the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 14 Jun 2021
| 14 Jun 2021
Quantifying organic matter and functional groups in particulate matter filter samples from the southeastern United States – Part 2: Spatiotemporal trends
Satoshi Takahama
Andrew T. Weakley
Bruno M. Debus
Stephanie L. Shaw
Eric S. Edgerton
Taekyu Joo
Nga L. Ng
Ann M. Dillner
Organic species within atmospheric particles vary widely in molecular structure. The variety of molecules that comprise the aerosol make it rich in information about its sources and chemical life cycle but also make particulate organic matter (OM) difficult to characterize and quantify. In Part 1 of this pair of papers, we described a direct method for measuring the composition and concentrations of OM in aerosol samples that is compatible with routine monitoring of air quality. This method uses Fourier transform infrared (FT-IR) spectrometry of filter-based aerosol samples to quantify bonds, or functional groups, that represent the majority of organic composition. Summation of these functional groups gives OM. In this paper, functional group and OM concentrations are directly measured in 8 years of aerosol samples collected at two rural and two urban sites in the Southeastern Aerosol Research and Characterization (SEARCH) network. FT-IR spectrometry with a multivariate calibration is used to quantify the concentrations of aliphatic C−H (aCH), carboxylic acid (COOH), oxalate (oxOCO; representing carboxylates), non-acid and non-oxalate carbonyl (naCO), and alcohol O−H (aCOH) in approximately 3500 filter samples collected every third day from 2009 through 2016. In addition, measurements are made on samples from all days in 2016. To the best of our knowledge, this is the longest time period over which this type of analysis has been applied, and this work also demonstrates the application of a more chemically complete and less destructive method than in prior work using alternate techniques. A decline in the total OM is observed from 2011 to 2016 due to a decrease in the more oxygenated functional groups (carboxylic acid and oxalate) and is attributed to anthropogenic SO2 and/or volatile organic compound (VOC) emissions reductions. The trend in OM composition is consistent with those observed using more time- and labor-intensive analytical techniques. Concurrently, the fractional contributions of aCOH and naCO to OM increased, which might be linked to monoterpene-derived secondary OM, with plausible influences from decreasing NOx and/or increasing O3 concentrations. In addition, this work demonstrates that OM to organic carbon () ratios in the southeastern US (SE US) did not appreciably change over the study time period as a result of these competing functional group contributions to OM. Monthly observations support the sources suggested by these overall trends, including evidence of strong biogenic and photo-oxidation influences. Daily samples from 2016 further elucidate the consistent impact of meteorology and biomass burning events on shorter-term OM variability, including prescribed burning in the winter or spring and wildfires in the autumn, although these sources did not appear to be strong contributors to long-term OM or composition trends in the SE US. These shorter-term and spatial observations reinforce the results of the broader dataset and serve to evaluate the applicability of FT-IR spectrometry measurement to trends analysis on various timescales relevant to routine monitoring of aerosol composition.
- Article
(2494 KB) - Full-text XML
- Companion paper
-
Supplement
(4299 KB) - BibTeX
- EndNote





1.1 A functional group perspective on atmospheric aerosol organic matter
The composition of atmospheric aerosol organic matter (OM) is rich in information about the sources and processes leading to particle phase pollution (Rogge et al., 1993). However, OM is not directly quantified in monitoring networks and many intensive field campaigns. While organic carbon (OC) concentrations are often measured in such cases, OC captures only the variability in carbon atom concentrations, and the contributions of oxygen and hydrogen atoms are assumed based on a single ratio. However, this ratio varies substantially in atmospherically relevant molecules (Turpin and Lim, 2001); within the ambient atmosphere, the ratios are also estimated to vary strongly between seasons and sites (Simon et al., 2011). Thus, a direct method for regularly and directly quantifying OM in aerosol samples would improve the accuracy of measured OM concentrations and trends.
Much of the work on OM mass has focused on speciation of individual compounds, so that only a portion of OM mass is measured (using chromatography and mass spectrometry, MS, techniques, for example, by Gao et al., 2006), or modeling approaches based on concentrations of speciated chemical tracers (Budisulistiorini et al., 2015; Kleindienst et al., 2010). Comprehensive OM quantification can be afforded by other techniques that are not readily applicable in routine measurements because of expense and labor requirements (for example, proton transfer reaction MS and aerosol MS; Ditto et al., 2018; Salvador et al., 2016; Schum et al., 2018; Xu et al., 2015a; Zhang et al., 2018). For routine monitoring and many field campaigns, measuring bulk chemical properties of OM could be an appropriate middle ground between estimating OM from OC measurements and speciating a small fraction of the OM by mass. Organic functional groups (FGs) have been explored as such a middle ground, quantifying the composition of OM from distinct sources (Coury and Dillner, 2009; Russell et al., 2011; Takahama et al., 2011), as well as measuring the age or degree of oxidation of sampled OM (Turpin and Lim, 2001; Ruthenburg et al., 2014; Russell, 2003).
The conceptualization of OM in terms of FGs differs from that of molecular composition, as used with chromatography methods, and is more akin to direct spectral results of hard ionization mass spectrometry. Multiple FGs are present in every molecule, so the contributions of FGs to each sample are indicative of OM composition; a greater fraction of oxygenated FG content can indicate more aged material (Moretti et al., 2008; Chhabra et al., 2011). By studying the FG composition of OM within atmospheric aerosol samples, patterns emerge in the bulk OM composition, from which sources and formation processes can be revealed (Corrigan et al., 2013; Liu et al., 2018; Russell et al., 2009; Takahama et al., 2011).
Fourier transform infrared (FT-IR) spectrometry is used to quantify FG concentrations in aerosol filter samples (Reggente et al., 2019; Russell et al., 2009; Coury and Dillner, 2008; Maria et al., 2002). The analysis can be performed on polytetrafluoroethylene (PTFE) filters without damaging the sample; this enables the filters to be archived and/or used for other analyses, making FT-IR spectrometry suitable for monitoring networks and field campaigns that collect PTFE filter samples.
1.2 Unique and changing atmospheric chemistry and climate of the southeast US
The hot, humid climate in the southeastern (SE) US creates a unique environment for mixing and reaction of anthropogenic and biogenic aerosol chemical components, including within liquid water, as well as the generation of oxidants (Hansen et al., 2003). Stagnation events occur both in the summer and winter, causing build-up of atmospheric pollutants (Hansen et al., 2003). In addition, aerosol composition and emissions sources of the SE US are distinct. The Southeastern Aerosol Research and Characterization (SEARCH) network was designed to study atmospheric chemistry in the SE US, at urban–rural pairs from 1998 through 2016 (Fig. 1; Blanchard et al., 2013a; Edgerton et al., 2005, 2006; Hansen et al., 2003). The configuration of sites varied over the years, but the longest-operated sites include the urban–rural pairs in Alabama and Georgia (Fig. 1).
Figure 1Map of the SEARCH network showing urban and rural sites: YRK is Yorkville, GA (rural); JST is Jefferson St., Atlanta, GA (urban); CTR is Centreville, AL (rural); BHM is Birmingham, AL (urban). The map was generated using the borders function created for MATLAB (Greene et al., 2019).
The Yorkville, Georgia (YRK), site was located ∼55 km from a major urban area (Atlanta) and was surrounded by pastures and nearby forest (∼100–300 m from samplers). Some agricultural emissions might be expected at YRK, while the rural Alabama site, Centreville (CTR), was surrounded by forest (Edgerton et al., 2005). The CTR site was ∼85 km from its paired urban area (Birmingham or BHM), with only a few residences in the area; the CTR site therefore represents a more rural location than YRK. Analysis methods were advanced and comprehensive, including real-time gas-phase measurements, light- and mass-based measurements of total particles, a variety of particle-phase composition measurements (trace elements, inorganic salts, OC, and elemental carbon), and supporting meteorological variables (Hansen et al., 2003).
A partial characterization of SE US OM in previous years can be drawn from studies within the SEARCH network data and the many field campaigns carried out in the region (Carlton et al., 2018; Edgerton et al., 2005; Kim et al., 2015; Liu et al., 2018; Mao et al., 2018; US Environmental Protection Agency, 2011; Xu et al., 2015a). The dominant contribution of secondary material to OM and a regional homogeneity of primary OM sources has been demonstrated (Chen et al., 2012). There are also unique emissions sources and secondary pollutants in the SE US; in particular, prevalent tree species emit high isoprene and monoterpene loadings relative to other regions (Guenther et al., 1994, 2012) and contribute substantially to the OM (Xu et al., 2015b; Zhang et al., 2018). Prescribed burns, wildfires, and residential wood burning smoke sources were demonstrated to be strong contributors of primary aerosol loadings at SEARCH sites (Blanchard et al., 2013a; Chen et al., 2012). Fossil fuel combustion contributes substantially to OC in the SE US, especially at urban sites: ∼50 % of primary and secondary OC at urban BHM was estimated to be from fossil sources, but at rural CTR, >80 % of primary and all of secondary OC was from modern sources such as trees (in the early 2000s; Blanchard et al., 2008). In addition, other emissions from fossil fuel combustion, including NOx, SO2, and primary OM also affect OM concentrations by modulating aerosol surface area, aerosol water content, aerosol acidity, and oxidant concentrations (Nguyen et al., 2015; Carlton et al., 2010; Al-Naiema et al., 2018). Since the late 1980s, controls on anthropogenic emissions have resulted in volatile organic compound (VOC), NOx, OC, SO2 , and other species concentration declines (Blanchard et al., 2014, 2016; Hand et al., 2012a; Hidy et al., 2014). More recent work by Chen et al. (2020; in 2016 at YRK) confirmed that secondary OM from biogenic carbon sources continues to be important in the SE US.
Several SE US studies have highlighted decreasing OM concentrations due to the interactions between anthropogenic and biogenic pollutants (Goldstein et al., 2009). In particular, declining SO2 emissions are likely to be driving declines in biogenic OM concentrations (Marais et al., 2017; Xu et al., 2015b). The result of declining NOx concentrations on OM in the SE US has also varied among studies (Pye et al., 2019; Travis et al., 2016; Wolff et al., 2013; Zhang et al., 2018), with trade-offs between the availabilities of major oxidants and volatilities of products at varying NOx concentrations. These changes in chemical climatology make the SE US a unique location for studying a method to characterize OM composition, as well as examining the effects of declining anthropogenic emissions on atmospheric chemistry.
1.3 Research statement
We herein present the application of FT-IR spectrometry to 8 years of routinely collected SEARCH network PTFE ambient aerosol filter samples to examine trends in OM concentrations, and their FG composition from 2009 to 2016. These quantities have not been directly measured in SEARCH network data prior to this work. FG calibration models described by Boris et al. (2019; Part 1 of this study, which describes the method development) were applied to SEARCH spectra in the current study to accomplish the following goals: (1) examine the yearly, seasonal, and daily trends in OM and its FG composition; (2) characterize the trends in directly measured over the 8 years at all sites; and (3) evaluate the applicability of the FT-IR spectrometry method for routine OM characterization. The comparison of the results to other FG studies, SEARCH network data, and data from other studies of SE US atmospheric chemistry provided context for these measurements and suggested explanations for the observed trends.
SEARCH network filter samples of atmospheric particulate matter with ≤2.5 µm aerodynamic diameter (PM2.5) were obtained from Atmospheric Research & Analysis, Inc. (Cary, NC). Samples from every third day in 2009–2015 and every day in 2016, from five sites in the SE US, were analyzed during this study (Sect. 2.1) along with additional samples collected at one SEARCH site in 2018 (Sect. 2.2). Additional datasets are used to support findings (Sect. 2.3). The samples were analyzed using FT-IR absorption spectrometry (Sect. 2.4), and concentrations of organic FGs were calculated using multivariate calibration models (Boris et al., 2019). Organic matter (OM) concentrations were calculated for each sample as a linear combination of the FG concentrations measured (Sect. 2.7). Spatial and temporal trends were explored in the resulting OM concentrations, , and contributing FG quantities (Sect. 2.8).
2.1 SEARCH aerosol samples and network data
Filter samples of ambient aerosol were collected in the SEARCH network on MTL 47 mm PTFE filters with 2 µm pore size (Measurement Technology Laboratories, https://mtlcorp.com/filters, last access: 16 May 2021) using Partisol Plus 2025 samplers (Rupprecht & Patashnick/Fisher Scientific, http://www.thermofisher.com/, last access: 16 May 2021). All samples were collected over 24 h at 16.7 L min−1 (24.0 m3; see Sect. 2.5 for outlier handling). Filters were shipped and stored at <4 ∘C (from Aerosol Research and Analysis, Inc., ARA, in Morrisville, NC) to minimize loss of volatile species. SEARCH samples from 2009–2016 were used in the present study (samples were also collected from 1999 to mid-2008 but were not refrigerated during storage, and thus may have a compromised organic composition). Although the paired methods paper reports only analyzing samples from 1 in 3 months per year, to better elucidate trends, samples from every 1 in 3 d in all months between 2009 and 2015 (a sampling schedule matching that for thermal optical reflectance, TOR, analysis for OC measurements) were used in this study; samples for all days were included during 2016. The FT-IR analysis of the additional months was performed at least 1 year after the initial set of samples was analyzed. During this additional analysis, samples collected in 2011, 2012, and 2014 at Centreville, AL, were erroneously kept at room temperature, rather than kept cold, between ∼August and November 2018. At each site, ∼100 (68–123) samples were analyzed by FT-IR spectrometry per year from 2009 to 2015. In total, 3508 samples from 2009 to 2016 and 1267 samples from 2016 were analyzed. The SEARCH network ended sampling in 2016 at BHM on 18 October. Three sites in addition to those in this work were closed before 2016.
Multiple chemical and physical parameters were measured by the SEARCH network (discussed in detail by Hansen et al., 2003; filter-based measurements by Edgerton et al., 2005; and continuous gas- and particle-phase measurements by Edgerton et al., 2006). Gravimetric analysis of PM2.5 mass and x-ray fluorescence of trace metal concentrations were performed using the same Teflon filters analyzed by FT-IR spectrometry. TOR analysis for OC and elemental carbon (EC) concentrations was performed using 37 mm quartz filters (and OC concentrations were blank-corrected by subtracting network-wide annual mean field blank OC concentrations). Ions including , , and were measured using ion chromatography from 47 mm PTFE filters; negative artifacts of and were quantified from 47 mm nylon and cellulose filters, respectively.
