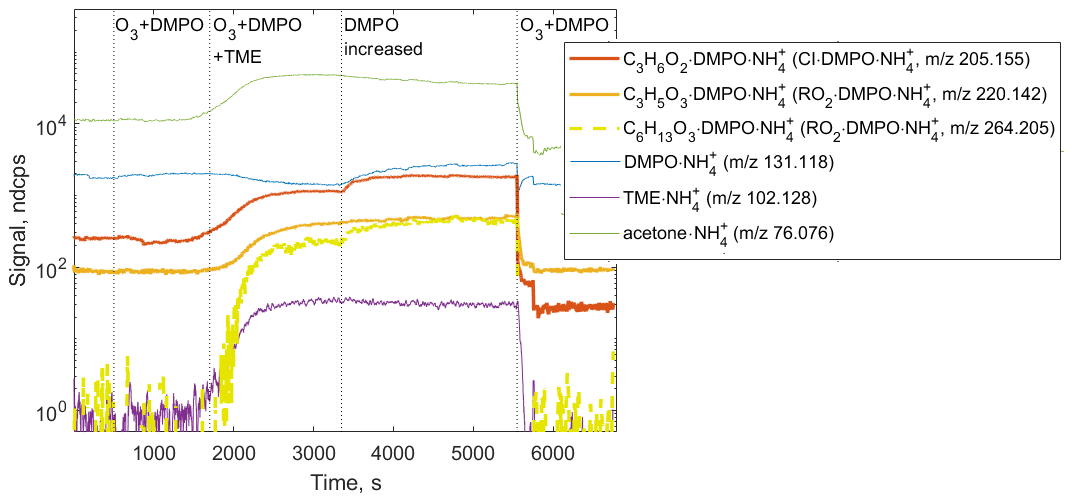
Figure 6Ion tracers observed by CIMS in a TME ozonolysis experiment as a function of the reactant conditions. Reactant concentrations are [TME] ; ; molecule cm−3.
In addition, DMPO is known to be highly reactive towards oxygen-centered radicals such as RO2 and to form stable radical adducts with them (Fig. 1):
We observed the formation of SCI • DMPO and RO2 • DMPO adducts in both CIMS (e.g., , 205.155 and , 220.142) and H3O+ CIMS (e.g., C9H17NO3•H+, 188.128 and C9H16NO4•H+, 203.116) under different experimental conditions (Fig. 6 and Fig. S8 in the Supplement). SCI • DMPO and RO2 • DMPO were only detected when TME, ozone, and DMPO were present in the experimental setup. Acetone, also formed via the ozonolysis of TME, was observed in the presence of TME and ozone and was not affected by the addition of DMPO (Fig. 6 and Fig. S8 in the Supplement). OH radicals formed via the decomposition of SCI can, in turn, react with TME, leading to the formation of another RO2 species, OH−C6H12OO•. This radical was detected as the C6H13O3•DMPO adduct (C12H24NO4, 264.205; Fig. 6). One of the benefits of CIMS is the possibility of quantifying compounds for which authentic standards are not available, using a voltage scanning procedure based on collision-induced dissociation (Zaytsev et al., 2019). Using this method, DMPO adducts with SCIs and RO2 were detected at high sensitivities: 2400 ndcps ppbv−1 for SCI • DMPO and 2000 ndcps ppbv−1 for RO2 • DMPO (Table S1 in the Supplement). Sensitivities were experimentally determined in each ozonolysis experiment and depended on the operational conditions of the PTR3 instrument. Detection limits for SCI • DMPO and RO2 • DMPO adducts were 3.4×107 and 1.6×108 molecule cm−3, respectively. These limits of detection were calculated for an integration time of 30 s as 3 standard deviations of the measured background divided by the derived sensitivity.
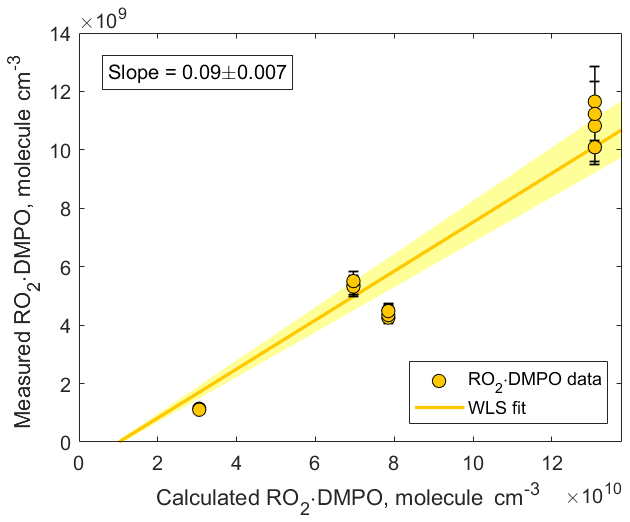
Figure 7Correlation plot comparing measured and calculated concentrations of CH3C(=O)CH2OO • DMPO. The adducts were detected using CIMS as CH3C(=O)CH2OO • DMPO • ( 220.142). The slope was calculated using weighted least squares (WLS). The 95 % confidence interval was estimated via a Monte Carlo simulation (N=5000) and is shown using yellow shading.
