the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 12 Aug 2021
| 12 Aug 2021
Impact of ozone and inlet design on the quantification of isoprene-derived organic nitrates by thermal dissociation cavity ring-down spectroscopy (TD-CRDS)
Patrick Dewald
Raphael Dörich
Jan Schuladen
Jos Lelieveld
We present measurements of isoprene-derived organic nitrates (ISOP-NITs) generated in the reaction of isoprene with the nitrate radical (NO3) in a 1 m3 Teflon reaction chamber. Detection of ISOP-NITs is achieved via their thermal dissociation to nitrogen dioxide (NO2), which is monitored by cavity ring-down spectroscopy (TD-CRDS). Using thermal dissociation inlets (TDIs) made of quartz, the temperature-dependent dissociation profiles (thermograms) of ISOP-NITs measured in the presence of ozone (O3) are broad (350 to 700 K), which contrasts the narrower profiles previously observed for, for example, isopropyl nitrate (iPN) or peroxy acetyl nitrate (PAN) under the same conditions. The shape of the thermograms varied with the TDI's surface-to-volume ratio and with material of the inlet walls, providing clear evidence that ozone and quartz surfaces catalyse the dissociation of unsaturated organic nitrates leading to formation of NO2 at temperatures well below 475 K, impeding the separate detection of alkyl nitrates (ANs) and peroxy nitrates (PNs). The use of a TDI consisting of a non-reactive material suppresses the conversion of isoprene-derived ANs at 473 K, thus allowing selective detection of PNs. The potential for interference by the thermolysis of nitric acid (HNO3), nitrous acid (HONO) and O3 is assessed.
- Article
(4423 KB) - Full-text XML
-
Supplement
(820 KB) - BibTeX
- EndNote





Understanding the atmospheric fate of nitrogen oxide (NO) and nitrogen dioxide (NO2) is critical as both trace gases have a great impact on air quality and human health (Crutzen and Lelieveld, 2001; Lelieveld et al., 2015). Ambient measurements of trace gases that function as NOx reservoirs or sinks (where NOx = NO + NO2) are thus needed to provide insight into NOx removal and transport. Organic compounds with nitrate functionality can serve as NOx reservoirs in the troposphere (Thornton et al., 2002; Horowitz et al., 2007) and are generally categorized as peroxy nitrates (PNs, RO2NO2, with peroxy acetyl nitric anhydride (PAN) being its most abundant representative in the troposphere) and alkyl (aliphatic) nitrates (ANs, RONO2). PNs are formed via the reaction of organic peroxy radicals (RO2) with NO2 (Reaction R1); ANs are formed via the minor (termolecular) channel of the reaction of RO2 with NO (Reaction R2a). The competitive bimolecular process leads to alkoxy radicals (RO, Reaction R2b). During the daytime, RO2 is formed mainly by the oxidation of volatile organic compounds (VOCs) by hydroxyl radicals (OH) in air (Reaction R3), with ozonolysis important at night (Reaction R4):
At night-time, when OH radicals and NO are significantly less abundant, the NO3 radical can initiate the oxidation of many VOCs that contain a double bond (Ng et al., 2017). NO3, formed in the oxidation of NO2 by O3 (Reaction R5), is photolysed rapidly by sunlight (Reaction R6) and also reacts efficiently with NO (Reaction R7) so that it is generally of minor importance during the day (Wayne et al., 1991).
NO3 readily undergoes reactions with many unsaturated organic trace gases of biogenic origin including isoprene, monoterpenes and sesquiterpenes to form organic nitrates in high yields (Ng et al., 2017; Wennberg et al., 2018; Mellouki et al., 2021). The focus of this work is the formation of organic nitrates in its reaction (in air) with the C5-diene isoprene (ISOP, Reaction R8):
where ISOP-NIT represents an isoprene-derived nitrate. Approximately 500 Tg yr−1 of isoprene is released to the atmosphere (Guenther et al., 2012), and a large fraction of the organic nitrates formed at night-time is attributed to the reaction between NO3 and isoprene (Reaction R8) (Carlton et al., 2009). The atmospheric oxidation of isoprene involving OH, O3 and NO3 as oxidizing agents is complex and leads to a huge variety of products (Ng et al., 2017; Wennberg et al., 2018) including multifunctional, unsaturated nitrates such as O2NOCH2C(CH3)=CHCHO, O2NOCH2C(CH3)=CHCH2OH or O2NOCH2C(CH3)=CHCH2OOH among other secondary oxidation products like dinitrates or epoxides (Wu et al., 2021).
Studies of the NO3-induced oxidation of isoprene in air report AN yields between 60 % and 100 % (Barnes et al., 1990; Berndt and Boge, 1997; Perring et al., 2009; Kwan et al., 2012; Schwantes et al., 2015; IUPAC, 2021; Wu et al., 2021; Brownwood et al., 2021), and the NO3-induced oxidation of isoprene is responsible for a dominant fraction of organic nitrates observed in rural environments with strong biogenic emissions (Beaver et al., 2012). The major fate of isoprene-derived organic nitrates formed in the boundary layer is deposition onto particulate matter to form nitric acid (HNO3) or secondary organic aerosols (SOAs) leading to largely irreversible removal of NOx from the gas phase (Ng et al., 2008; Rollins et al., 2009; Fry et al., 2018; Hamilton et al., 2021).