2.2 Additional dataset: January and February 2018 at the Atlanta site
Daily samples were collected at JST from 15 to 21 January, 29 January to 4 February, and 12 to 25 February 2018. Samples were collected in a similar manner to those collected in the SEARCH network including sampling for 24 h with a Partisol 2025 particle sampler loaded with MTL 47 mm diameter PTFE filters (as described above).
2.3 Supporting datasets
In addition to chemical data, meteorological variables were gathered at the SEARCH network sites. However, precipitation data were not available. Precipitation observations were therefore retrieved from the National Oceanographic and Atmospheric Administration website (https://www.climate.gov/maps-data/dataset/past-weather-zip-code-data-table, last access: 16 May 2021). Satellite imagery was used to determine whether smoke plumes from wildfires and/or prescribed burns were observed on each sample date; these images were retrieved from National Aeronautics and Space Administration WorldView (https://worldview.earthdata.nasa.gov/, last access: 16 May 2021) using the Aqua MODIS Corrected Reflectance base layer.
2.4 FT-IR spectrometry analyses
All filter samples and standards were analyzed using a Bruker Tensor II FT-IR spectrometer (Bruker Optics, Inc.; http://www.bruker.com/, last access: 16 May 2021) operated in transmission mode. The spectrometer was equipped with a liquid-nitrogen-cooled mercury cadmium telluride detector. Filters were placed into a sample chamber within the spectrometer that was custom-built (Debus et al., 2019); the chamber was flushed continuously with air scrubbed of H2O and CO2 (model VCDA air purge system, Puregas, LLC, http://www.puregas.com/, last access: 16 May 2021; <10 % humidity). The FT-IR spectrometry analyses have been described in more detail elsewhere (Takahama et al., 2013). Spectra used in the present work include the wavenumber range of 4000 to 1500 cm−1; the range of 1500 to 400 cm−1 was excluded due to strong absorption by the PTFE filter substrate (Mayo et al., 2004) and highly specific absorption for each chemical. Samples were re-analyzed after 1–2 years to demonstrate the stability of the samples and resulting spectra and concentrations; handling, analysis, and long-term storage did not substantially affect the spectra, but a small bias (−5 % yr−1 to −10 % yr−1 after the first 2 years) was observed, likely due to storage and/or transport of samples (Boris et al., 2019). Outliers were detected and removed to minimize the influence of questionable data on trends (Boris et al., 2019).
2.5 Outlier detection and handling
Outlier samples and field blanks, such as ripped filters or swapped samples, were identified using the following criteria. First, when available, if the observed flow rate, duration, or total volume was not 100±5 % of the expected value (e.g., <23.75 of 24.0 expected hours), the sample was excluded from the dataset; samples with three or more null flow observations were assumed to be inaccurately sampled and were also excluded. In addition, field blanks with clear collected aerosol material were removed. Samples with spectra that were obviously collected incorrectly were also excluded (e.g., no filter was loaded into the chamber and the spectrum contained no expected absorption features). Samples with the following characteristics were also flagged and removed if the sample was definitely collected incorrectly (e.g., an anomalous FT-IR spectrum and poor predictions): (1) a spectrum corresponding to a high TOR OC concentration but low infrared absorption or (2) a high error in prediction after calibration. Overall, approximately 5 % () of the ambient 2009–2016 1:3 d samples were removed from the dataset; approximately 5 % () of the ambient 2016 daily samples were removed. Samples missing TOR OC concentrations or other network measurements were still included in the results.
2.6 Functional group calibration models
FG concentrations were quantified in the ambient samples using multivariate calibration models (methods are described in detail by Boris et al. (2019). The calibration models were constructed using laboratory standards of 17 organic compounds and 3 inorganic interferents, as discussed in our methods paper. Five FGs were quantified and reported using separate FT-IR spectrometry and partial least-squares (PLS) calibration models: aliphatic C−H (aCH), carboxylic acid (COOH), carboxylate represented by oxalate (oxOCO), non-acid non-oxalate carbonyl (naCO), and alcohol O−H (aCOH). One model was constructed for each FG with the exception of naCO, for which a linear regression between carboxylic acids and total carbonyls was used to determine the concentration (Takahama et al., 2013; Boris et al., 2019). The spectra and masses were regressed using a multivariate PLS algorithm (Wold et al., 2001) to obtain a set of regression coefficients describing each FG (Reggente et al., 2019; Ruthenburg et al., 2014). Sample concentrations of each FG were then determined by applying the regression coefficients to each sample FT-IR spectrum. Because there were no directly comparable FG concentrations for these samples, models were evaluated using a variety of metrics, such as TOR OC and residual OM concentrations (the latter is calculated by subtracting a weighted sum of major inorganic ionic and elemental aerosol components and elemental carbon from PM2.5 mass for each sample, as described by Hand et al., 2012b).
2.7 Calculation of OC and OM concentrations
Concentrations of OC and OM were calculated as linear combinations of the measured FG concentrations (Boris et al., 2019). OC concentrations were calculated as the summation of carbon masses contributed by each quantified FG, assuming the following ratios of carbon atoms per bond: aCH=0.5C (i.e., on average, C−H bonds were assumed to be ), COOH=1C, oxOCO=1C, naCO=1C, and aCOH=0.5. The same ratios were used by Russell (2003) and Boris et al. (2019) and were supported by modeling work (Takahama and Ruggeri, 2016). In contrast, Ruthenburg et al. (2014) assumed that aCOH contributed no carbon (assumed to be primary or secondary). OM concentrations were similarly calculated as the sum of each of the FG contributions to O, H, and carbon mass. The same assumed ratios for carbon contributions were applied, plus all associated O and H atoms. ratios were calculated from these OM and OC concentrations; contributions of each functional group to were calculated as the contributions of O and H mass to OC, with carbon mass contribution to the OC set equal to one (as in previous representations; e.g., Takahama and Ruggeri, 2016).
2.8 Method detection limits and sample uncertainty
The method detection limit (MDL) for each FG was estimated as the difference between the 95th and 50th percentiles of FG concentrations (UC Davis Air Quality Research Center, 2017) measured from 20 laboratory blanks and 616 field blanks. The MDLs of the calculated OM and OC concentrations were estimated using the same blanks, as the root of the sum of squares of the FG MDLs (Boris et al., 2019). Samples with FG or OM concentrations less than the value of the respective MDLs were censored as described by Boris et al. (2019). For all FGs, >85 % of sample concentrations were above the MDL (97 % oxOCO, 95 % aCH, 86 % aCOH, 92 % COOH, and 89 % naCO).
The precision of the method (including aerosol sampling, differences in filter substrates, handling, and FT-IR spectrometry analysis) was evaluated by comparing measurements from two colocated sampling sites within the SEARCH network. This “sampling uncertainty” was calculated (Hyslop and White, 2008, 2009) and FG concentrations from samples collected at the JST site and its colocated site, cJST, during 2009–2011, October 2015, and 2016.
2.9 Annual, monthly, and daily concentrations and trend values
Median values were used to report annual and monthly functional group and OM concentrations to minimize the impact of extreme values. Theil–Sen rank-invariant regression (Theil, 1992; Sen, 1968) was used to determine the slopes of multi-year trends in measured variables. The slopes were expressed as change in concentration per year () or change in fractional FG contribution to OM per year (% OM yr−1). Confidence intervals about each slope were calculated as 95th and 5th percentiles of bootstrapped slopes and intercepts (n=2500; the method for confidence interval estimation was carried out following the percentiles method for Theil–Sen regressions described by Wilcox, 1998).
Declining trends in aerosol OM concentrations were observed in the SE US in urban and rural locations (Table 1; Sect. 3.1). The OM composition also changed, with distinct declines in contributions from COOH and oxOCO but enhancements from naCO and aCOH (Table S1 in the Supplement, Sects. 3.2 and 3.3). The OM was somewhat regional in character, as suggested in other SEARCH network research (Hidy et al., 2014): coefficients of determination (R2) between urban and rural site OM concentrations were 0.69 (Georgia sites) and 0.56 (Alabama sites). Calculated ratios were similar in magnitude to previously observed SE US values, but multi-year trends differed from those estimated using TOR OC and other measurements (Sect. 3.4). The seasonality of the OM composition, as well as changes in the seasonality over the 2009–2016 period (Sect. 3.5), supported mechanisms suggested for the longer-term trends, with more highly oxygenated FG concentrations declining more during the summer, and less oxygenated FG abundances increasing more in shoulder seasons. Median annual FT-IR spectra demonstrated trends supporting those of the FGs. The daily 2016 dataset (Sect. 3.6) was used to demonstrate clear patterns in OM concentration build-up and washout, especially related to smoke, and differences in OM dynamics by season.
A comparison was made to determine whether the sampling frequency was representative of the trends in a complete dataset: FG and OM concentrations of the 2016 dataset, which included all days, were contrasted with those of the 1:3 d dataset (Sect. S7 in the Supplement). While the lack of daily samples added noise to the data, there was no apparent bias in OM or FG concentrations.
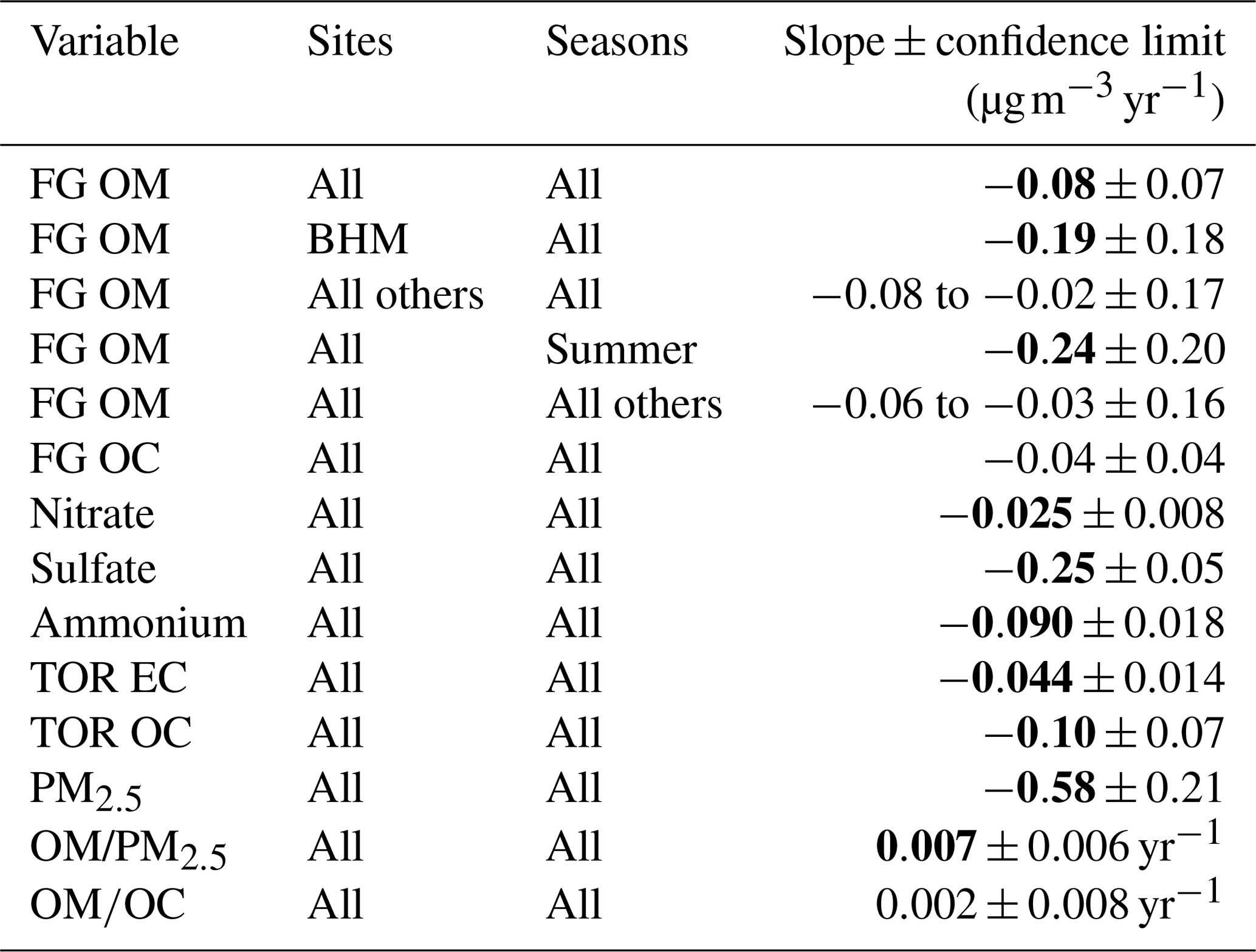
Table 1Trends in variables measured using FT-IR spectrometry. Slopes were calculated using Theil–Sen regression for data between 2011 and 2016 (Sect. 2.8). For FG OM and FG OC, the seasons and sites with the most extreme slopes are shown separately from “all others”. Confidence limits were bootstrapped at 95 % confidence. Slope values in bold are greater than the confidence limits. Note that units for and trends are yr−1.
3.1 Trends of OM: corroboration of chemical climatology change as a result of anthropogenic emission declines
Overall, decreasing concentrations of OM measured by FT-IR spectrometry (“FG OM”) were observed between 2009 and 2016 in the SE US ( , −8 % total). This change in OM concentration has a pattern similar to that of the concentrations of anthropogenic inorganic pollutant species, which also decreased in abundance over the study period, as shown in Sect. 2. Declines in concentrations were observed for total PM2.5, particle , , elemental carbon (EC), and in SEARCH data and other SE US aerosol samples (Blanchard et al., 2013a; Feng et al., 2020; Malm et al., 2017; Marais et al., 2017). OM concentrations increased during the 2008–2010 recovery from an economic recession (Feng et al., 2020; de Foy et al., 2016; Hand et al., 2019; Hidy et al., 2014), and thus multi-year trends discussed in further paragraphs will be expressed for the latter 6-year period of 2011 through 2016 (Table 1). The resulting slope in OM ( , which is equivalent to −2.9 % yr−1 or −18 % total) is similar in magnitude to those estimated over previous decades (e.g., −1.9 % yr−1 OM 1998–2013 using SEARCH data, Marais et al., 2017).