In addition, we compared the measured concentrations of RO2 • DMPO adducts with the concentrations predicted by the kinetic model (Fig. 7). The observed values were an order of magnitude lower than the modeled ones. Similar to experiments described in Sect. 3.1, several factors can contribute to this discrepancy: (1) gas-wall partitioning of RO2 species and RO2 • DMPO adducts in the experimental setup and inside the PTR3 instrument; (2) uncertainty in the sensitivity at which RO2 • DMPO adducts were detected; (3) potential fragmentation of RO2 • DMPO • product ions; and (4) uncertainty in the reaction rate coefficient . In our model, we assumed that the largest fraction of the RO2 species was scavenged by DMPO. This assumption is valid if is larger than cm3 molecule−1 s−1. Otherwise, other loss channels for peroxy radicals, especially the RO2+RO2 reaction, become more important (Fig. S9 in the Supplement). Additional experiments performed under different conditions and an intercomparison with established methods (i.e., LIF) are needed to further estimate the measurement capability of the proposed analytical method.
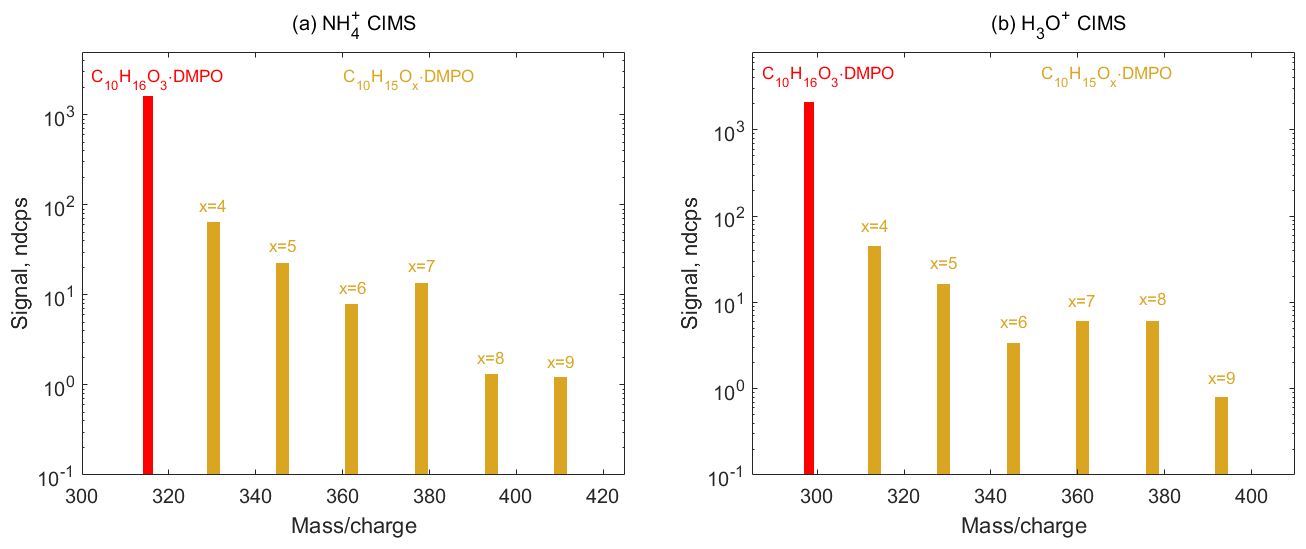
Figure 8Mass spectra of SCI • DMPO (red) and RO2 • DMPO (yellow) adducts in α-pinene ozonolysis experiments observed using (a) CIMS and (b) H3O+ CIMS. Primary RO2 species (C10H15O4) are formed via CI isomerization and can in turn undergo various autoxidation reactions resulting in the formation of several organic peroxy radicals (C10H15Ox, x=5–9), which were detected as adducts with the spin trap DMPO via and H3O+ CIMS.