Individual isoprene nitrates have been measured selectively in the atmosphere by mass-spectrometric methods (Wolfe et al., 2007; Wu et al., 2021). An alternative detection scheme, in which the sum of all atmospheric PNs (ΣPNs) and ANs (ΣANs) are separately measured, takes advantage of their different C–N bond energy by combining thermal dissociation to NO2 (Reactions R9 and R10) with detection of the latter with means of laser-induced fluorescence (TD-LIF) or cavity ring-down spectroscopy (TD-CRDS).
Several instruments using thermal dissociation inlets consisting of fused silica (quartz) with residence times between tens to hundreds of milliseconds have been described in the literature (Day et al., 2002; Paul et al., 2009; Wild et al., 2014; Sobanski et al., 2016; Thieser et al., 2016; Keehan et al., 2020). In these instruments, quantitative conversion of PAN is reported for temperatures between 375 and 420 K. Generally, the temperature dependence of the instrument's response to ANs has been tested using mostly saturated organic nitrates such as isopropyl nitrate (iPN) and isobutyl nitrate, which are dissociated to NO2 at temperatures between 500 and 675 K. The temperatures at which PNs and ANs are quantitatively converted to NO2 thus differ by ∼ 200 K and are largely independent of their organic backbone (Kirchner et al., 1999; Wild et al., 2014) allowing separate measurement of the sum of all alkyl nitrates (ΣANs) and of the sum of all peroxy nitrates (ΣPNs). These observations have provided the basis for analysis of field data in which an unknown mixture of PNs and ANs are present. We note, however, that most of the first-generation ANs formed in the NO3 + isoprene system still contain a double bond (Barnes et al., 1990; Skov et al., 1992; Schwantes et al., 2015), which renders them more reactive towards oxidizing agents than, for example, iPN. A well-characterized thermogram for aliphatic nitrates derived from the oxidation of, for example, isoprene is thus a pre-requirement for extracting the mixing ratios of PNs and ANs from ambient measurements when using a TD inlet. To date, only one such thermogram has been presented (Brownwood et al., 2021) which appears to be the result of a single experiment (i.e. no variation of experimental conditions) using a sample that was not stable over time. The thermogram also features slopes before and after the AN transition temperature, which is consistent with the ideal behaviour of, for example, iPN.
In this study, we generated ISOP-NITs by reacting isoprene and NO3 in a Teflon simulation chamber and used a custom-built, five-channel cavity ring-down spectrometer (CRDS) (Sobanski et al., 2016) to analyse the organic nitrates formed. In the presence of O3 we find that ISOP-NIT does not behave like the saturated analogue iPN in our quartz TD inlet and we characterized the processes (both gas-phase and surface-catalysed processes) that lead to the observed behaviour. We also examined the potential role of surface-catalysed dissociation of HNO3 and nitrous acid (HONO) to NO2 as well as the effect of humidity as a potential bias to measurements of PNs and ANs.
2.1 Simulation chamber
In order to analyse organic nitrates formed from the NO3 + isoprene system under realistic operational conditions for the five-channel CRDS (e.g. normal sample flow rates), we constructed a dynamic, flow-through simulation chamber “SCHARK” (Simulation CHamber for Atmospheric Reactions and Kinetics) of volume 1 m3 (cubic, all sides ∼ 1 m long) made of PFA foil of 0.005 in. (∼ 0.13 mm) thickness (Ingeniven). The chamber is operated at ambient pressure and temperature; a magnetically coupled, Teflon-coated propeller-type stirrer situated in the centre of the chamber floor ensures continuous mixing of the air. The trace-gas inlets and sampling ports were located at opposite corners of the cubic chamber to reduce the potential of sampling gas that had not yet mixed. The PFA foil is surrounded by a 120 × 120 × 120 cm cube constructed of four Perspex and two steel walls; the interspace (0.7 m3) is permanently flushed with 1 SLPM (L (STP) min−1) of dry synthetic “zero air” in order to avoid contamination through permeation of trace gases present in the laboratory air. The Perspex walls serve as observation windows and were covered with light-tight material during the experiments described here.
Zero air was provided by passing pressurized air through a commercial air purifier (CAP 180, Fuhr GmbH). Humidification of the air was achieved with a permeation source (MH-110-24-F-4, Perma Pure LLC) filled with deionized water. Typical total flow rates of 15 or 23 SLPM zero air into the chamber result in exchange rates kexch of 2.7 or 4.2 × 10−4 s−1, i.e. lifetimes of gases in the chamber of ∼ 40–60 min. Note that in “flow-through” operation, the concentrations of trace gases in the chamber are controlled both by chemical processes and by the rate of flow into (and out of) the chamber so that “steady state” is achieved on the order of hours.