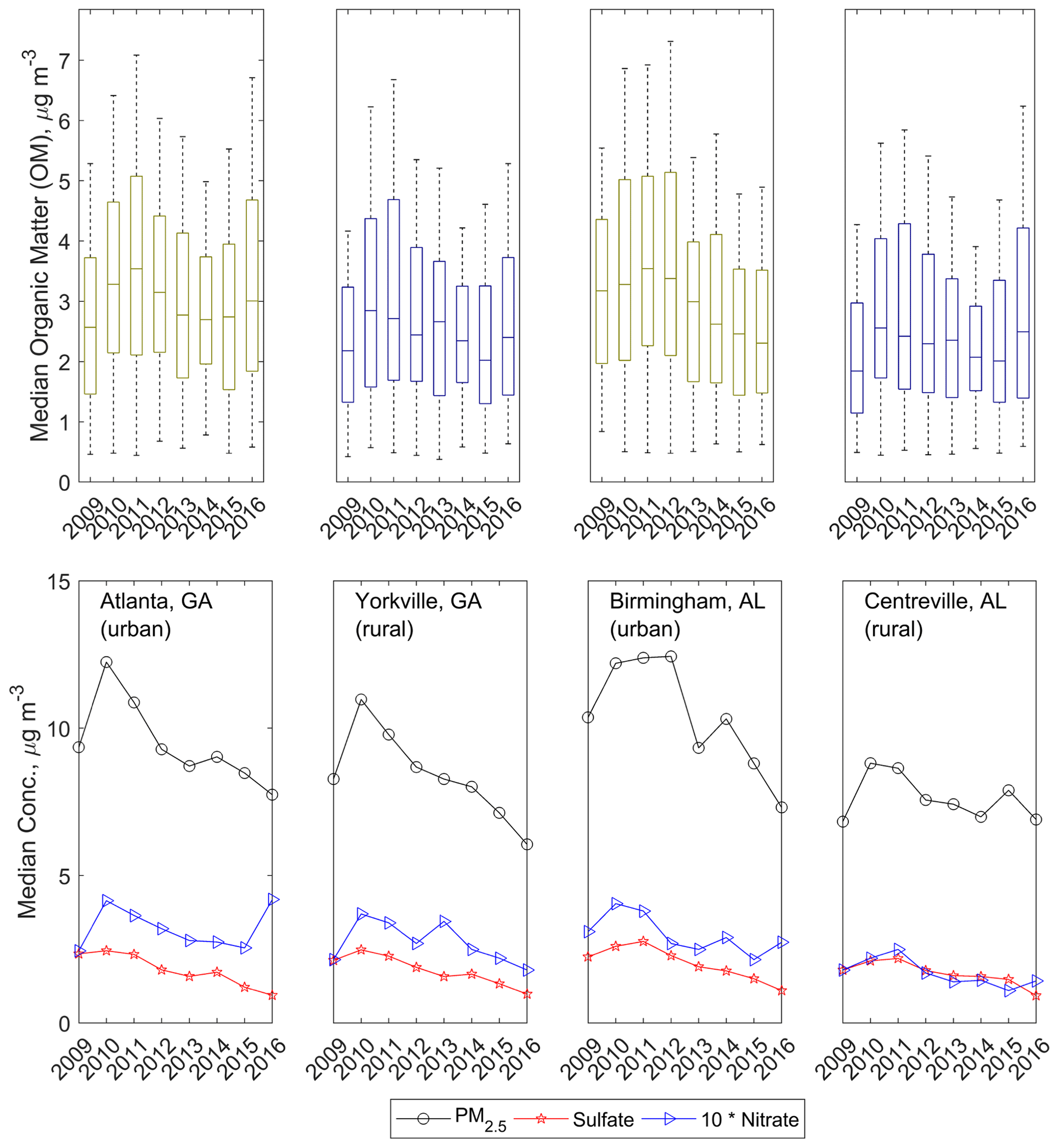
Figure 2The top row shows annual median OM concentrations quantified via FT-IR spectrometry; colors indicate rural (dark blue) or urban (olive) site locale. The bottom row shows annual median total PM2.5 concentration, particle , and particle concentrations as measured in the SEARCH network. Column-wise subpanels correspond to individual sites, as indicated in the bottom row.
Other work has demonstrated an anthropogenic component to the changes in particle composition: concentrations of particulate non-polar organic species also decreased between 2006 and 2010 (Blanchard et al., 2014), as did non-methane VOC concentrations between 1999 and 2008 (Hidy et al., 2014). A slight increase (0.007±0.006 yr−1) in the contribution of OM to PM2.5 concentrations was observed in the SEARCH network (Fig. 2; Table 1), which has been attributed especially to the strong decline in concentrations and therefore the enhanced contribution of OM to PM2.5 (Marais et al., 2017).
The overall decline in OM concentrations was observable for all four sites in the annual medians (Fig. 2), with the largest at BHM and the smallest at the most rural of the sites studied, CTR (only the BHM site-specific OM trend was statistically significant; Table 1), supporting the anthropogenic origin of the decline in OM. In contrast to other pollutant trends, mean 24 h mixing ratios of SEARCH network O3 increased slightly over the studied time period (approximately +0.03 ppb yr−1), with strong enhancements in springs and strong declines in summers (+0.79 ppb yr−1 in springs over all sites; summers −1.17, autumns +0.10, and winters +0.49 ppb yr−1). Coincident with O3 declines, summertime OM concentration declines were strongest ( µ g m−3), supporting O3 as an influential constituent in controlling OM concentrations (Sect. 3.2).
Decreasing anthropogenic PM2.5 concentrations are expected to be offset by increasing wildfire emissions globally (Mehta et al., 2018) and in the SE US especially (Ford et al., 2018). However, overall OM and PM2.5 concentrations in the present study period have declined, demonstrating that any enhancement in overall OM due to biomass burning emissions had not yet exceeded the impact of regulatory success in decreasing air pollution emissions. In agreement with this, smoke OM also did not appear to change appreciably between 2000 and 2011 (Blanchard et al., 2013a).
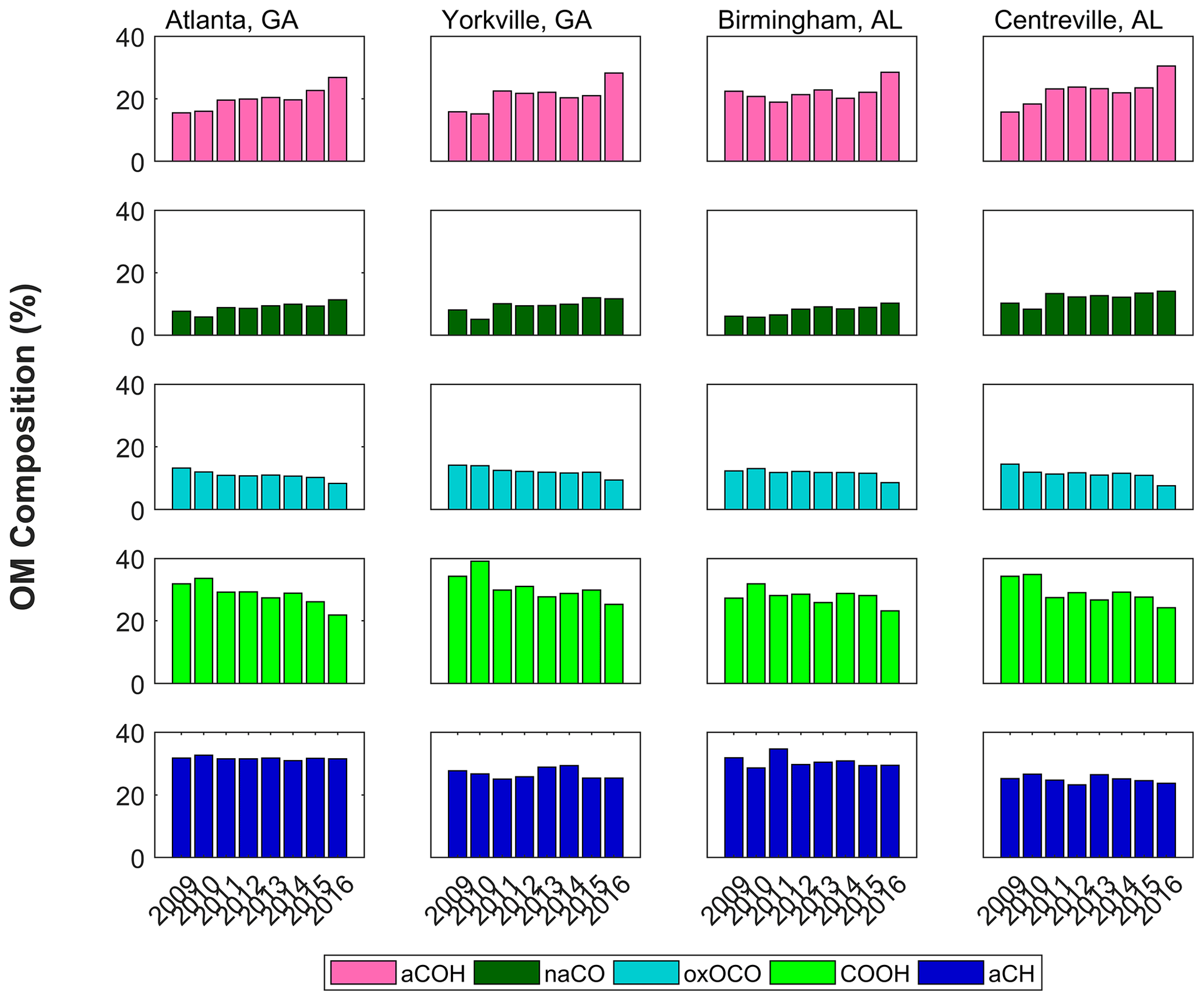
Figure 3Annual median FG fractions of OM composition as quantified via FT-IR spectrometry. Column-wise subplots correspond to individual sites.
3.2 Multi-year trends of OM composition using functional groups: observations
Further clues as to the changes in atmospheric chemistry leading to the overall decrease in OM can be drawn from the FG composition measured using FT-IR spectrometry. The FGs did not decline uniformly with OM over the 2011–2016 time period; rather, the composition changed, suggesting a change in dominant sources and atmospheric processes. As shown in Fig. 3, the contributions of the more oxygenated FGs, COOH and oxOCO, decreased, while those of the moderately oxygenated FGs, aCOH and naCO, became slightly enriched (aCH contributions did not change significantly). The FG composition in the SE US was dominated by aCH and COOH, each contributing between 24 % and 35 % of OM. The aCOH contributed between 16 % and 30 %, while oxOCO and naCO contributed less than 15 % over all sites and years (Fig. 3).
Trends in FG measurements by season and site helped to elucidate influences on atmospheric chemistry. Site-specific trends demonstrated in general that urban area samples changed most over time (with the exception of oxOCO, but in agreement with overall OM trends), likely reflecting the availability of carbon sources and/or oxidants near cities. Larger changes in more oxygenated FG concentrations occurred in summers, dominated by the larger magnitude declines in COOH (0.12±0.07 ; Table S1) and oxOCO (). In contrast, the positive changes in naCO and aCOH concentrations were steepest in the spring (0.018±0.019 ; 0.03±0.04 ) followed by autumn (0.009±0.020 ; 0.01±0.04 ; although these increases were not statistically significant, their fractional changes were). This indicates that the losses of more oxygenated OM concentrations occurred mainly in summer, while the increases in the less oxygenated OM contributions occurred in the shoulder seasons.
The multi-annual trends in FG abundance, as well as those through 2016, were mirrored in the sample FT-IR spectra (Fig. 4).
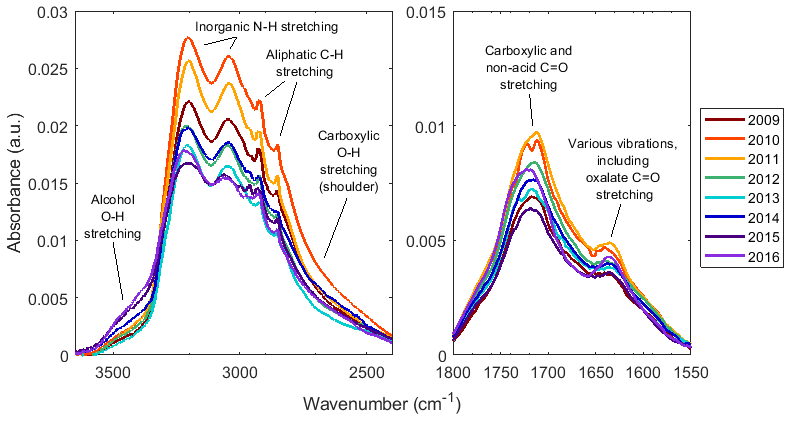
Figure 4Annual median baseline corrected spectra of samples collected at JST. Colored lines, as indicated in the legend, demonstrate a progression of absorbance over time. For baseline correction and blank spectrum subtraction details, see Sect. S5.
To further explain the changing composition over time, Fig. 4 shows the annual median spectra for one site over time. An absorbance peak at approximately 3500 cm−1 corresponding to the aCOH O−H stretching absorption, increased, while the shoulder between ∼2650 and 2900 cm−1 corresponding to COOH O−H stretching decreased in absorbance. In the right subplot of Fig. 4, the C=O stretching region (from naCO, but also COOH groups) absorbance increased from 2009 to 2011 and demonstrated a general decline after 2011. Similarly, the absorbance peaks due to inorganic N−H stretches and aCH (the sharp peaks between ∼2800 and 3000 cm−1 correspond to aCH C−H stretching absorption) were greatest in 2010, and that due to vibrations including oxOCO C=O stretching at ∼1630 cm−1 was greatest in 2011. Note that although their absorption spectra are quite different (Hay and Myneni, 2007), COOH and oxOCO are related: the origin of oxOCO in OM is likely deprotonation of COOH and coordination with another cation such as Na+ or (Ortiz-Montalvo et al., 2014) or within metal complexes (Sorooshian et al., 2013). These same trends were also generally shown by spectra from other sites (Sect. S5).