Finally, we employed spin trapping for the detection of SCIs and organic peroxy radicals formed via the ozonolysis of larger cyclic alkenes, such as α-pinene. Decomposition of the α-pinene primary ozonide yields four different C10 SCIs, all of which have the same molecular formula C10H16O3 (Newland et al., 2018). These SCIs can further isomerize to form the primary peroxy radicals C10H15O4 and OH. Autoxidation of C10H15O4−RO2 species can, in turn, result in the formation of several more oxygenated peroxy radicals C10H15Ox, x=5–9 (Zhao et al., 2018). Signals of SCI • DMPO (C10H16O3 • DMPO) and RO2 • DMPO (C10H15Ox•DMPO, x=4–9) adducts were observed in both and H3O+ CIMS (Fig. 8 and Figs. S10 and S11 in the Supplement). OH radicals formed via the decomposition of SCI can in turn react with α-pinene, leading to the formation of the OH-derived RO2 species C10H17O3 and the subsequent autoxidation RO2 species C10H17O5 (Berndt et al., 2016). These radicals were detected as the RO2•DMPO adducts (Figs. S10 and S11 in the Supplement). This demonstrates that the analytical method allows for the simultaneous detection of a wide range of atmospheric radicals, including those with high oxygen contents (an O:C ratio of up to 0.9) that are formed via the autoxidation pathway, and can be used to study the kinetics of these species in the laboratory.
In summary, we experimentally demonstrated the measurement of speciated, short-lived, highly reactive atmospheric compounds such as Criegee intermediates, organic peroxy radicals, and hydroxyl radicals that are formed during the ozonolysis of alkenes. The analysis was carried out using chemical derivatization techniques, including spin trapping, while the detection of the formed radical adducts and closed-shell secondary ozonides was achieved via H3O+ and CIMS. The detected adducts and secondary ozonides have unique mass defects and can therefore be clearly separated from other compounds observed in the mass spectrum. Implementing chemical derivatization agents with low proton affinities allows the stabilized Criegee intermediates to be fully scavenged and quantified without depleting CIMS reagent ions. We show that spin traps can be used to effectively scavenge atmospheric radicals and reactive intermediates by demonstrating their high reactivity with radicals in the gas phase, using the TEMPO + OH reaction as an example. Using the spin trap DMPO, SCIs and RO2 species can be simultaneously detected, while the observed adducts can be quantified without directly calibrating them. Detection limits of spin trap and chemical derivatization agent adducts of 1.4×107 molecule cm−3 for SCIs and 1.6×108 molecule cm−3 for RO2 for an integration time of 30 s were estimated for the instrumentation used here. These limits suggest that the techniques would also work when sampling ambient air. In particular, this method enables any CIMS instrument to detect radicals and SCIs. Since spin traps such as DMPO and TEMPO are reactive towards a plethora of atmospheric radicals and reactive intermediates, including RO2, SCIs, and OH, the implementation of such spin traps results in the effective suppression of radical secondary chemistry and thus the elimination of potential chemical interferences. The direct method for speciated SCIs and RO2 measurements provides a means to study the atmospheric chemistry of these compounds. We stress that the quantification of RO2 species was done under well-defined laboratory conditions using the collision-induced dissociation (CID) technique, such that the estimated sensitivities are likely unique to the electric fields, pressures, and flows of the CIMS instrument. Further validation of the proposed analytical methods in more complex environments that are closer to the ambient conditions and an intercomparison with established methods (i.e., LIF) are needed.
For future applications of the method in field and laboratory experiments, various modifications of the experimental setup can be implemented to improve its measurement capability. We plan to synthesize and test new chemical derivatization agents that are optimized for gas-phase measurements in terms of their vapor pressures and selective reactivities and by labeling with atomic isotopes to simplify mass spectrometric detection and improve detection limits. With labeled spin traps, the identification of reactive intermediates may be greatly simplified and detection limits may be further improved, as the spin trap can provide a unique signature in the complex mass spectrum and move the observed to a region with very low background.
Data used within this work are available upon request. Please email Alexander Zaytsev (zaytsev@g.harvard.edu).
The supplement related to this article is available online at: https://doi.org/10.5194/amt-14-2501-2021-supplement.
FNK initially conceived the work. AZ and MB developed the laboratory setups, performed the experiments, and analyzed the CIMS data. AZ performed the quantum-chemical calculations and wrote the manuscript. AN and HF provided data and analysis for the LP-LIF experiments. DAK supported the study with PTR-MS measurements. All authors were involved in helpful discussions and contributed to the manuscript.