Ozone mixing ratios in SCHARK were measured by sampling 2 SLPM through a ∼ 3 m long section of 0.25 in. (outer diameter, OD) PFA tubing to a commercial ozone monitor (2B Technologies, model 205) with a detection limit of ∼ 1 part per billion by volume (ppbv) and 5 % uncertainty. O3 measurements were also used to establish the time required (under standard flow conditions) to achieve complete mixing within the chamber (< 1 min) and to derive the exchange rate by monitoring the exponential rise or decay of O3 when its supply was switched on or off (Fig. S1 in the Supplement). O3 (up to 600 ppbv) was generated by passing a fraction of the air flowing into the chamber through a UV-transparent cuvette (∼ 70 cm3) illuminated by a low-pressure Hg lamp (PenRay) that dissociated O2 (to O atoms and thus O3) at 185 nm.
A known flow of isoprene entered the chamber as a dilute sample from a 12 L stainless-steel storage canister (Landefeld GmbH) which was prepared manometrically from evaporation of pure isoprene (Acros Organics, 98 %) and mixed with helium (5.0, Westfalen). The isoprene concentration in the storage canister was quantified indirectly by measuring the NO3 reactivity via flow-tube CRDS (Liebmann et al., 2017; Dewald et al., 2020) and was found to be 46.5 ppmv, in agreement (within 15 %) with the manometrically derived mixing ratio. A gas sample of isopropyl nitrate (Sigma Aldrich, 58 ppmv in N2 5.0, Westfalen) was prepared in a similar fashion.
Two methods of in situ NO3 generation were employed. In the first, NO3 was produced in the chamber via the reaction of NO2 with O3 (Reaction R5), whereby O3 was generated as described above and NO2 was taken from a bottled sample (Air Liquide, 1 ppmv in N2). Typical concentrations of NO2 and O3 were 6–10 and 100–160 ppbv, respectively. Alternatively, NO3 was generated in the thermal decomposition of N2O5 (Reaction R11) which was eluted into the chamber by passing a regulated flow of N2 over N2O5 crystals held at temperatures between −78 and −70 ∘C.
N2O5 was synthesized by the sequential, gas-phase oxidation of NO (5 % in N2, Westfalen) in an excess of O3 (Davidson et al., 1978) and trapped at −78 ∘C (acetone and dry ice). In this case, O3 was obtained by electrical discharge through oxygen (5.0, Westfalen) using a commercial generator (Ozomat Com, Anseros). Note that the latter method enables us to generate NO3 in the chamber in an O3-free environment.
2.2 Detection of organic nitrates by cavity ring-down spectroscopy (CRDS)
Simultaneous measurements of the mixing ratios of NO2, NO3, N2O5, ΣANs and ΣPNs in the SCHARK chamber were made using a five-channel cavity ring-down spectrometer (CRDS) that has been described in detail (Sobanski et al., 2016), and only a brief summary of key features of the instrument is given here. Each of the five cavities consists of FEP-coated (FEPD 121, DuPont) stainless-steel tubes which are equipped with two high-reflectivity mirrors (see below) supported 90 cm apart (L). The volumes in front of the mirrors are purged with dry synthetic air, which results in a reduction of the effective optical path length from 90 to 62.1 cm (d). The standard expression Eq. (1) is used to derive in-cavity concentrations [X] from the difference in ring-down constant in the absence (k0) and presence (k) of an absorber X:
where c is the speed of light and σeff is the effective cross-section derived from the overlap of the laser emission and the NO2 (Vandaele et al., 1998) or NO3 absorption spectrum (Orphal et al., 2003).
Three of the cavities are operated at 409 nm for detection of NO2, whereby 409 nm light is provided by a square-wave-modulated (2500 Hz) laser diode. The three 409 nm cavities, thermostated to 303 K and typically operated at a pressure of ∼ 733 hPa, sampled from SCHARK at a total flow rate of 6 SLPM, which initially passes through a 2.3 m long PFA inlet (1.5 m with OD 0.25 in. and 0.8 m with OD 0.125 in.) before being split into three equal flows. One flow is directed to a cavity via an unheated, 60 cm long PFA tube (0.375 in. OD) to measure NO2. The other two flows are directed through thermal dissociation inlets (TDIs) in which PNs and ANs are converted to NO2. At the given conditions (i.e. flow rate, pressure, residence time in the heated section), keeping our TDI at temperatures close to 448 K results in quantitative conversion of PNs to NO2 so that the cavity sampling via this inlet measures the sum of PNs + NO2. Heating our second TDI to ≈ 650 K results in the complete conversion of ANs to NO2 so that the sum of ANs + PNs + NO2 can be measured as described in the literature cited in the introduction. The choice of material for these TD inlets has a profound influence on the results obtained, as described below.
The standard deviation (2σ) of consecutive baseline measurements defines the limits of detection (LODs), which are 38, 44 and 90 pptv for [NO2], [ΣPNs] and [ΣANs] respectively under laboratory conditions. The total uncertainty for the NO2 measurement is 9 %, which includes uncertainty in the (effective) NO2 cross-sections. For the measurements of ANs and PNs the associated uncertainties are highly dependent on the concentrations of other trace gases and the corrective procedure accounting for radical recombination effects (Sobanski et al., 2016).