The impacts of filter storage on sample FG concentrations were examined. As stated above, filters were stored and shipped at <4 ∘C to minimize changes to aerosol composition and mass. Within the annual median trends in contributions to OM (Fig. 3), the 2016 annual median composition is notably different from that in 2015. The 2016 samples were freshest when analyzed via FT-IR spectrometry; to evaluate possible analytical causes of this difference, storage or sample handling losses were considered. SEARCH samples from 2011 and 2016 were re-analyzed via FT-IR spectrometry 1–2 years after the original analysis date (performed and reported in Part 1 of this paper series, Boris et al., 2019). Fresh (2016) samples were subject to some loss of OM, likely due to storage and handling (in 28 % of the samples, OM concentrations showed losses greater than the sampling uncertainty of 0.39 µg m−3 after 2 years). Most of this loss was due to a decline in aCOH (in 52 % of the samples, aCOH concentrations showed losses greater than the sampling uncertainty of 0.13 µg m−3 in the same 2-year period). Thus, considering the shift between 2015 and 2016 OM concentrations (and contributing FGs), we cannot rule out the possibility that the difference could have been explained by losses during storage. However, as an explanation for the overall multi-annual decline in COOH, oxOCO, and OM, results after longer-term storage do not support the losses of organic species from filters. After 2 years of storage for older samples (2011), the changes in concentrations for OM, COOH, and oxOCO were within sampling uncertainties for 85 %, 84 %, and 89 % of samples, respectively (sampling uncertainties were 0.15 µg m3 for COOH and 0.04 µg m3 for oxOCO). In addition, winter 2018 OM composition demonstrated a continuation of trends in naCO and aCOH (January and February; Fig. S5 in the Supplement). While a positive trend was not observed in naCO and aCOH fractions of OM in all seasons with 2016 excluded (2011–2015), the trends during spring 2011–2015 were still positive (aCOH: 0.030±0.007 % of OM; naCO: 0.018±0.003 % of OM; Table S1). Given these observations, the trends in FG concentrations and composition appear to be robust. In addition, confidence is added to the measurements by the increasing 2009/10 trend in OM concentrations, which demonstrates an opposing trend to the declines in later years (due to the 2008 recession; Sect. 3.1).
3.3 Multi-year trends of OM composition using functional groups: interpretation
These annual and seasonal FG measurements, mirrored by the FT-IR spectra themselves, concisely demonstrate trends in OM composition. They provide observations useful in identifying OM sources and formation processes and tie together studies of SE US aerosol with more limited scopes in time or chemical analysis. The following paragraphs provide interpretation of the trends in FG contributions to OM informed by the current results, as well as a variety of laboratory, modeling, and campaign-based studies relevant to the SE US OM composition. While FGs are not specific to any particular emission sources or processes, their variations can provide insight into atmospheric processes. Therefore, past FT-IR spectrometry source attribution studies with factors from positive matrix factorization (PMF; e.g., Corrigan et al., 2013; Russell et al., 2009; Schwartz et al., 2010; Takahama et al., 2011) are considered in particular to help guide the discussion of trends.
3.3.1 Interpretation of multi-year decrease in COOH and oxOCO contributions to OM
The most distinct trend in FG composition is a decline of COOH and oxOCO abundance over time (−0.9 ; −0.59 ; Table S1). Since oxOCO has been studied less in previous work, and fewer literature sources are available, we focus here on COOH. COOH is associated with highly oxidized material (e.g., Chen et al., 2021), and past FT-IR spectrometry studies have shown that elevated COOH contributions often originate from aged combustion emissions, particularly from fossil fuels (at Whistler, British Columbia: ∼30 %, Schwartz et al., 2010, and ∼45 %, Takahama et al., 2011; ∼42 % in southern central California, Liu et al., 2011; ∼33 % in Houston area oil combustion, Russell et al., 2009; and ∼40 %–45 % in a heavily forested region of Finland, Corrigan et al., 2013). This suggests the contribution of anthropogenic emissions to the declines in OM observed in the present work. Two FT-IR factors found in SE US aerosol to be elevated in COOH contributions, aged combustion and mixed organic aerosol, were also correlated with concentrations (Liu et al., 2018), further suggesting a shared fossil fuel combustion source and/or oxidation process between the regional declining OM and . In the present work, FT-IR COOH and oxOCO concentrations were correlated with those of (R2=0.48 and R2=0.54, respectively), and both species declined greatest at an urban site (BHM). Based on this and previous FG measurements, the decreasing trends in COOH and oxOCO may thus be attributed directly to regionally declining emissions of anthropogenic VOCs and/or indirectly to SO2 emissions declines (discussed further in Sect. S1.2; see also Marais et al., 2017; Schindelka et al., 2013; Xu et al., 2015b; Zheng et al., 2020).
3.3.2 Interpretation of multi-year increase in aCOH and naCO contributions OM
The other distinct trend in FG composition is the relative enhancement in the aCOH and naCO over time (+1.2 ; +0.45 ; Table S1). These trends oppose the decline in abundance observed for most measured anthropogenic pollutants, including NOx, SO2, and the contribution of to PM2.5, and may indicate a shift in the dominant sources and/or processes contributing to OM. An aggregation of field and lab observations from the literature regarding the trends in aCOH and naCO point to oxidized monoterpenes as a source of carbon and possible chemical formation processes by low NOx oxidants (Sect. S1.3). FT-IR spectrometry and aerosol mass spectrometry results from the SE US have demonstrated an association between less oxidized OM and monoterpene emissions (Corrigan et al., 2013; Schwartz et al., 2010; Takahama et al., 2011). The seasonalities of aCOH and naCO concentration changes also support contributions from monoterpene emissions from shoulder season events (Geron and Arnts, 2010; Kim, 2001). The production of secondary biogenic OM varies with NOx concentrations in the SE US (Liu et al., 2018; Xu et al., 2018; Zhang et al., 2018), and modeling studies (Mao et al., 2018) and SEARCH data suggest that the region is shifting from NOx-dominated to O3-dominated (and thus •OH) chemistry. These changes in oxidant and oxidation product distributions (Pullinen et al., 2020; Pye et al., 2019; Ziemann and Atkinson, 2012) could be causing the relative enhancement of less oxygenated FGs. Even while the reaction of monoterpenes with O3 is slower than •OH and at peak oxidant concentrations (e.g., Ziemann and Atkinson, 2012), oxidation by O3 can be sustained when these other oxidants are found in lower abundance (Peräkylä et al., 2014) and generate secondary OM with lower volatility (Watne et al., 2017).
Smoke is another plausible contributor of the measured aCOH and naCO since emissions from prescribed burns, wildfires, and residential wood burning contribute substantially to SE US aerosol (Peräkylä et al., 2014), and aCOH and naCO have been associated with biomass burning (Corrigan et al., 2013; Liu et al., 2018, 2009; Russell et al., 2011; Takahama et al., 2011). However, neither the enhancement in smoke over the study period (prescribed and wildland fires may actually have decreased in acreage; Melvin, 2012, 2015, 2018) nor a clear enhancement in these FGs during fire periods could be demonstrated (Sects. 3.6; S1.3.1). This may be related to the detection method used for identifying smoke plumes (Sect. 2.3) or to effects of dilution or aging on the FG contributions (Russell et al., 2011). Likewise, residential wood burning was concluded not to be a responsible factor because the expected wintertime increase was not in line with FG trend seasonality. Other sources of aCOH and naCO, including isoprene epoxydiol reactions, organonitrate hydrolysis, and O3 enhancements due to climatological changes, are also possible (Sect. S1.3.1).
3.4 Annual and seasonal trends in the Ratio
Seasonal and annual median values of varied between ∼2.0 and 2.4 (Fig. 5a), greater than some typical values used for TOR OC conversion in many networks (e.g., 1.8 or 1.4; Blanchard et al., 2013b; Malm et al., 2017) but similar to those used in more current work (Marais et al., 2017). The FT-IR-derived values are similar to those measured in previous work on SE US aerosol: an of ∼2.15 was observed by El-Zanan et al. (2009) using masses of solvent extracts and a mass balance method, ∼1.9 was observed using re-suspension and high-resolution aerosol mass spectrometry by Sun et al. (2011b), ∼1.8–2.2 was observed by Hand et al. (2019) for 2009–2016 IMPROVE network data using a multiple regression method, and 2.24 was observed using direct aerosol mass spectrometry by Kim et al. (2015). Especially with respect to values, it must be noted that some molecular bonds are not captured using FT-IR spectrometry: for example, the organic and groups that might be particularly relevant in discussions of SE US aerosol are not included in the OM contribution. However, the majority of OM and OC masses are captured (Boris et al., 2019).

Figure 5, expressed as (a) seasonal median ratios at urban and rural grouped sites and (b) annual median functional group contributions to ratios at each site.
During the summertime, when photochemistry is enhanced, the degree of OM oxidation is generally expected to be greater than in months with less solar insolation (Blanchard et al., 2008). Likewise, a greater degree of oxidation is anticipated at rural sites due to aging of urban pollutants. Median ratios were greater in summers than in winters and also greater at rural than at urban sites, in agreement with these expectations (Fig. 5).
The competing impacts of the changes in FG concentrations resulted in a trend in the that was not statistically significant ( yr−1 2011–2016; Fig. 5b; Sect. S2). In parallel to absolute trends of FGs (Sect. 3.2), material contributed to by COOH and oxOCO declined during this period, while there was an enhancement in aCOH and naCO concentrations (the aCH contribution was statistically unchanged; yr−1). Trends in for each of the sites and seasons were also not significant. Using a similar measurement technique, Bürki et al. (2020) showed no trend in for annual medians of the 2011 and 2013 IMPROVE network sites, including at St. Marks, Florida. However, the FG contributions did not appear to vary, in contrast to the findings of the current calibration models and SEARCH dataset.
Differing from FG trends, the work of Hand et al. (2019) using a multiple linear regression method demonstrated an increase in of SE US aerosol between 2011 and 2014, and a decrease between 2014 and 2016. These trends were attributed to a change in the character of OM or OC that were ubiquitous throughout the US and over all seasons, to analytical challenges in the various instrumental or collection methods, or to calculation and algorithm challenges of reconstructed fine mass composition. The trend in residual OM and TOR OC also increased with time ( yr−1 between 2011 and 2016; see Sect. 2.6 for residual OM definition), further suggesting that FG provides different results from determined using TOR OC and IMPROVE data. Using the residual OC concentrations, the median value of was also only 1.65, lower than anticipated based on other methods (see above; e.g., El-Zanan et al., 2009; Hand et al., 2019).
Efforts to apply multiple linear regression methods to 2011–2016 SEARCH data similar to Hand et al. (2019) suggested a systematic discrepancy in the results: derived regression coefficients were not physically meaningful with respect to expected chemistry (Simon et al., 2011). Possible explanations for the lack of physically meaningful results included volatilization of nitrate (although nitrate was expressed as the sum of the Teflon and backup nylon filters; Edgerton et al., 2005b), inadequate accounting for particle water (although this was approximated, as in Part 1 of this work, Boris et al., 2019) and/or TOR OC concentration abnormalities due to hardware degradation or changes in methodology (Hand et al., 2019). These limitations notably did not prohibit calculation of using FT-IR spectrometry measurements, since the OM and OC are estimated directly from FT-IR spectra of samples and collected chemical standards.
In addition to , FG measurements were used to calculate the carbon oxidation state (OSC) as a metric of degree of oxidation, demonstrating a lack of trend, as with ratios. Values of OSC were within the range of semi-volatile or low-volatility oxygenated OM but were more oxidized than aerosol from urban sources or biomass burning (Kroll et al., 2011; Sect. S3).
3.5 Trends and changes in seasonal OM composition
Summertime aerosol carbon in the SE US is predominantly from biogenic sources, with particularly strong seasonality in isoprene emissions (Guenther et al., 1994; Xu et al., 2015a). The greater photochemistry and stagnant conditions in the summertime in the SE US are also favorable for enhanced secondary OM production, with high OM concentrations originating from urban and remote sources (Hidy et al., 2014; supporting meteorological observations are summarized in Sect. S13; select wind roses are shown in Sect. S6). Absolute FG OM concentrations were greater during the long SE US warm season than in colder months (Fig. 6), despite the higher boundary layer during the warmer months in the SE US (Seidel et al., 2012). While the seasonality of all FG concentrations generally matched that of OM, with greater abundances in the summer and autumn than winter for all years, the relative contribution of COOH to OM was significantly greater in the summer (Figs. 6; S8). The greater summertime OM concentration and COOH fraction of OM support enhanced oxidation and isoprene emission as a prominent source of COOH in warmer months (in line with Sect. 3.3.1 findings).