The authors declare that they have no conflict of interest.
Martin Breitenlechner acknowledges support from the Austrian Science Fund. Hendrik Fuchs and Anna Novelli acknowledge support from the European Research Council (ERC). Daniel Knopf acknowledges support from the US National Science Foundation. Jesse Kroll acknowledges support from the US National Science Foundation.
This research has been supported by the Harvard Global Institute (A New Strategy for the US and China: Joint Research on Air Pollution and Climate Using Innovative Airborne Instrumentation), the Austrian Science Fund (FWF (grant no. J-3900), the H2020 European Research Council (SARLEP (grant agreement no. 681529)), the National Science Foundation (grant nos. AGS-1446286 and AGS-1638672).
This paper was edited by Keding Lu and reviewed by three anonymous referees.
Bagryanskaya, E. G. and Marque, S. R.: Scavenging of organic C-centered radicals by nitroxides, Chem. Rev., 114, 5011–5056, https://doi.org/10.1021/cr4000946, 2014.
Berndt, T., Jokinen, T., Sipilä, M., Mauldin III, R. L., Herrmann, H., Stratmann, F., Junninen, H., and Kulmala, M.: H2SO4 formation from the gas-phase reaction of stabilized Criegee Intermediates with SO2: Influence of water vapour content and temperature, Atmos. Environ., 89, 603–612, https://doi.org/10.1016/j.atmosenv.2014.02.062, 2014.
Berndt, T., Richters, S., Jokinen, T., Hyttinen, N., Kurtén, T., Otkjær, R. V., Kjaergaard, H. G., Stratmann, F., Herrmann, H., Sipilä, M., Kulmala, M., and Ehn, M.: Hydroxyl radical-induced formation of highly oxidized organic compounds, Nat. Commun., 7, 13677, https://doi.org/10.1038/ncomms13677, 2016.
Berndt, T., Herrmann, H., and Kurten, T.: Direct Probing of Criegee Intermediates from Gas-Phase Ozonolysis Using Chemical ionization Mass Spectrometry, J. Am. Chem. Soc., 139, 13387–13392, https://doi.org/10.1021/jacs.7b05849, 2017.
Berndt, T., Mentler, B., Scholz, W., Fischer, L., Herrmann, H., Kulmala, M., and Hansel, A.: Accretion product formation from ozonolysis and OH radical reaction of α-pinene: mechanistic insight and the influence of isoprene and ethylene, Environ. Sci. Technol., 52, 11069–11077, https://doi.org/10.1021/acs.est.8b02210, 2018.
Berndt, T., Hyttinen, N., Herrmann, H., and Hansel, A.: First oxidation products from the reaction of hydroxyl radicals with isoprene for pristine environmental conditions, Commun. Chem., 2, 1–10, https://doi.org/10.1038/s42004-019-0120-9, 2019.
Breitenlechner, M., Fischer, M., Hainer, M., Heinritzi, M., Curtius, M., and Hansel, A.: PTR3: An instrument for Studying the Lifecycle of Reactive Organic Carbon in the Atmosphere, Anal. Chem., 89, 5824–5831, https://doi.org/10.1021/acs.analchem.6b05110, 2017.
Cantrell, C. A., Stedman, D. H., and Wendel, G. J.: Measurement of atmospheric peroxy radicals by chemical amplification, Anal. Chem., 56, 1496–1502, https://doi.org/10.1021/ac00272a065, 1984.
Chhantyal-Pun, R., Welz, O., Savee, J. D., Eskola, A. J., Lee, E. P. F., Blacker, L., Hill, H. R., Ashcroft, M., Khan, M. A. H., Lloyd-Jones, G. C., Evans, L., Rotavera, B., Huang, H., Osborn, D. L., Mok, D. K. W., Dyke, J. M., Shallcross, D. E., Percival. C. J., Orr-Ewing, A. J., and Taatjes, C. A.: Direct Measurements of Unimolecular and Bimolecular Reaction Kinetics of the Criegee Intermediate (CH3)2COO, J. Phys. Chem. A, 121, 4–15, https://doi.org/10.1021/acs.jpca.6b07810, 2016.
Drozd, G. T. and Donahue, N. M.: Pressure Dependence of Stabilized Criegee Intermediate Formation from a Sequence of Alkenes, J. Phys. Chem. A, 115, 4381–4387, https://doi.org/10.1021/jp2001089, 2011.