For simplicity, we refer to the three cavities as the “NO2 cavity” (room-temperature inlet), the “PN cavity” (TD inlet at circa 473 K in which ΣPNs + NO2 are measured) and the “AN cavity” (TD inlet at circa 673 K in which ΣANs + ΣPNs + NO2 are measured).
The remaining two cavities of the CRDS were operated at 662 nm (laser modulation at 625 Hz) for detection of NO3. While one cavity is thermostated to 303 K (and detects NO3 only), the second one (as well as an FEP-coated glass reactor located upstream) is thermostated to 373 K so that N2O5 is stoichiometrically converted NO3 and the summed mixing ratio of NO3 and N2O5 is obtained. The two 662 nm cavities sampled air from SCHARK at a total flow rate of 15 SLPM through a ∼ 1.5 m in. (OD) PFA tube. Corrections to the mixing ratios were made to account for loss of NO3 and N2O5 during transport to and through the cavities. Using the method described in Sobanski et al. (2016), NO3 transmission was found to be 89 % in both cavities. The NO3 and N2O5 measurements are not central to this study but allowed the quantitative surveillance of NO3 (and indirect N2O5) consumption by isoprene.
Figure 1 shows three types of thermal dissociation inlets (TDIs) used to convert organic nitrates to NO2. In the original version of this instrument (Sobanski et al., 2016) the ΣPNs and ΣAN cavities sampled via 12 mm ID quartz TD inlets (TDI-1), with a length of 55 cm, the first ∼ 10 cm of which was wrapped with heating wire. In order to reduce bias caused, for example, by the reformation of the organic nitrate after its thermal dissociation, this section was filled with glass beads (Sigma-Aldrich G9268, ø ∼ 0.5 mm) to provide a surface for heterogeneous loss of radicals. The glass beads were supported on a 2 cm thick glass frit and reduce the pressure downstream by ≈ 28 hPa compared to TDI-1. Problems associated with temperature-dependent flow resistance through these small beads and the need for an extra filter (upstream) to prevent their transport into the inlet lines when flows were temporarily reversed (e.g. during instrument shut-down) led us to switch to larger beads (Merck, ø = 3 mm), and these were used throughout this study in TDI-1. TDI-2 is made of a quartz glass tube with the same dimensions as TDI-1 but features a longer