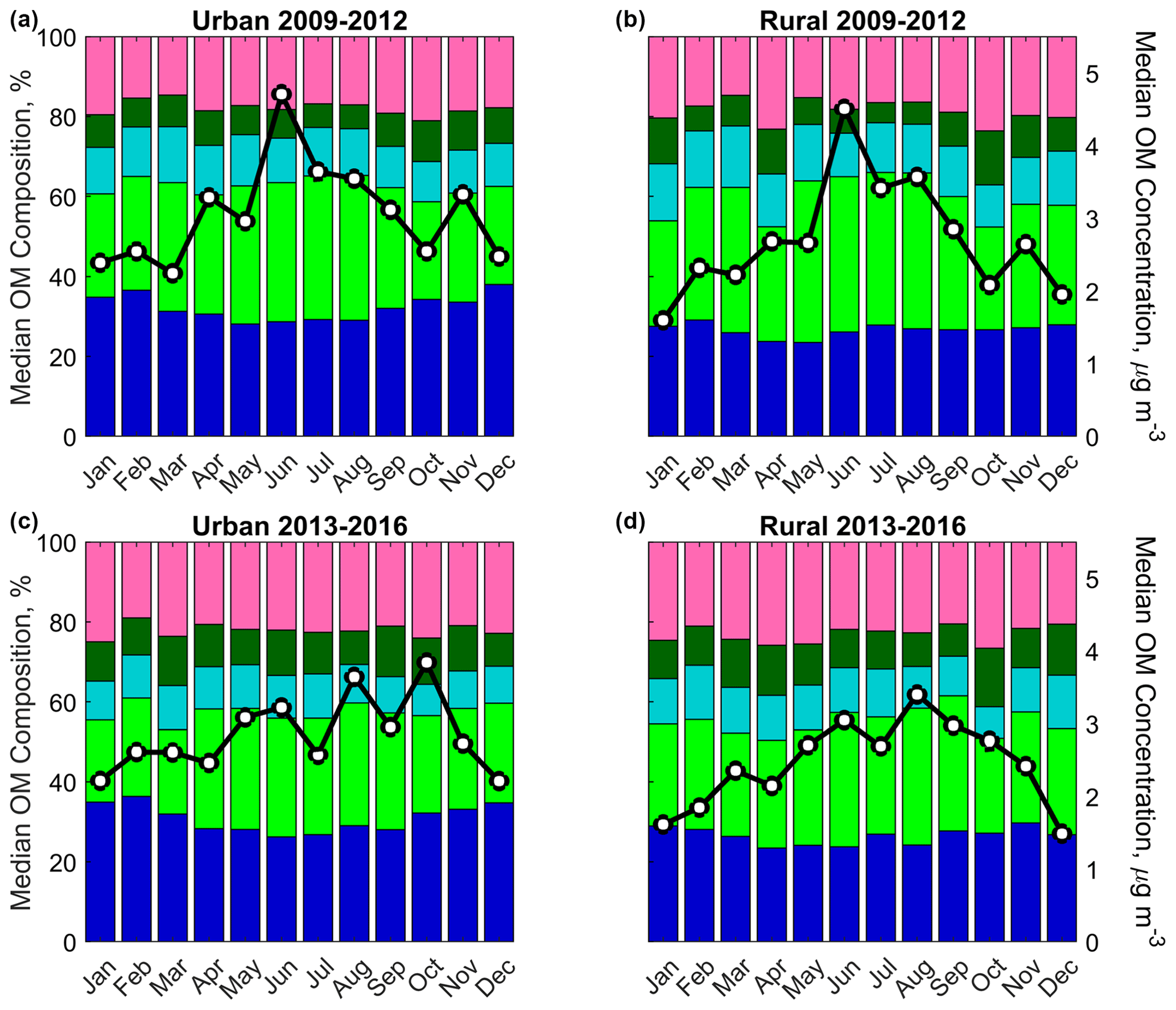
Figure 6Monthly FG composition and OM concentration during 2009–2012 (a, b) and 2013–2016 (c, d) and urban (a, c) and rural (b, d) subcategories. Stacked color bars represent the monthly median FG contributions to OM concentration (see Fig. 3 for FG legend), while black lines with white markers represent the monthly median OM concentrations.
In contrast, markers of fossil fuel combustion and wood burning have been measured at greater concentrations in the winter and spring in the SE US (Blanchard et al., 2014; Sun et al., 2011a). FT-IR spectrometry results mirrored these observations: the relative contribution of aCH to OM was greatest in the wintertime (Fig. S8), and the urban/rural ratios of OM concentrations were greater in the cold months (∼1.2–1.4 December–February) than the warm months (1.0–1.2 June–August; Georgia sites; Fig. S11). Indicative of a seasonality of the carbon emissions instead of secondary production (Sun et al., 2011a), the same seasonality was not observed in urban/rural ratios of concentration (Fig. S12). The impact of aged combustion emissions was also demonstrated by the elevated cold-season particle concentrations observed (2013–2016 monthly medians; e.g., Fig. S15; Kleeman et al., 2005). These results suggest that anthropogenic and combustion sources were more prominent during the colder months in the SE US and that the contribution of the FG aCH was indicative of those sources.
An annual peak in monthly median OM concentrations was observed in June in the earlier years, but late summer/early autumn months in the later years (Fig. 6; with notable variation between years: 2009 and 2011 have the most prominent June peaks, but 2016 also has a small June peak, and the most prominent late summer or autumn peak was in 2013; Fig. S8). The June peak was also observed in all FG concentrations 2009–2012, while greater aCOH, naCO, and aCH concentrations were observed in autumn 2013–2016 (Fig. S9). Since the seasonality of monoterpene-related OM is much less pronounced than isoprene OM in the SE US (as measured via aerosol MS; Xu et al., 2018), this seasonality of aCOH and naCO particularly supports the hypothesis put forward in Sect. 3.3.2 (which was based on other FT-IR spectrometry work; Russell et al., 2011) that these FGs are predominantly attributable to monoterpene oxidation products.
3.6 The 2016 daily dataset: observations of warm season stagnation and cold-season combustion emissions
FT-IR spectra were collected for all sites and each day in 2016 until site closure (Sect. 2.1). This rich dataset allowed identification of trends across seasons and additional comparison between fire and non-fire periods. Weekly trends were also observed in OM and FG concentrations using this higher-resolution data. While weekend concentrations were lowest only for NOx, species including all FGs, OM, TOR OC and EC, x-ray fluorescence K, and CO were lowest in concentration on a midweek day, generally Tuesdays (medians during winters and summers, 2013–2016, at urban and rural sites). This observation may be related to feedbacks with regional rainfall (Bell et al., 2008), although there was no clear pattern between weekly concentration minima and hygroscopicity of species (Sect. S14).
Lower than normal precipitation and higher than normal temperatures in 2016 led to stronger drought conditions and wildfires, especially during the latter part of the year, although prior years were also dry in the SE US (Konrad and Knox, 2018). OM concentrations were dynamic, with build-up and flushing out of material on a more rapid timescale in the cold season than in the warmer months. The source of the dynamic OM concentrations appeared to differ by season as well, with smoke and secondary OM the most likely controls on OM variation in the cold and warm seasons, respectively.
While some work has demonstrated a difference in the and in fire-impacted samples (Corrigan et al., 2013; Russell et al., 2011), no FG “fingerprint” for smoke was apparent in the present work. The differences between FG contributions for which smoke was visible in satellite images versus those with no indication of smoke impact were inconsistent (Sect. S1.3.1; Fig. S1): in 2016 samples with suspected fire impact, and were significantly higher, and , , and were significantly lower than in samples with no indication of fire impact; in all samples from 2011 to 2016, only the observations for and (higher in suspected fire impact samples) were significant. The lack of “fingerprint” may be due to the subjective nature of the satellite categories used in this analysis (Sect. 2.3), dilution or other obfuscating factors such as aging of the smoke plume, type of burn (flaming, smoldering, crop type, etc.), or the environmental conditions. These results contradict the alternative hypothesis that the multi-annual increases in naCO and aCOH concentrations are due to smoke, despite the relationships identified in prior FT-IR spectrometry factor analysis work (Sect. 3.3.2).
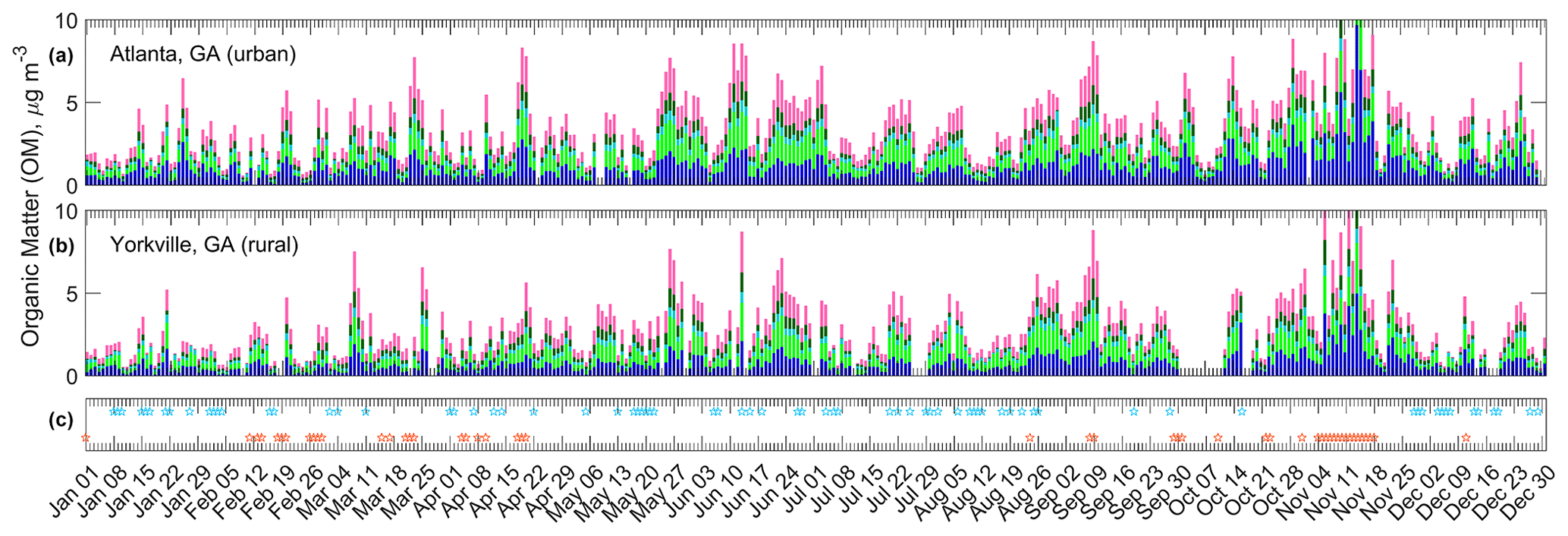
Figure 7Absolute OM and FG concentrations during 2016 for the urban (a) and rural (b) Georgia sites. Some values (three at each site) extend beyond the vertical axis scale so that variation can be observed within low and typical OM concentration ranges. Events related to OM concentration dynamics are represented in (c): precipitation events correspond to blue markers; fires correspond to orange markers. See Fig. 2 for legend FG colors.
Winter and spring (1 January through 20 April; Fig. 7) were distinct in that smoke plumes visible in satellite imagery were often coincident with high OM concentrations. Prescribed burning likely explains this observation: it is widespread in the SE US between January and April on days when conditions are appropriate (Larkin et al., 2014). Events in precipitation and related meteorological variables typically coincided with decreases in OM concentrations after elevated OM periods.
In contrast, during the late spring and summer (21 April–25 August), the contribution of OM from prescribed burning sources is expected to be minimal because burns of most types are not permitted in Alabama and Georgia (Alabama Forestry Commission, 2020; Georgia Environmental Protection Division, 2020). Convective activity in the summer also brings regular rain (Diem and Mote, 2005), and local wildfires are uncommon relative to other seasons (Larkin et al., 2014). Visible satellite imagery showed no smoke plumes during the 2016 late spring and summer. Summer OM events were likely caused by secondary OM formation with carbon originating from biogenic sources (Sun et al., 2011a), as discussed in Sect. 3.5, or from aged smoke transported from wildfires in the western US. The high OM events were longer in duration than those in the winter and spring: springtime events were on the order of a few days and were punctuated by frontal passages, while summertime events sometimes lasted 1.5 weeks, interrupted by less frequent meteorological events (the latter was also observed by Liu et al. (2018).
Periods of OM build-up also occurred during the autumn (26 August–31 October), but smoke plumes were again visible using satellite imagery, indicating that local fires may have contributed to OM. Wood smoke appeared to be ubiquitous during the winter season in the SE US (defined here as 31 October to 31 December) based on observed elevated in the winters (Fig. S17 and supported in prior work; Chen et al., 2012), although the overlap of other combustion sources such as vehicle emissions is probable. Severe wildfires in the southeastern Appalachian Mountains occurred at the end of October and into mid-November, due in part to a drought (Konrad and Knox, 2018), resulting in the highest seasonal OM concentrations observed in 2016. In agreement with the lack of smoke “fingerprint” in the absolute concentrations of FGs, in the 2009–2016 1:3 d dataset no correlations between FG concentrations and non-soil K were observed (; non-soil K calculated from XRF K and Si concentrations; Blanchard et al., 2016a). Additional precision in identifying smoke impact on FG composition may be possible by performing factor analysis (Russell et al., 2011).
Multi-annual, seasonal, and daily trends in OM concentrations and composition were examined in PM2.5 from the SE US using FT-IR spectrometry and PLS regression. OM concentrations declined at all SEARCH sites from 2011 to 2016, due mainly to decreasing concentrations of COOH and oxOCO, while the relative contributions to OM of aCOH and naCO increased. Annual median FT-IR spectra corroborated the trends in FG concentrations, as did monthly and seasonal observations. Monthly median OM concentrations were generally higher in the warmer months, in line with predominant biogenic secondary OM sources; this was additionally corroborated by the greater COOH contribution to OM in the summers. A greater aCH contribution to OM was observed in the winters, as were higher urban/rural ratios of OM, suggesting more anthropogenic carbon emissions during the cold season. The two observed FG trends were suggested to originate from separate phenomena. Declines in the more oxygenated FGs were attributed to declining secondary OM from anthropogenic VOC emissions and/or SO2 concentrations; the latter directly affects concentrations and implies one of a handful of indirect mechanisms suggested elsewhere (Blanchard et al., 2016; Marais et al., 2017; Xu et al., 2015b). These hypothesized sources of the more oxygenated FG declines were supported by several observations: (a) summertime SE US OM measured in other studies had predominant fractions from -associated OC and isoprene (Blanchard et al., 2016; Xu et al., 2015b); (b) COOH was prominent in mixed oxidized OM and fossil fuel combustion OM in prior FT-IR spectrometry studies (Sect. 3.3.1); (c) the maximal declines in OM and oxidized FG (oxOCO and COOH) concentrations were in summertime, suggesting an isoprene carbon source and/or a photochemical formation mechanism; and (d) the decline spatially matches that of , another secondary product of the atmosphere. While there are multiple plausible hypotheses for the source of the aCOH and naCO fractions of OM, which trended separately from the more oxygenated FGs, the enhancement in low-NOx secondary OM from monoterpene emissions is most likely based on prior factor-based FT-IR spectrometry literature on these two FGs. Increasing aCOH and naCO concentrations corresponded seasonally with monoterpene emission events and enhancements in O3 concentrations, suggesting either a link between NOx and O3 in this pathway, or that O3 may be independently involved in the increase in FG concentrations.
estimated from functional group measurements did not significantly change over time due to the competing enhancement or decline of the oxygenated FGs. values were greater in summers and at rural sites (as anticipated), and the magnitudes were in agreement with previous work using a similar methodology (Bürki et al., 2020). In contrast, the trend in ratios previously estimated by incorporating TOR OC measurements and other techniques (Hand et al., 2019) differed from that observed in the current study. Additional comparisons of measurements should be made.