Drozd, G. T., Kroll, J. H., and Donahue, N. M.: 2,3-Dimethyl-2-butene (TME) Ozonolysis: Pressure Dependence of Stabilized Criegee Intermediates and Evidence of Stabilized Vinyl Hydroperoxides, J. Phys. Chem. A, 115, 161–166, https://doi.org/10.1021/jp108773d, 2011.
Edwards, G. D., Cantrell, C. A., Stephens, S., Hill, B., Goyea, O., Shetter, R. E., Mauldin III, R. L., Kosciuch, E., Tanner, D. J., and Eisele, F. L.: Chemical Ionization Mass Spectrometer Instrument for the Measurement of Tropospheric HO2 and RO2, Anal. Chem., 75, 5317–5327, https://doi.org/10.1021/ac034402b, 2003.
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., Scalmani, G., Barone, V., Mennucci, B., Petersson, G. A., Nakatsuji, H., Caricato, M., Li, X., Hratchian, H. P., Izmaylov, A. F., Bloino, J., Zheng, G., Sonnenberg, J. L., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Montgomery Jr., J. A., Peralta, J. E., Ogliaro, F., Bearpark, M., Heyd, J. J., Brothers, E., Kudin, K. N., Staroverov, V. N., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A., Burant, J. C., Iyengar, S. S., Tomasi, J., Cossi, M., Rega, N., Millam, J. M., Klene, M., Knox, J. E., Cross, J. B., Bakken, V., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R. E., Yazyev, O., Austin, A. J., Cammi, R., Pomelli, C., Ochterski, J. W., Martin, R. L., Morokuma, K., Zakrzewski, V. G., Voth, G. A., Salvador, P., Dannenberg, J. J., Dapprich, S., Daniels, A. D., Farkas, O., Foresman, J. B., Ortiz, J. V., Cioslowski, J., and Fox, D. J.: Gaussian 09, Revision D.01, Gaussian, Inc.: Wallingford, CT, USA, 2009.
Fuchs, H., Holland, F., and Hofzumahaus, A.: Measurement of tropospheric RO2 and HO2 radicals by a laser-induced fluorescence instrument, Rev. Sci. Instrum., 79, 084104, https://doi.org/10.1063/1.2968712, 2008.
Fuchs, H., Novelli, A., Rolletter, M., Hofzumahaus, A., Pfannerstill, E. Y., Kessel, S., Edtbauer, A., Williams, J., Michoud, V., Dusanter, S., Locoge, N., Zannoni, N., Gros, V., Truong, F., Sarda-Esteve, R., Cryer, D. R., Brumby, C. A., Whalley, L. K., Stone, D., Seakins, P. W., Heard, D. E., Schoemaecker, C., Blocquet, M., Coudert, S., Batut, S., Fittschen, C., Thames, A. B., Brune, W. H., Ernest, C., Harder, H., Muller, J. B. A., Elste, T., Kubistin, D., Andres, S., Bohn, B., Hohaus, T., Holland, F., Li, X., Rohrer, F., Kiendler-Scharr, A., Tillmann, R., Wegener, R., Yu, Z., Zou, Q., and Wahner, A.: Comparison of OH reactivity measurements in the atmospheric simulation chamber SAPHIR, Atmos. Meas. Tech., 10, 4023–4053, https://doi.org/10.5194/amt-10-4023-2017, 2017.
Giorio, C., Campbell, S. J., Bruschi, M., Tampieri, F., Barbon, A., Toffoletti, A., Tapparo, A., Paijens, C., Wedlake, A. J., Grice, P., Howe, D. J., and Kalberer, M.: Online Quantification of Criegee Intermediates of α-Pinene Ozonolysis by Stabilization with Spin Traps and Proton-Transfer Reaction Mass Spectrometry Detection, J. Am. Chem. Soc., 139, 3999–4008, https://doi.org/10.1021/jacs.6b10981, 2017.
Hansel, A., Scholz, W., Mentler, B., Fischer, L., and Berndt, T.: Detection of RO2 radicals and other products from cyclohexene ozonolysis with NH and acetate chemical ionization mass spectrometry, Atmos. Environ., 186, 248–255, https://doi.org/10.1016/j.atmosenv.2018.04.023, 2018.
Hawkins, C. L. and Davies, M. J.: Detection and characterisation of radicals in biological materials using EPR methodology, Biochim. Biophys. Acta, 1840, 708–721, https://doi.org/10.1016/j.bbagen.2013.03.034, 2014.