heated section (20 cm) and is free of additional surfaces like glass beads or frits. TDI-2 is thus similar to many other thermal dissociation inlets described in the literature (see above). TDI-3 is constructed from a 55 cm long PFA tube (0.375 in. OD), where the first 20 cm is heated. The melting point of PFA is lower than the temperature required to thermally dissociate ANs, so TDI-3 could only be used for the measurement of PNs + NO2. The temperature of the external wall of the TD inlets was measured with a K-type thermocouple situated at the centre of the heated section, which was insulated with mineral wool. At a flow rate of 2 SLPM and an operating pressure of 733 mbar, approximate residence times in the inlets without glass beads are 0.20 s (in TDI-2 at 650 K) and 0.13 s (TDI-3 at 450 K) when assuming a homogeneous temperature distribution equal to that measured on the outer wall of the tubing.
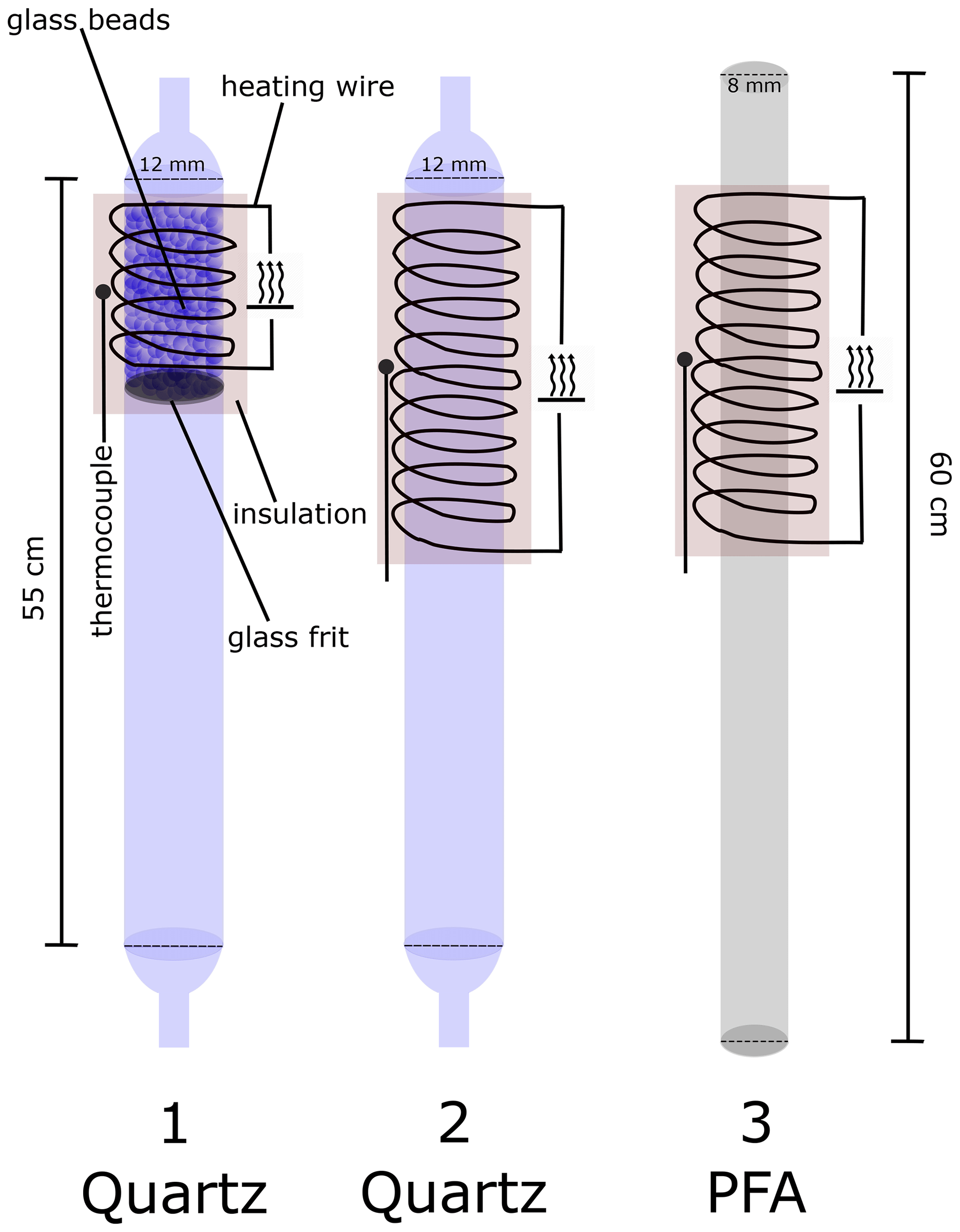
Figure 2 shows the result of an experiment in which 150 sccm NO2 (1 ppmv in air) was flowed into SCHARK along with isoprene (7 sccm of 46.5 ppmv in He) and 24 SLPM zero air of which 5 SLPM was passed over the low-pressure Hg-lamp zero air to generate O3 (∼ 96 ppbv). NO2 was sampled (as usual) via the room-temperature PFA inlet, the ΣPNs (473 K) and ΣAN cavities (673 K) both sampled via TDI-1 (quartz tube with glass beads). O3 was added at 09:30 and NO2 at 10:00 (all times are local times, LT).
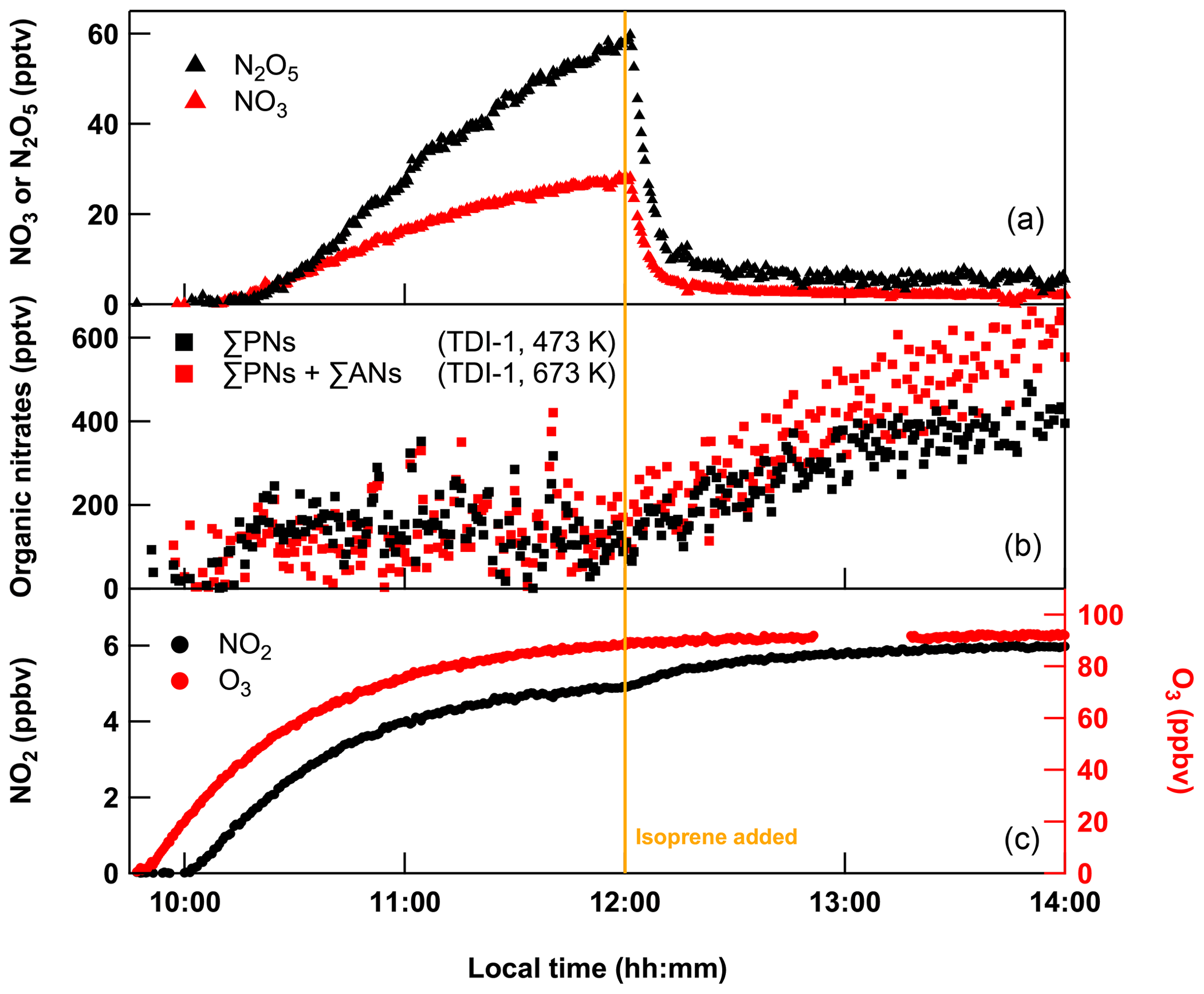
Figure 2Evolution of the mixing ratios of NO2, ΣPNs, ΣPNs + ΣANs, N2O5 and O3 when flowing 150 sccm NO2 (from 1 ppmv bottle), 7 sccm isoprene (from a 46.5 ppmv cylinder) and 24 SLPM of zero air (of which 5 SLPM was passed over a PenRay lamp) into SCHARK. Isoprene was added at 12:00 LT after the system was close to steady state. Note that the NO2 and N2O5 mixing ratios were subtracted from the organic nitrate signals (b).
Just prior to the addition of isoprene at 12:00 LT, the system is close to steady state with ∼ 5 ppbv NO2 and 92 ppbv O3. After subtraction of the measured N2O5 mixing ratios, a residual signal of ∼ 100 pptv is detected in both the PN and AN channels, which may be caused, as discussed below, by interference of HNO3 in the AN channel and a memory effect of the glass beads in the PN channel (Sect. 3.1 and 3.2). Note that after addition of isoprene, both NO3 and N2O5 are reduced drastically (NO3 ∼ 3 pptv, N2O5 ∼ 5 pptv) and the thermal dissociation of N2O5 no longer contributes to NO2 signals in the PN and AN channels (Sobanski et al., 2016; Thieser et al., 2016).