Events such as fires and stagnation, which have been suggested by chemical tracers and meteorological data elsewhere (Hidy et al., 2014; Zhang et al., 2010; Zheng et al., 2002), can be visualized in the daily OM and FG concentrations. While trends in absolute OM concentrations were often dominated by anthropogenic and/or biogenic secondary OM, variability in the shorter term (as demonstrated by daily data in 2016) was often related to smoke and precipitation events. During the winter and spring (January through April), smoke from prescribed burning activities appears to have contributed to many of the higher OM concentration samples. Summertime (April through August) OM concentrations showed periods of build-up due to secondary biogenic OM followed by rainout. Severe fires in November generated the greatest 2016 daily OM concentrations, but elevated aCH contributions to the OM persisted throughout November and December, perhaps suggesting residential wood burning emissions (which are a substantial source of OM in the SE US; Chen et al., 2012; Sun et al., 2011b). A fire “fingerprint” of FG composition was not, however, apparent in the present work, despite the clear relationship with overall OM concentrations, perhaps due to dilution or other obfuscating factors.
This work additionally served to evaluate the applicability of FT-IR spectrometry and PLS-based FG models for routine OM characterization, air quality monitoring networks, or extended studies. As also validated in Part 1 of this set of papers (Boris et al., 2019), the measurements demonstrate general similarity in magnitude to other OM characterization studies.
This work adds to the growing literature on SE US aerosol formation, including its evolution as anthropogenic emissions decrease. These FT-IR spectrometry and PLS analysis results corroborate the presence of multiple fractions of OM as observed in other work (Liu et al., 2018; Xu et al., 2015b), but highlight the different trajectories of those fractions within the 2011–2016 time frame. The agreement between FG data and other work supports the application of FT-IR and PLS methodology to routine (multi-annual) studies, which has not been investigated before. In particular, results demonstrated the success of pairing the concise information provided by FGs with shorter-term studies that used more intensive techniques (such as aerosol mass spectrometry or organic molecular tracer analyses). The daily patterns observed in the present work also demonstrate the utility of characterizing FG OM to easily elucidate shorter-term carbonaceous aerosol dynamics, including the impacts of fires and meteorological events.
The functional group, OM, and OC concentrations with uncertainties, as well as raw spectra, are available at https://doi.org/10.25338/B8SG73 (Dillner et al., 2019).
The supplement related to this article is available online at: https://doi.org/10.5194/amt-14-4355-2021-supplement.
AMD and SLS conceived of the project. AMD provided leadership for the project, including mentoring and supervising AJB in data analysis, interpretation, and final review prior to publication. AJB performed the data analysis and interpretation, created the figures, and wrote and edited the manuscript. ST provided additional mentoring to AJB on data analysis and interpretation and editing of the manuscript. ATW and BMD provided input on the data analysis and reviewed the paper. ESE and SLS provided SEARCH network filters and data. TJ and NLN provided additional samples for the Atlanta site after the SEARCH network was shut down and reviewed the paper.
The authors declare that they have no conflict of interest.
Funding for this project was generously provided by the Electric Power Research Institute, with equipment and logistical support from Atmospheric Research & Analysis, Inc. Taekyu Joo and Nga L. Ng acknowledge support from CAREER AGS-1555034. Special thanks to Kelsey Seibert, who skillfully managed the collection of FT-IR spectra and sample handling. Thank you to Matthew Coates, Martin Esparza-Sanchez, Carley Fredrickson, Nathaniel Hopper, and Alex Williams, who worked to collect calibration standards and perform various essential data analysis functions. The authors also acknowledge Jenny Hand for her guidance on calculation, Charlotte Burki for her work in calculating the ratios via regression methods, and Sean Raffuse for his support with fire detection methods and data.
This research has been supported by the Electric Power Research Institute (grant no. 10003745). Taekyu Joo and Nga L. Ng have been supported from CAREER AGS-1555034.
This paper was edited by Charles Brock and reviewed by two anonymous referees.
Alabama Forestry Commission: Current Burn Restrictions: https://forestry.alabama.gov/Pages/Fire/BurnRestrictions.aspx, last access: 16 May 2021.
Al-Naiema, I. M., Hettiyadura, A. P. S., Wallace, H. W., Sanchez, N. P., Madler, C. J., Cevik, B. K., Bui, A. A. T., Kettler, J., Griffin, R. J., and Stone, E. A.: Source apportionment of fine particulate matter in Houston, Texas: insights to secondary organic aerosols, Atmos. Chem. Phys., 18, 15601–15622, https://doi.org/10.5194/acp-18-15601-2018, 2018.
Bell, T. L., Rosenfeld, D., Kim, K.-M., Yoo, J.-M., Lee, M.-I., and Hahnenberger, M.: Midweek increase in US summer rain and storm heights suggests air pollution invigorates rainstorms, J. Geophys. Res., 113, D02209, https://doi.org/10.1029/2007JD008623, 2008.
Blanchard, C. L., Hidy, G. M., Tanenbaum, S., Edgerton, E., Hartsell, B., and Jansen, J.: Carbon in southeastern US aerosol particles: Empirical estimates of secondary organic aerosol formation, Atmos. Environ., 42, 6710–6720, https://doi.org/10.1016/j.atmosenv.2008.04.011, 2008.
Blanchard, C. L., Tanenbaum, S., and Hidy, G. M.: Source attribution of air pollutant concentrations and trends in the Southeastern Aerosol Research and Characterization (SEARCH) network, Environ. Sci. Technol., 47, 13536–13545, https://doi.org/10.1021/es402876s, 2013a.
Blanchard, C. L., Hidy, G. M., Tanenbaum, S., Edgerton, E. S., and Hartsell, B. E.: The Southeastern Aerosol Research and Characterization (SEARCH) study: Temporal trends in gas and PM concentrations and composition, 1999–2010, J. Air Waste Manage., 63, 247–259, https://doi.org/10.1080/10962247.2012.748523, 2013b.
Blanchard, C. L., Chow, J. C., Edgerton, E. S., Watson, J. G., Hidy, G. M., and Shaw, S.: Organic aerosols in the southeastern United States: Speciated particulate carbon measurements from the SEARCH network, 2006–2010, Atmos. Environ., 95, 327–333, https://doi.org/10.1016/j.atmosenv.2014.06.050, 2014.
Blanchard, C. L., Hidy, G. M., Shaw, S., Baumann, K., and Edgerton, E. S.: Effects of emission reductions on organic aerosol in the southeastern United States, Atmos. Chem. Phys., 16, 215–238, https://doi.org/10.5194/acp-16-215-2016, 2016.
Boris, A. J., Takahama, S., Weakley, A. T., Debus, B. M., Fredrickson, C. D., Esparza-Sanchez, M., Burki, C., Reggente, M., Shaw, S. L., Edgerton, E. S., and Dillner, A. M.: Quantifying organic matter and functional groups in particulate matter filter samples from the southeastern United States – Part 1: Methods, Atmos. Meas. Tech., 12, 5391–5415, https://doi.org/10.5194/amt-12-5391-2019, 2019.
Budisulistiorini, S. H., Li, X., Bairai, S. T., Renfro, J., Liu, Y., Liu, Y. J., McKinney, K. A., Martin, S. T., McNeill, V. F., Pye, H. O. T., Nenes, A., Neff, M. E., Stone, E. A., Mueller, S., Knote, C., Shaw, S. L., Zhang, Z., Gold, A., and Surratt, J. D.: Examining the effects of anthropogenic emissions on isoprene-derived secondary organic aerosol formation during the 2013 Southern Oxidant and Aerosol Study (SOAS) at the Look Rock, Tennessee ground site, Atmos. Chem. Phys., 15, 8871–8888, https://doi.org/10.5194/acp-15-8871-2015, 2015.
Bürki, C., Reggente, M., Dillner, A. M., Hand, J. L., Shaw, S. L., and Takahama, S.: Analysis of functional groups in atmospheric aerosols by infrared spectroscopy: method development for probabilistic modeling of organic carbon and organic matter concentrations, Atmos. Meas. Tech., 13, 1517–1538, https://doi.org/10.5194/amt-13-1517-2020, 2020.
Carlton, A. G., Pinder, R. W., Bhave, P. V., and Pouliot, G. A.: To what extent can biogenic SOA be controlled?, Environ. Sci. Technol., 44, 3376–3380, https://doi.org/10.1021/es903506b, 2010.
Carlton, A. G., de Gouw, J., Jimenez, J. L., Ambrose, J. L., Attwood, A. R., Brown, S., Baker, K. R., Brock, C., Cohen, R. C., Edgerton, S., Farkas, C. M., Farmer, D., Goldstein, A. H., Gratz, L., Guenther, A., Hunt, S., Jaeglé, L., Jaffe, D. A., Mak, J., McClure, C., Nenes, A., Nguyen, T. K., Pierce, J. R., de Sa, S., Selin, N. E., Shah, V., Shaw, S., Shepson, P. B., Song, S., Stutz, J., Surratt, J. D., Turpin, B. J., Warneke, C., Washenfelder, R. A., Wennberg, P. O., and Zhou, X.: Synthesis of the southeast atmosphere studies: Investigating fundamental atmospheric chemistry questions, B. Am. Meteorol. Soc., 99, 547–567, https://doi.org/10.1175/BAMS-D-16-0048.1, 2018.
Chen, Y., Zheng, M., Edgerton, E. S., Ke, L., Sheng, G., and Fu, J.: PM2.5 source apportionment in the southeastern US: Spatial and seasonal variations during 2001–2005, J. Geophys. Res., 117, 1–12, https://doi.org/10.1029/2011JD016572, 2012.
Chen, Y., Takeuchi, M., Nah, T., Xu, L., Canagaratna, M. R., Stark, H., Baumann, K., Canonaco, F., Prévôt, A. S. H., Huey, L. G., Weber, R. J., and Ng, N. L.: Chemical characterization of secondary organic aerosol at a rural site in the southeastern US: insights from simultaneous high-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS) and FIGAERO chemical ionization mass spectrometer (CIMS) measurements, Atmos. Chem. Phys., 20, 8421–8440, https://doi.org/10.5194/acp-20-8421-2020, 2020.
Chen, Y., Guo, H., Nah, T., Tanner, D. J., Sullivan, A. P., Takeuchi, M., Gao, Z., Vasilakos, P., Russell, A. G., Baumann, K., Huey, L. G., Weber, R. J., and Ng, N. L.: Low-Molecular-Weight Carboxylic Acids in the Southeastern U.S.: Formation, Partitioning, and Implications for Organic Aerosol Aging, Environ. Sci. Technol., 55, 6688–6699, https://doi.org/10.1021/acs.est.1c01413, 2021.
Chhabra, P. S., Ng, N. L., Canagaratna, M. R., Corrigan, A. L., Russell, L. M., Worsnop, D. R., Flagan, R. C., and Seinfeld, J. H.: Elemental composition and oxidation of chamber organic aerosol, Atmos. Chem. Phys., 11, 8827–8845, https://doi.org/10.5194/acp-11-8827-2011, 2011.
Corrigan, A. L., Russell, L. M., Takahama, S., Äijälä, M., Ehn, M., Junninen, H., Rinne, J., Petäjä, T., Kulmala, M., Vogel, A. L., Hoffmann, T., Ebben, C. J., Geiger, F. M., Chhabra, P., Seinfeld, J. H., Worsnop, D. R., Song, W., Auld, J., and Williams, J.: Biogenic and biomass burning organic aerosol in a boreal forest at Hyytiälä, Finland, during HUMPPA-COPEC 2010, Atmos. Chem. Phys., 13, 12233–12256, https://doi.org/10.5194/acp-13-12233-2013, 2013.
Coury, C. and Dillner, A. M.: A method to quantify organic functional groups and inorganic compounds in ambient aerosols using attenuated total reflectance FTIR spectroscopy and multivariate chemometric techniques, Atmos. Environ., 42, 5923–5932, https://doi.org/10.1016/j.atmosenv.2008.03.026, 2008.
Coury, C. and Dillner, A. M.: ATR-FTIR characterization of organic functional groups and inorganic ions in ambient aerosols at a rural site, Atmos. Environ., 43, 940–948, https://doi.org/10.1016/j.atmosenv.2008.10.056, 2009.
Debus, B., Takahama, S., Weakley, A. T., Seibert, K., and Dillner, A. M.: Long-Term Strategy for Assessing Carbonaceous Particulate Matter Concentrations from Multiple Fourier Transform Infrared (FT-IR) Instruments: Influence of Spectral Dissimilarities on Multivariate Calibration Performance, Appl. Spectrosc., 73, 271–283, https://doi.org/10.1177/0003702818804574, 2019.
de Foy, B., Lu, Z., and Streets, D. G.: Impacts of control strategies, the Great Recession and weekday variations on NO2 columns above North American cities, Atmos. Environ., 138, 74–86, https://doi.org/10.1016/j.atmosenv.2016.04.038, 2016.
Diem, J. E. and Mote, T. L.: Interepochal changes in summer precipitation in the Southeastern United States: Evidence of possible urban effects near Atlanta, Georgia, J. Appl. Meteorol., 44, 717–730, https://doi.org/10.1175/JAM2221.1, 2005.
Dillner, A. M., Boris, A. J., Takahama, S., Weakley, A. T., Debus, B., Frederickson, C. D., Esparza-Sanchez, M., Burke, C., Reggente, M., Shaw, S. L., and Edgerton, E. S.: Data from: Organic functional group and organic matter concentrations from FT-IR measurements of particulate matter samples in the Southeastern Aerosol Research and Characterization (SEARCH) network from 2009–2016, Dryad [data set], https://doi.org/10.25338/B8SG73, 2019.
Ditto, J. C., Barnes, E. B., Khare, P., Takeuchi, M., Joo, T., Bui, A. A. T., Lee-Taylor, J., Eris, G., Chen, Y., Aumont, B., Jimenez, J. L., Ng, N. L., Griffin, R. J., and Gentner, D. R.: An omnipresent diversity and variability in the chemical composition of atmospheric functionalized organic aerosol, Communications Chemistry, 1, 75, https://doi.org/10.1038/s42004-018-0074-3, 2018.