Horie, O., Schäfer, C., and Moortgat, G. K.: High reactivity of hexafluoro acetone toward criegee intermediates in the gas-phase ozonolysis of simple alkenes, Int. J. Chem. Kinet., 31, 261–269, https://doi.org/10.1002/(SICI)1097-4601(1999)31:4<261::AID-KIN3>3.0.CO;2-Z, 1999.
Hornbrook, R. S., Crawford, J. H., Edwards, G. D., Goyea, O., Mauldin III, R. L., Olson, J. S., and Cantrell, C. A.: Measurements of tropospheric HO2 and RO2 by oxygen dilution modulation and chemical ionization mass spectrometry, Atmos. Meas. Tech., 4, 735–756, https://doi.org/10.5194/amt-4-735-2011, 2011.
Hunter, E. P. and Lias, S. G.: Evaluated Gas Phase Basicities and Proton Affinities of Molecules: An Update, J. Phys. Chem. Ref. Data, 27, 413–656, https://doi.org/10.1063/1.556018, 1998.
Isaacman-Van Wertz, G., Massoli, P., O'Brien, R. E., Nowak, J. B., Canagaratna, M. R., Jayne, J. T., Worsnop, D. R., Su, L., Knopf, D. A., Misztal, P. K., Arata, C., Goldstein, A. H., and Kroll, J. H.: Using advanced mass spectrometry techniques to fully characterize atmospheric organic carbon: current capabilities and remaining gaps, Faraday Discuss., 200, 579–598, https://doi.org/10.1039/c7fd00021a, 2017.
Isaacman-Van Wertz, G., Massoli, P., O'Brien, R., Lim, C., Franklin, J. P., Moss, J. A., Hunter, J. F., Nowak, J. B., Canagaratna, M. R., Misztal, P. K., Arata, C., Roscioli, J. R., Herndon, S. T., Onasch, T. B., Lambe, A. T., Jayne, J. T., Su, L., Knopf, D. A., Goldstein, A. H., Worsnop, D. R., and Kroll, J. H.: Chemical evolution of atmospheric organic carbon over multiple generations of oxidation, Nat. Chem., 10, 462–468, https://doi.org/10.1038/s41557-018-0002-2, 2018.
Jenkin, M. E., Saunders, S. M., Pilling, M. J.: The tropospheric degradation of volatile organic compounds: a protocol for mechanism development, Atmos. Environ., 31, 81–104, https://doi.org/10.1016/S1352-2310(96)00105-7, 1997.
Johnson, D. and Marston, G.: The gas-phase ozonolysis of unsaturated volatile organic compounds in the troposphere, Chem. Soc. Rev., 37, 699–716, https://doi.org/10.1039/b704260b, 2008.
Khan, M. A. H., Percival, C. J., Caravan, R. L., Taatjes, C. A., and Shallcross, D. E.: Criegee intermediates and their impacts on the troposphere, Environ. Sci., 20, 437, https://doi.org/10.1039/C7EM00585G, 2018.
Lewis, T. R., Blitz, M. A., Heard, D. E., and Seakins, P. W.: Direct evidence for a substantive reaction between the Criegee intermediate, CH2OO, and the water vapour dimer, Phys. Chem. Chem. Phys., 17, 4859–4863, https://doi.org/10.1039/C4CP04750H, 2015.
Long, B., Bao, J. L., and Truhlar, D. G.: Unimolecular reaction of acetone oxide and its reaction with water in the atmosphere, P. Natl. Acad. Sci. USA, 115, 6135–6140, https://doi.org/10.1073/pnas.1804453115, 2018.
Lou, S., Holland, F., Rohrer, F., Lu, K., Bohn, B., Brauers, T., Chang, C. C., Fuchs, H., Häseler, R., Kita, K., Kondo, Y., Li, X., Shao, M., Zeng, L., Wahner, A., Zhang, Y., Wang, W., and Hofzumahaus, A.: Atmospheric OH reactivities in the Pearl River Delta – China in summer 2006: measurement and model results, Atmos. Chem. Phys., 10, 11243–11260, https://doi.org/10.5194/acp-10-11243-2010, 2010.
Mihelcic, D., Müsgen, P., and Ehhalt, D. H.: An improved method of measuring tropospheric NO2 and RO2 by matrix isolation and electron spin resonance, J. Atmos. Chem., 3, 341–361, https://doi.org/10.1007/BF00122523, 1985.