At 14:00 LT, the cavity sampling from the 673 K TD inlet indicated ∼ 610 pptv for the summed mixing ratio of ΣANs + ΣPNs, whereas the cavity sampling from the 473 K TD inlet (ΣPNs) indicated ∼ 400 pptv. Since the signal in the ΣAN channel includes both the contribution of peroxy and alkyl nitrates, this implies that only 210 pptv (34 %) of the detected products can be attributed to alkyl nitrates, which is inconsistent with the high yields (60 %–100 %) of ANs that result from the reaction of NO3 with isoprene (Barnes et al., 1990; Berndt and Boge, 1997; Perring et al., 2009; Rollins et al., 2009; Kwan et al., 2012; Schwantes et al., 2015; IUPAC, 2021; Brownwood et al., 2021). Compared to ANs, we expect the mixing ratios of, for example, PAN, O2NOCH2C(CH3)=CHC(O)O2NO2 or methacryloyl peroxynitrate (MPAN) in this system to be negligible as their precursors such as O2NOCH2C(CH3)=CHCHO (Jenkin et al., 2015) or methacrolein (Kwok et al., 1996; Berndt and Boge, 1997; Schwantes et al., 2015) are oxidized only inefficiently in the dark. The formation of PNs only takes place once isoprene has been depleted so that secondary oxidation of the above-mentioned aldehydes by OH or NO3 leading to further acyl-peroxy radicals (which form PNs) become at least competitive to the primary oxidation of isoprene. This is however never the case in the present experiments as isoprene is continuously flowed into the chamber and remains according to model calculations (see below) at a level of ≈ 11.4 ppbv. Given the high abundance of O3 and isoprene in this system, ozonolysis of the latter together with the associated formation and decomposition of Criegee intermediates to acetylperoxy radicals CH3C(O)O2 (Nguyen et al., 2016; Vansco et al., 2020) should make PAN the most important, potential contributor to a signal in the PN cavity. However, according to the branching ratios given in Nguyen et al. (2016), this reaction path is a minor one and CH3C(O)O2 (and thus PAN) should be formed in negligible amounts.