Edgerton, E. S., Hartsell, B. E., Saylor, R. D., Jansen, J. J., Hansen, D. A., and Hidy, G. M.: The Southeastern Aerosol Research and Characterization Study: Part II. Filter-based measurements of fine and coarse particulate matter mass and composition, J. Air Waste Manage., 55, 1527–1542, https://doi.org/10.1080/10473289.2005.10464744, 2005.
Edgerton, E. S., Hartsell, B. E., Saylor, R. D., Jansen, J. J., Hansen, D. A., and Hidy, G. M.: The Southeastern Aerosol Research and Characterization Study, part 3: Continuous measurements of fine particulate matter mass and composition, J. Air Waste Manage., 56, 1325–1341, https://doi.org/10.1080/10473289.2006.10464585, 2006.
El-Zanan, H. S., Zielinska, B., Mazzoleni, L. R., and Hansen, D. A.: Analytical determination of the aerosol organic mass-to-organic carbon ratio, J. Air Waste Manage., 59, 58–69, https://doi.org/10.3155/1047-3289.59.1.58, 2009.
Feng, J., Chan, E., and Vet, R.: Air quality in the eastern United States and Eastern Canada for 1990–2015: 25 years of change in response to emission reductions of SO2 and NOx in the region, Atmos. Chem. Phys., 20, 3107–3134, https://doi.org/10.5194/acp-20-3107-2020, 2020.
Ford, B., Val Martin, M., Zelasky, S. E., Fischer, E. V., Anenberg, S. C., Heald, C. L., and Pierce, J. R.: Future fire impacts on smoke concentrations, visibility, and health in the contiguous United States, GeoHealth, 2, 229–247, https://doi.org/10.1029/2018GH000144, 2018.
Gao, S., Surratt, J. D., Knipping, E. M., Edgerton, E. S., Shahgholi, M., and Seinfeld, J. H.: Characterization of polar organic components in fine aerosols in the southeastern United States: Identity, origin, and evolution, J. Geophys. Res., 111, 1–27, https://doi.org/10.1029/2005JD006601, 2006.
Georgia Environmental Protection Division: Summer Open Burning Ban: https://epd.georgia.gov/air-protection-branch/open-burning-rules-georgia/summer-open-burning-ban, last access: 16 May 2021.
Geron, C. D. and Arnts, R. R.: Seasonal monoterpene and sesquiterpene emissions from Pinus taeda and Pinus virginiana, Atmos. Environ., 44, 4240–4251, https://doi.org/10.1016/j.atmosenv.2010.06.054, 2010.
Goldstein, A. H., Koven, C. D., Heald, C. L., and Fung, I. Y.: Biogenic carbon and anthropogenic pollutants combine to form a cooling haze over the southeastern United States, P. Natl. Acad. Sci. USA, 106, 8835–8840, https://doi.org/10.1073/pnas.0904128106, 2009.
Greene, C. A., Thirumalai, K., Kearney, K. A., Delgado, J. M., Schwanghart, W., Wolfenbarger, N. S., Thyng, K. M., Gwyther, D. E., Gardner, A. S., and Blankenship, D. D.: The Climate Data Toolbox for MATLAB, Geochem. Geophys. Geosyst., 20, 3774–3781, https://doi.org/10.1029/2019GC008392, 2019.
Guenther, A., Zimmerman, P., and Wildermuth, M.: Natural volatile organic compound emission rate estimates for US woodland landscapes, Atmos. Environ., 28, 1197–1210, https://doi.org/10.1016/1352-2310(94)90297-6, 1994.
Guenther, A. B., Jiang, X., Heald, C. L., Sakulyanontvittaya, T., Duhl, T., Emmons, L. K., and Wang, X.: The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 5, 1471–1492, https://doi.org/10.5194/gmd-5-1471-2012, 2012.
Hand, J. L., Schichtel, B. A., Malm, W. C., and Pitchford, M. L.: Particulate sulfate ion concentration and SO2 emission trends in the United States from the early 1990s through 2010, Atmos. Chem. Phys., 12, 10353–10365, https://doi.org/10.5194/acp-12-10353-2012, 2012a.
Hand, J. L., Schichtel, B. A., Pitchford, M., Malm, W. C., and Frank, N. H.: Seasonal composition of remote and urban fine particulate matter in the United States: Composition of remote and urban aerosols, J. Geophys. Res., 117, 1–22, https://doi.org/10.1029/2011JD017122, 2012b.
Hand, J. L., Prenni, A. J., Schichtel, B. A., Malm, W. C., and Chow, J. C.: Trends in remote PM2.5 residual mass across the United States: Implications for aerosol mass reconstruction in the IMPROVE network, Atmos. Environ., 203, 141–152, https://doi.org/10.1016/j.atmosenv.2019.01.049, 2019.
Hansen, D. A., Edgerton, E. S., Hartsell, B. E., Jansen, J. J., Kandasamy, N., Hidy, G. M., and Blanchard, C. L.: The Southeastern Aerosol Research and Characterization Study: Part 1 – overview, J. Air Waste Manage., 53, 1460–1471, https://doi.org/10.1080/10473289.2003.10466318, 2003.
Hay, M. B. and Myneni, S. C. B.: Structural environments of carboxyl groups in natural organic molecules from terrestrial systems. Part 1: Infrared spectroscopy, Geochim. Cosmochim. Ac., 71, 3518–3532, https://doi.org/10.1016/j.gca.2007.03.038, 2007.
Hidy, G. M., Blanchard, C. L., Baumann, K., Edgerton, E., Tanenbaum, S., Shaw, S., Knipping, E., Tombach, I., Jansen, J., and Walters, J.: Chemical climatology of the southeastern United States, 1999–2013, Atmos. Chem. Phys., 14, 11893–11914, https://doi.org/10.5194/acp-14-11893-2014, 2014.
Hyslop, N. and White, W.: An evaluation of interagency monitoring of protected visual environments (IMPROVE) collocated precision and uncertainty estimates, Atmos. Environ., 42, 2691–2705, https://doi.org/10.1016/j.atmosenv.2007.06.053, 2008.
Hyslop, N. P. and White, W. H.: Estimating precision using duplicate measurements, J. Air Waste Manage., 59, 1032–1039, https://doi.org/10.3155/1047-3289.59.9.1032, 2009.
Kim, J.-C.: Factors controlling natural VOC emissions in a southeastern US pine forest, Atmos. Environ., 35, 3279–3292, https://doi.org/10.1016/S1352-2310(00)00522-7, 2001.
Kim, P. S., Jacob, D. J., Fisher, J. A., Travis, K., Yu, K., Zhu, L., Yantosca, R. M., Sulprizio, M. P., Jimenez, J. L., Campuzano-Jost, P., Froyd, K. D., Liao, J., Hair, J. W., Fenn, M. A., Butler, C. F., Wagner, N. L., Gordon, T. D., Welti, A., Wennberg, P. O., Crounse, J. D., St. Clair, J. M., Teng, A. P., Millet, D. B., Schwarz, J. P., Markovic, M. Z., and Perring, A. E.: Sources, seasonality, and trends of southeast US aerosol: an integrated analysis of surface, aircraft, and satellite observations with the GEOS-Chem chemical transport model, Atmos. Chem. Phys., 15, 10411–10433, https://doi.org/10.5194/acp-15-10411-2015, 2015.
Kleeman, M. J., Ying, Q., and Kaduwela, A.: Control strategies for the reduction of airborne particulate nitrate in California's San Joaquin Valley, Atmos. Environ., 39, 5325–5341, https://doi.org/10.1016/j.atmosenv.2005.05.044, 2005.
Kleindienst, T. E., Lewandowski, M., Offenberg, J. H., Edney, E. O., Jaoui, M., Zheng, M., Ding, X., and Edgerton, E. S.: Contribution of primary and secondary sources to organic aerosol and PM2.5 at SEARCH network sites, J. Air Waste Manage., 60, 1388–1399, https://doi.org/10.3155/1047-3289.60.11.1388, 2010.
Konrad II, C. E. and Knox, P.: The Southeastern Drought and Wildfires of 2016, NOAA Southeast Regional Climate Center, 2018.
Kroll, J. H., Donahue, N. M., Jimenez, J. L., Kessler, S. H., Canagaratna, M. R., Wilson, K. R., Altieri, K. E., Mazzoleni, L. R., Wozniak, A. S., Bluhm, H., Mysak, E. R., Smith, J. D., Kolb, C. E., and Worsnop, D. R.: Carbon oxidation state as a metric for describing the chemistry of atmospheric organic aerosol, Nat. Chem., 3, 133–139, https://doi.org/10.1038/nchem.948, 2011.
Larkin, N. K., Raffuse, S. M., and Strand, T. M.: Wildland fire emissions, carbon, and climate: US emissions inventories, Forest Ecol. Manag., 317, 61–69, https://doi.org/10.1016/j.foreco.2013.09.012, 2014.
Liu, J., Russell, L. M., Ruggeri, G., Takahama, S., Claflin, M. S., Ziemann, P. J., Pye, H. O. T., Murphy, B. N., Xu, L., Ng, N. L., McKinney, K. A., Budisulistiorini, S. H., Bertram, T. H., Nenes, A., and Surratt, J. D.: Regional similarities and NOx-related increases in biogenic secondary organic aerosol in summertime southeastern United States, J. Geophys. Res. Atmos., 123, 10620–10636, https://doi.org/10.1029/2018JD028491, 2018.
Liu, S., Takahama, S., Russell, L. M., Gilardoni, S., and Baumgardner, D.: Oxygenated organic functional groups and their sources in single and submicron organic particles in MILAGRO 2006 campaign, Atmos. Chem. Phys., 9, 6849–6863, https://doi.org/10.5194/acp-9-6849-2009, 2009.
Liu, S., Day, D. A., Shields, J. E., and Russell, L. M.: Ozone-driven daytime formation of secondary organic aerosol containing carboxylic acid groups and alkane groups, Atmos. Chem. Phys., 11, 8321–8341, https://doi.org/10.5194/acp-11-8321-2011, 2011.
Malm, W. C., Schichtel, B. A., Hand, J. L., and Collett, J. L.: Concurrent temporal and spatial trends in sulfate and organic mass concentrations Mmeasured in the IMPROVE monitoring program: Trends in sulfate and organic mass, J. Geophys. Res. Atmos., 122, 10462–10476, https://doi.org/10.1002/2017JD026865, 2017.
Mao, J., Carlton, A., Cohen, R. C., Brune, W. H., Brown, S. S., Wolfe, G. M., Jimenez, J. L., Pye, H. O. T., Lee Ng, N., Xu, L., McNeill, V. F., Tsigaridis, K., McDonald, B. C., Warneke, C., Guenther, A., Alvarado, M. J., de Gouw, J., Mickley, L. J., Leibensperger, E. M., Mathur, R., Nolte, C. G., Portmann, R. W., Unger, N., Tosca, M., and Horowitz, L. W.: Southeast Atmosphere Studies: learning from model-observation syntheses, Atmos. Chem. Phys., 18, 2615–2651, https://doi.org/10.5194/acp-18-2615-2018, 2018.
Marais, E. A., Jacob, D. J., Turner, J. R., and Mickley, L. J.: Evidence of 1991–2013 decrease of biogenic secondary organic aerosol in response to SO2 emission controls, Environ. Res. Lett., 12, 054018, https://doi.org/10.1088/1748-9326/aa69c8, 2017.
Maria, S. F., Russell, L. M., Turpin, B. J., and Porcja, R. J.: FTIR measurements of functional groups and organic mass in aerosol samples over the Caribbean, Atmos. Environ., 36, 5185–5196, https://doi.org/10.1016/S1352-2310(02)00654-4, 2002.
Mayo, D. W., Miller, F. A., and Hannah, R. W. (Eds.): Course Notes on the Interpretation of Infrared and Raman Spectra: Mayo/Raman Spectra, John Wiley & Sons, Inc., Hoboken, NJ, USA, https://doi.org/10.1002/0471690082, 2004.
Mehta, M., Singh, N., and Anshumali: Global trends of columnar and vertically distributed properties of aerosols with emphasis on dust, polluted dust and smoke-inferences from 10 year long CALIOP observations, Remote Sens. Environ., 208, 120–132, https://doi.org/10.1016/j.rse.2018.02.017, 2018.
Melvin, M. A.: 2012 National Prescribed Fire Use Survey Report, Coalition of Prescribed Fire Councils, Inc., Technical Report, 01-12, 24 pp., available at: https://forestry.alabama.gov/Pages/Fire/Forms/2012_National_Fire_Use_Survey_Report.pdf (last access: 26 May 2021), 2012.
Melvin, M. A.: 2015 National Prescribed Fire Use Survey Report, Coalition of Prescribed Fire Councils, Inc., Technical Report, 02-15, 22 pp., available at: https://gfmc.online/wp-content/uploads/USA-2015-Prescribed-Fire-Use-Survey-Report.pdf (last access: 26 May 2021), 2015.
Melvin, M. A.: 2018 National Prescribed Fire Use Survey Report, Coalition of Prescribed Fire Councils, Inc., Technical Report, 03-18, 29 pp., available at: https://southernfireexchange.org/wp-content/uploads/2018-Prescribed-Fire-Use-Survey-Report-1.pdf (last access: 26 May 2021), 2018.
Moretti, F., Tagliavini, E., Decesari, S., Facchini, M. C., Rinaldi, M., and Fuzzi, S.: NMR Determination of Total Carbonyls and Carboxyls: A tool for tracing the evolution of atmospheric oxidized organic aerosols, Environ. Sci. Technol., 42, 4844–4849, https://doi.org/10.1021/es703166v, 2008.
Nguyen, T. K. V., Capps, S. L., and Carlton, A. G.: Decreasing aerosol water is consistent with OC trends in the Southeast US, Environ. Sci. Technol., 49, 7843–7850, https://doi.org/10.1021/acs.est.5b00828, 2015.
Ortiz-Montalvo, D. L., Häkkinen, S. A. K., Schwier, A. N., Lim, Y. B., McNeill, V. F., and Turpin, B. J.: Ammonium addition (and aerosol pH) has a dramatic impact on the volatility and yield of glyoxal secondary organic aerosol, Environ. Sci. Technol., 48, 255–262, https://doi.org/10.1021/es4035667, 2014.