Murray, R. W., Story, P. R., and Loan, L. D.: Ozonides from Aldehydic Zwitterions and Acetone, J. Am. Chem. Soc., 87, 3025–3026, https://doi.org/10.1021/ja01091a054, 1965.
Neeb, P., Sauer, F., Horie, O., and Moortgat, G. K.: Formation of hydroxymethyl hydroperoxide and formic acid in alkene ozonolysis in the presence of water vapour, Atmos. Environ., 31, 1417–1423, https://doi.org/10.1016/S1352-2310(96)00322-6, 1997.
Newland, M. J., Rickard, A. R., Alam, M. S., Vereecken, L., Munoz, A., Rodenasd, M., and Bloss, W. J.: Kinetics of stabilised Criegee intermediates derived from alkene ozonolysis: reactions with SO2, H2O and decomposition under boundary layer conditions, Phys. Chem. Chem. Phys., 17, 4076–4088, https://doi.org/10.1039/C4CP04186K, 2015.
Newland, M. J., Rickard, A. R., Sherwen, T., Evans, M. J., Vereecken, L., Muñoz, A., Ródenas, M., and Bloss, W. J.: The atmospheric impacts of monoterpene ozonolysis on global stabilised Criegee intermediate budgets and SO2 oxidation: experiment, theory and modelling, Atmos. Chem. Phys., 18, 6095–6120, https://doi.org/10.5194/acp-18-6095-2018, 2018.
Nosaka, Y. and Nosaka, A. Y.: Generation and detection of reactive oxygen species in photocatalysis, Chem. Rev., 117, 11302–11336, https://doi.org/10.1021/acs.chemrev.7b00161, 2017.
Novelli, A., Hens, K., Tatum Ernest, C., Martinez, M., Nölscher, A. C., Sinha, V., Paasonen, P., Petäjä, T., Sipilä, M., Elste, T., Plass-Dülmer, C., Phillips, G. J., Kubistin, D., Williams, J., Vereecken, L., Lelieveld, J., and Harder, H.: Estimating the atmospheric concentration of Criegee intermediates and their possible interference in a FAGE-LIF instrument, Atmos. Chem. Phys., 17, 7807–7826, https://doi.org/10.5194/acp-17-7807-2017, 2017.
Nozière, B. and Vereecken, L.: Direct Observation of Aliphatic Peroxy Radical Autoxidation and Water Effects: An Experimental and Theoretical Study, Angew. Chem., 58, 13976–13982, https://doi.org/10.1002/anie.201907981, 2019.
Pagonis, D., Krechmer, J. E., de Gouw, J., Jimenez, J. L., and Ziemann, P. J.: Effects of gas–wall partitioning in Teflon tubing and instrumentation on time-resolved measurements of gas-phase organic compounds, Atmos. Meas. Tech., 10, 4687–4696, https://doi.org/10.5194/amt-10-4687-2017, 2017.
Reiner, T., Hanke, M., and Arnold, F.: Atmospheric peroxy radical measurements by ion molecule reaction-mass spectrometry: A novel analytical method using amplifying chemical conversion to sulfuric acid, J. Geophys. Res.-Atmos., 102, 1311–1326, https://doi.org/10.1029/96JD02963, 1997.
Roberts, J. G., Voinov, M. A., Schmidt, A. C., Smirnova, T. I., and Sombers, L. A.: The hydroxyl radical is a critical intermediate in the voltammetric detection of hydrogen peroxide, J. Am. Chem. Soc., 138, 2516–2519, https://doi.org/10.1021/jacs.5b13376, 2016.
Samuni, A., Goldstein, S., Russo, A., Mitchell, J. B., Krishna, M. C., and Neta, P.: Kinetics and mechanism of hydroxyl radical and OH-adduct radical reactions with nitroxides and with their hydroxylamines, J. Am. Chem. Soc., 124, 8719–8724, https://doi.org/10.1021/ja017587h, 2002.
Saunders, S. M., Jenkin, M. E., Derwent, R. G., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): tropospheric degradation of non-aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 161–180, https://doi.org/10.5194/acp-3-161-2003, 2003.
Sipilä, M., Jokinen, T., Berndt, T., Richters, S., Makkonen, R., Donahue, N. M., Mauldin III, R. L., Kurtén, T., Paasonen, P., Sarnela, N., Ehn, M., Junninen, H., Rissanen, M. P., Thornton, J., Stratmann, F., Herrmann, H., Worsnop, D. R., Kulmala, M., Kerminen, V.-M., and Petäjä, T.: Reactivity of stabilized Criegee intermediates (sCIs) from isoprene and monoterpene ozonolysis toward SO2 and organic acids, Atmos. Chem. Phys., 14, 12143–12153, https://doi.org/10.5194/acp-14-12143-2014, 2014.