In order to identify the origin of the unexpectedly high ΣPN signal when NO3 and isoprene are mixed in the dark, thermograms of the NO3 + isoprene system were recorded in an experiment where ∼ 2.8 ppbv of isoprene-derived nitrates (as measured with TDI-2 at 625 K) was generated by flowing NO2 (200 sccm of 1 ppmv) and isoprene (9.8 sccm of 46.5 ppmv) in 15 SLPM dry synthetic air (with 5 SLPM over the Hg lamp for generation of ∼ 150 ppbv O3). Similar to the experiment in Fig. 2, N2O5 mixing ratios are expected to be suppressed to a few parts per trillion by volume under these conditions so that its thermal dissociation (to NO2) did not contribute to the ΣPN and ΣAN signals. Using this chemical system, we simultaneously measured ISOP-NIT thermograms once steady state established using TDI-1 (quartz, glass beads, 10 cm heated section) and TDI-2 (quartz, no glass beads, 20 cm heated section) both initially held at 703 K. Subsequently, both TDIs were cooled to ambient temperature over a period of ∼ 1.75 h. The ΣAN signals from this experiment are plotted against the inlet temperature in Fig. 3a to generate the ISOP-NIT thermogram. This is displayed along with an isopropyl-nitrate thermogram (iPN, red data points) measured using the same inlets under the same flow conditions but using iPN diluted to 5.5 ppbv in dry synthetic air sampled directly through a PFA line (together with 1.5 ppbv NO2 impurity) to the instrument.
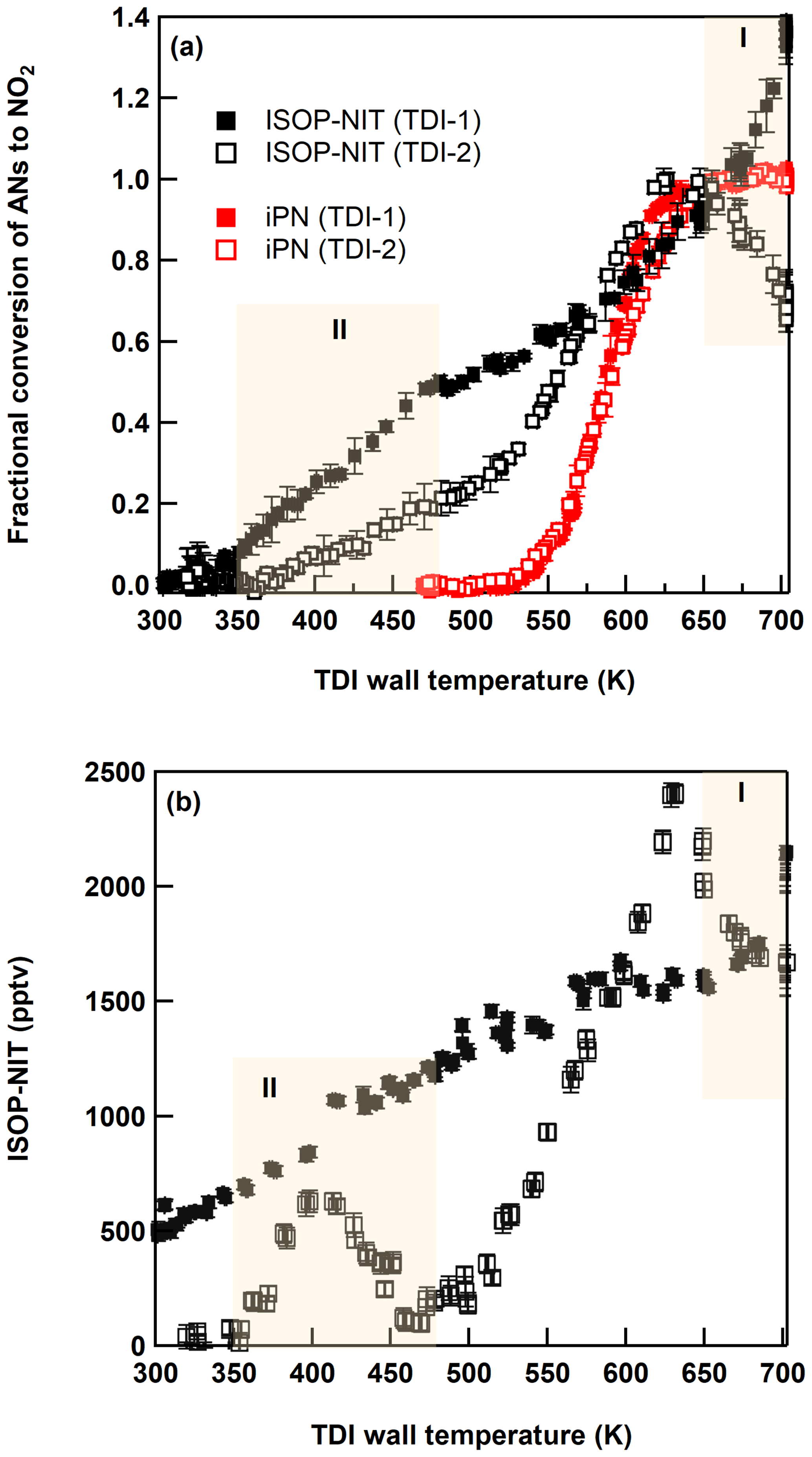
Figure 3(a) Thermograms (relative to the signal at 650 K) of isoprene-derived organic nitrates (ISOP-NIT) and isopropyl nitrate (iPN) obtained with TDI-1 (solid symbols) and TDI-2 (open symbols) under dry conditions. (b) Absolute thermograms of ISOP-NIT obtained with TDI-1 (solid symbols) and TDI-2 (open symbols) under humid conditions (RH = 34 %, 22 ∘C). Regions (I and II) with unexpected detection of NO2 are shaded yellow. Error bars denote standard deviation (1σ, 30 s) of the signal.