Peräkylä, O., Vogt, M., Tikkanen, O.-P., Laurila, T., Kajos, M. K., Rantala, P. A., Patokoski, J., Aalto, J., Yli-Juuti, T., Ehn, M., Sipilä, M., Paasonen, P., Rissanen, M., Nieminen, T., Taipale, R., Keronen, P., Lappalainen, H. K., Ruuskanen, T. M., Rinne, J., Kerminen, V.-M., Kulmala, M., Bäck, J., and Petäjä, T.: Monoterpenes' oxidation capacity and rate over a boreal forest: temporal variation and connection to growth of newly formed particles, Boreal Environ. Res., 19, 293–310, 2014.
Pullinen, I., Schmitt, S., Kang, S., Sarrafzadeh, M., Schlag, P., Andres, S., Kleist, E., Mentel, T. F., Rohrer, F., Springer, M., Tillmann, R., Wildt, J., Wu, C., Zhao, D., Wahner, A., and Kiendler-Scharr, A.: Impact of NOx on secondary organic aerosol (SOA) formation from α-pinene and β-pinene photooxidation: the role of highly oxygenated organic nitrates, Atmos. Chem. Phys., 20, 10125–10147, https://doi.org/10.5194/acp-20-10125-2020, 2020.
Pye, H. O. T., D'Ambro, E. L., Lee, B. H., Schobesberger, S., Takeuchi, M., Zhao, Y., Lopez-Hilfiker, F., Liu, J., Shilling, J. E., Xing, J., Mathur, R., Middlebrook, A. M., Liao, J., Welti, A., Graus, M., Warneke, C., de Gouw, J. A., Holloway, J. S., Ryerson, T. B., Pollack, I. B., and Thornton, J. A.: Anthropogenic enhancements to production of highly oxygenated molecules from autoxidation, P. Natl. Acad. Sci. USA, 116, 6641–6646, https://doi.org/10.1073/pnas.1810774116, 2019.
Reggente, M., Dillner, A. M., and Takahama, S.: Analysis of functional groups in atmospheric aerosols by infrared spectroscopy: systematic intercomparison of calibration methods for US measurement network samples, Atmos. Meas. Tech., 12, 2287–2312, https://doi.org/10.5194/amt-12-2287-2019, 2019.
Rogge, W. F., Hildemann, L. M., Mazurek, M. A., Cass, G. R., and Simoneit, B. R. T.: Sources of fine organic aerosol. 2. Noncatalyst and catalyst-equipped automobiles and heavy-duty diesel trucks, Environ. Sci. Technol., 27, 636–651, https://doi.org/10.1021/es00041a007, 1993.
Russell, L. M.: Aerosol organic-mass-to-organic-carbon ratio measurements, Environ. Sci. Technol., 37, 2982–2987, https://doi.org/10.1021/es026123w, 2003.
Russell, L. M., Takahama, S., Liu, S., Hawkins, L. N., Covert, D. S., Quinn, P. K., and Bates, T. S.: Oxygenated fraction and mass of organic aerosol from direct emission and atmospheric processing measured on the R/V Ronald Brown during TEXAQS/GoMACCS 2006, J. Geophys. Res., 114, D00F05, https://doi.org/10.1029/2008JD011275, 2009.
Russell, L. M., Bahadur, R., and Ziemann, P. J.: Identifying organic aerosol sources by comparing functional group composition in chamber and atmospheric particles, P. Natl. Acad. Sci. USA, 108, 3516–3521, https://doi.org/10.1073/pnas.1006461108, 2011.
Ruthenburg, T. C., Perlin, P. C., Liu, V., McDade, C. E., and Dillner, A. M.: Determination of organic matter and organic matter to organic carbon ratios by infrared spectroscopy with application to selected sites in the IMPROVE network, Atmos. Environ., 86, 47–57, https://doi.org/10.1016/j.atmosenv.2013.12.034, 2014.
Salvador, C. M., Ho, T.-T., Chou, C. C.-K., Chen, M.-J., Huang, W.-R., and Huang, S.-H.: Characterization of the organic matter in submicron urban aerosols using a Thermo-Desorption Proton-Transfer-Reaction Time-of-Flight Mass Spectrometer (TD-PTR-TOF-MS), Atmos. Environ., 140, 565–575, https://doi.org/10.1016/j.atmosenv.2016.06.029, 2016.
Schindelka, J., Iinuma, Y., Hoffmann, D., and Herrmann, H.: Sulfate radical-initiated formation of isoprene-derived organosulfates in atmospheric aerosols, Faraday Discuss., 165, 237, https://doi.org/10.1039/c3fd00042g, 2013.
Schum, S. K., Zhang, B., Džepina, K., Fialho, P., Mazzoleni, C., and Mazzoleni, L. R.: Molecular and physical characteristics of aerosol at a remote free troposphere site: implications for atmospheric aging, Atmos. Chem. Phys., 18, 14017–14036, https://doi.org/10.5194/acp-18-14017-2018, 2018.
Schwartz, R. E., Russell, L. M., Sjostedt, S. J., Vlasenko, A., Slowik, J. G., Abbatt, J. P. D., Macdonald, A. M., Li, S. M., Liggio, J., Toom-Sauntry, D., and Leaitch, W. R.: Biogenic oxidized organic functional groups in aerosol particles from a mountain forest site and their similarities to laboratory chamber products, Atmos. Chem. Phys., 10, 5075–5088, https://doi.org/10.5194/acp-10-5075-2010, 2010.
Seidel, D. J., Zhang, Y., Beljaars, A., Golaz, J.-C., Jacobson, A. R., and Medeiros, B.: Climatology of the planetary boundary layer over the continental United States and Europe, J. Geophys. Res., 117, 1–15, https://doi.org/10.1029/2012JD018143, 2012.
Sen, P. K.: Estimates of the regression coefficient based on Kendall's tau, J. Am. Stat. Assoc., 63, 1379–1389, https://doi.org/10.1080/01621459.1968.10480934, 1968.
Simon, H., Bhave, P. V., Swall, J. L., Frank, N. H., and Malm, W. C.: Determining the spatial and seasonal variability in ratios across the US using multiple regression, Atmos. Chem. Phys., 11, 2933–2949, https://doi.org/10.5194/acp-11-2933-2011, 2011.
Sorooshian, A., Wang, Z., Coggon, M. M., Jonsson, H. H., and Ervens, B.: Observations of sharp oxalate reductions in stratocumulus clouds at variable altitudes: Organic acid and metal measurements during the 2011 E-PEACE campaign, Environ. Sci. Technol., 47, 7747–7756, https://doi.org/10.1021/es4012383, 2013.
Sun, Y., Zhang, Q., Zheng, M., Ding, X., Edgerton, E. S., and Wang, X.: Characterization and source apportionment of water-soluble organic matter in atmospheric fine particles (PM2.5) with high-resolution aerosol mass spectrometry and GC–MS, Environ. Sci. Technol., 45, 4854–4861, https://doi.org/10.1021/es200162h, 2011a.
Sun, Y. L., Zhang, Q., Schwab, J. J., Chen, W. N., Bae, M. S., Lin, Y. C., Hung, H. M., and Demerjian, K. L.: A case study of aerosol processing and evolution in summer in New York City, Atmos. Chem. Phys., 11, 12737–12750, https://doi.org/10.5194/acp-11-12737-2011, 2011b.
Takahama, S. and Ruggeri, G.: Technical note: Relating functional group measurements to carbon types for improved model–measurement comparisons of organic aerosol composition, Atmos. Chem. Phys., 17, 4433–4450, https://doi.org/10.5194/acp-17-4433-2017, 2017.
Takahama, S., Schwartz, R. E., Russell, L. M., Macdonald, A. M., Sharma, S., and Leaitch, W. R.: Organic functional groups in aerosol particles from burning and non-burning forest emissions at a high-elevation mountain site, Atmos. Chem. Phys., 11, 6367–6386, https://doi.org/10.5194/acp-11-6367-2011, 2011.
Takahama, S., Johnson, A., and Russell, L. M.: Quantification of carboxylic and carbonyl functional groups in organic aerosol infrared absorbance spectra, Aerosol Sci. Tech., 47, 310–325, https://doi.org/10.1080/02786826.2012.752065, 2013.
Theil, H.: A Rank-Invariant Method of Linear and Polynomial Regression Analysis, Parts I and II, in: Henri Theil's Contributions to Economics and Econometrics, vol. 23, Royal Netherlands Academy of Sciences, 345–381, 1992.
Travis, K. R., Jacob, D. J., Fisher, J. A., Kim, P. S., Marais, E. A., Zhu, L., Yu, K., Miller, C. C., Yantosca, R. M., Sulprizio, M. P., Thompson, A. M., Wennberg, P. O., Crounse, J. D., St. Clair, J. M., Cohen, R. C., Laughner, J. L., Dibb, J. E., Hall, S. R., Ullmann, K., Wolfe, G. M., Pollack, I. B., Peischl, J., Neuman, J. A., and Zhou, X.: Why do models overestimate surface ozone in the Southeast United States?, Atmos. Chem. Phys., 16, 13561–13577, https://doi.org/10.5194/acp-16-13561-2016, 2016.
Turpin, B. J. and Lim, H.-J.: Species contributions to PM2.5 mass concentrations: Revisiting common assumptions for estimating organic mass, Aerosol Sci. Tech., 35, 602–610, https://doi.org/10.1080/02786820119445, 2001.
U. C. Davis Air Quality Research Center: UCD IMPROVE Standard Operating Procedure no. 351: Data Processing and Validation, Davis, California, 2017.
US Environmental Protection Agency: Appendix L to Part 50 – Reference Method for the Determination of Fine Particulate Matter as PM2.5 in the Atmosphere, Federal Regulations, Title 40, Washington, D.C., 2011.
Watne, Å. K., Westerlund, J., Hallquist, Å. M., Brune, W. H., and Hallquist, M.: Ozone and OH-induced oxidation of monoterpenes: Changes in the thermal properties of secondary organic aerosol (SOA), J. Aerosol Sci., 114, 31–41, https://doi.org/10.1016/j.jaerosci.2017.08.011, 2017.
Wilcox, R. R.: A note on the Theil-Sen regression estimator when the regressor is random and the error term is heteroscedastic, Biometrical J., 40, 261–268, https://doi.org/10.1002/(SICI)1521-4036(199807)40:3<261::AID-BIMJ261>3.0.CO;2-V, 1998.
Wold, S., Sjöström, M., and Eriksson, L.: PLS-regression: A basic tool of chemometrics, Chemometr. Intell. Lab., 58, 109–130, https://doi.org/10.1016/S0169-7439(01)00155-1, 2001. .
Wolff, G. T., Kahlbaum, D. F., and Heuss, J. M.: The vanishing ozone weekday/weekend effect, J. Air Waste Manage., 63, 292–299, https://doi.org/10.1080/10962247.2012.749312, 2013.
Xu, L., Suresh, S., Guo, H., Weber, R. J., and Ng, N. L.: Aerosol characterization over the southeastern United States using high-resolution aerosol mass spectrometry: spatial and seasonal variation of aerosol composition and sources with a focus on organic nitrates, Atmos. Chem. Phys., 15, 7307–7336, https://doi.org/10.5194/acp-15-7307-2015, 2015a.
Xu, L., Guo, H., Boyd, C. M., Klein, M., Bougiatioti, A., Cerully, K. M., Hite, J. R., Isaacman-VanWertz, G., Kreisberg, N. M., Knote, C., Olson, K., Koss, A., Goldstein, A. H., Hering, S. V., de Gouw, J., Baumann, K., Lee, S.-H., Nenes, A., Weber, R. J., and Ng, N. L.: Effects of anthropogenic emissions on aerosol formation from isoprene and monoterpenes in the southeastern United States, P. Natl. Acad. Sci. USA, 112, 37–42, https://doi.org/10.1073/pnas.1417609112, 2015b.
Xu, L., Pye, H. O. T., He, J., Chen, Y., Murphy, B. N., and Ng, N. L.: Experimental and model estimates of the contributions from biogenic monoterpenes and sesquiterpenes to secondary organic aerosol in the southeastern United States, Atmos. Chem. Phys., 18, 12613–12637, https://doi.org/10.5194/acp-18-12613-2018, 2018.
Zhang, H., Yee, L. D., Lee, B. H., Curtis, M. P., Worton, D. R., Isaacman-VanWertz, G., Offenberg, J. H., Lewandowski, M., Kleindienst, T. E., Beaver, M. R., Holder, A. L., Lonneman, W. A., Docherty, K. S., Jaoui, M., Pye, H. O. T., Hu, W., Day, D. A., Campuzano-Jost, P., Jimenez, J. L., Guo, H., Weber, R. J., de Gouw, J., Koss, A. R., Edgerton, E. S., Brune, W., Mohr, C., Lopez-Hilfiker, F. D., Lutz, A., Kreisberg, N. M., Spielman, S. R., Hering, S. V., Wilson, K. R., Thornton, J. A., and Goldstein, A. H.: Monoterpenes are the largest source of summertime organic aerosol in the southeastern United States, P. Natl. Acad. Sci. USA, 115, 2038–2043, https://doi.org/10.1073/pnas.1717513115, 2018.
Zhang, X., Hecobian, A., Zheng, M., Frank, N. H., and Weber, R. J.: Biomass burning impact on PM2.5 over the southeastern US during 2007: integrating chemically speciated FRM filter measurements, MODIS fire counts and PMF analysis, Atmos. Chem. Phys., 10, 6839–6853, https://doi.org/10.5194/acp-10-6839-2010, 2010.
Zheng, M., Cass, G. R., Schauer, J. J., and Edgerton, E. S.: Source apportionment of PM2.5 in the southeastern United States using solvent-extractable organic compounds as tracers, Environ. Sci. Technol., 36, 2361–2371, https://doi.org/10.1021/es011275x, 2002.
Zheng, Y., Thornton, J. A., Ng, N. L., Cao, H., Henze, D. K., McDuffie, E. E., Hu, W., Jimenez, J. L., Marais, E. A., Edgerton, E., and Mao, J.: Long-term observational constraints of organic aerosol dependence on inorganic species in the southeast US, Atmos. Chem. Phys., 20, 13091–13107, https://doi.org/10.5194/acp-20-13091-2020, 2020.
Ziemann, P. J. and Atkinson, R.: Kinetics, products, and mechanisms of secondary organic aerosol formation, Chem. Soc. Rev., 41, 6582, https://doi.org/10.1039/c2cs35122f, 2012.