Stephens, P. J., Devlin, F. J., Chabalowski, C. F. N., and Frisch, M. J.: Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields, J. Phys. Chem., 98, 11623–11627, https://doi.org/10.1021/j100096a001, 1994.
Taatjes, C. A., Meloni, G., Selby, T. M., Trevitt, A. J., Osborn, D. L., Percival, C. J., and Shallcross, D. E.: Direct observation of the gas-phase Criegee intermediate (CH2OO), J. Am. Chem. Soc., 130, 11883–11885, https://doi.org/10.1021/ja804165q, 2008.
Taatjes, C. A., Welz, O., Eskola, A. J., Savee, J. D., Osborn, D. L., Lee, E. P. F., Dyke, J. M., Mok, D. W. K., Shallcross, D. E., and Percival, C. J.: Direct measurement of Criegee intermediate (CH2OO) reactions with acetone, acetaldehyde, and hexafluoroacetone, Phys. Chem. Chem. Phys., 14, 10391–10400, https://doi.org/10.1039/C2CP40294G, 2012.
Van Der Zee, J., Barr, D. P., and Mason, R. P.: ESR spin trapping investigation of radical formation from the reaction between hematin and tert-butyl hydroperoxide, Free Radical Bio. Med., 20, 199–206, https://doi.org/10.1016/0891-5849(95)02031-4, 1996.
Vereecken, L., Novelli, A., and Taraborrelli, D.: Unimolecular decay strongly limits the atmospheric impact of Criegee intermediates, Phys. Chem. Chem. Phys., 19, 31599–31612, https://doi.org/10.1039/C7CP05541B, 2017.
Watanabe, T., Yoshida, M., Fujiwara, S., Abe, K., Onoe, A., Hirota, M., and Igarashi, S.: Spin trapping of hydroxyl radical in the troposphere for determination by electron spin resonance and gas chromatography/mass spectrometry, Anal. Chem., 54, 2470–2474, https://doi.org/10.1021/ac00251a015, 1982.
Welz, O., Savee, J. D., Osborn, D. L., Vasu, S. S., Percival, C. J., Shallcross, D. E., and Taatjes, C. A.: Direct kinetic measurements of Criegee intermediate (CH2OO) formed by reaction of CH2I with O2, Science, 335, 204–207, https://doi.org/10.1126/science.1213229, 2012.
Wolfe, G. M., Marvin, M. R., Roberts, S. J., Travis, K. R., and Liao, J.: The Framework for 0-D Atmospheric Modeling (F0AM) v3.1, Geosci. Model Dev., 9, 3309–3319, https://doi.org/10.5194/gmd-9-3309-2016, 2016.
Womack, C. C., Martin-Drumel, M. A., Brown, G. G., Field, R. W., and McCarthy, M. C.: Observation of the simplest Criegee intermediate CH2OO in the gas-phase ozonolysis of ethylene, Sci. Adv., 1, e1400105, https://doi.org/10.1126/sciadv.1400105, 2015.
Wood, E. C. and Charest, J. R.: Chemical amplification – cavity attenuated phase shift spectroscopy measurements of atmospheric peroxy radicals, Anal. Chem., 86, 10266–10273, https://doi.org/10.1021/ac502451m, 2014.
Yuan, B., Koss, A. R., Warneke, C., Coggon, M., Sekimoto, K., and de Gouw, J. A.: Proton-Transfer-Reaction Mass Spectrometry: Applications in Atmospheric Sciences, Chem. Rev., 117, 13187–13229, https://doi.org/10.1021/acs.chemrev.7b00325, 2017.
Zaytsev, A., Breitenlechner, M., Koss, A. R., Lim, C. Y., Rowe, J. C., Kroll, J. H., and Keutsch, F. N.: Using collision-induced dissociation to constrain sensitivity of ammonia chemical ionization mass spectrometry ( CIMS) to oxygenated volatile organic compounds, Atmos. Meas. Tech., 12, 1861–1870, https://doi.org/10.5194/amt-12-1861-2019, 2019.
Zhao, Y., Thornton, J. A., and Pye, H. O.: Quantitative constraints on autoxidation and dimer formation from direct probing of monoterpene-derived peroxy radical chemistry, P. Natl. Acad. Sci. USA, 115, 12142–12147, https://doi.org/10.1073/pnas.1812147115, 2018.