For iPN, we observe a well-defined onset of thermal dissociation at ∼ 525 K with a plateau (maximum conversion) at ∼ 650 K as reported previously for this set-up (Sobanski et al., 2016). When measuring iPN, TDI-1 results in a slightly steeper thermogram than TDI-2 in the 575–650 K range, which may be related to changes in gas flow and heat transfer within the inlet caused by the glass beads. Neither TD-inlet type results in dissociation to NO2 at temperatures < 500 K. In contrast, the ISOP-NIT thermograms (normalized to the signal at the plateau at 625 K of TDI-2) indicate formation of NO2 over a much broader range of temperatures (350–700 K).
The effect of humidifying the air was examined in an almost identical experiment conducted with NO2 (150 sccm of 1 ppmv) and isoprene (7 sccm of 46.5 ppmv) in 15 SLPM synthetic air with relative humidity (in SCHARK) of 33.5 % at 22 ∘C. In this case, ∼ 2.3 ppbv ISOP-NIT was formed. The thermograms obtained with TDI-1 and TDI-2 under these conditions are depicted in Fig. 3b. The broad thermogram measured with TDI-1 is very similar to that obtained under dry conditions (Fig. 3a), although even at room temperature an additional NO2 signal of 500 pptv is detected. Sampling via TDI-2 yields an ISOP-NIT thermogram that has similar features to that obtained under dry conditions, although the peak at ∼ 400 K has well-defined minima on both flanks and is shifted to higher temperatures. In separate experiments, humidified synthetic air (RH = 40 %, 23 ∘C) and NO2 (10.8 ppbv) were sampled through a PFA line directly to the instrument. The thermogram (in the absence of isoprene or ISOP-NIT) using TDI-2 was recorded and can be found in the Supplement (Fig. S2) revealing that the presence of water and NO2 in the inlet is sufficient to reproduce some features displayed in Fig. 3b with TDI-2. It is well known that H2O and NO2 can react on surfaces to form HONO and HNO3 (Pitts et al., 1984; Finlayson-Pitts et al., 2003), and their formation in SCHARK was verified in Sect. 3.1. In the presence of H2O, the efficiency of conversion of ISOP-NIT to NO2 drops to about 5 % at ∼ 460 K. This is much less than under dry conditions whereby 20 % conversion of ISOP-NIT was observed between 375 and 475 K (Fig. 3a). Within a framework for surface-catalysed conversion of ISOP-NIT to NO2 presented below, this observation can be interpreted as arising from the competitive adsorption to the surface of nitrated hydroperoxides and H2O; i.e. H2O (which is vastly more abundant) reduces the surface coverage of the organic nitrate at the surface.
The results in Figs. 2 and 3 show that separate detection of ANs and PNs based on their thermal dissociation can be problematic for the NO3 + isoprene system. Identifying the cause of this and providing potential solutions to circumvent the problem is the aim of this work. To do this, we first focus on the “dry” experiment and highlight two regions of the thermograms in which large deviations from the expected behaviour are observed.
3.1 Thermograms of ISOP-NIT
3.1.1 Region I (T>648 K)
Figure 3a indicates that, at temperatures above 648 K (shaded region I), the behaviour of the two TDIs diverges significantly: while use of TDI-1 (glass beads) results in an increase in NO2 with increasing temperature, the use of TDI-2 leads to a decrease in the NO2 signal in the same temperature range. The increase in NO2 continues at temperatures above that required to convert ANs to NO2, which implies the presence of a NO2-containing trace gas where the NO2 moiety is more strongly bound than in ANs.
In order to assess to which extent this behaviour is potentially caused by inorganic trace gases that are not directly related to isoprene oxidation, an experiment with only NO2 (2.75 ppbv) and O3 (146 ppbv) in 23 SLPM dry synthetic air was performed. The steady-state concentration of N2O5 + NO3 was measured as 78 pptv. The resulting thermograms using TDI-1 and TDI-2 and after subtraction of the signal from the NO2 cavity (i.e. unheated inlet) are depicted in Fig. 4. No significant additional signal is observed below 475 K (region I) in either of the inlets. In region II (T > 675 K) on the other hand, we observed an increase (by ∼ 500 pptv) in the signal at 703 K with TDI-1, whereas ∼ 50 pptv is lost in TDI-2. In order to identify the trace gas(es) responsible for the signals observed in the system without isoprene, an iodide chemical ionization mass spectrometer (I-CIMS; Eger et al., 2019) described in the Supplement (Sect. S8) was coupled to the experiment. As shown in the Supplement (Fig. S3) both HNO3 and nitrous acid (HONO) were observed as soon as O3 and NO2 were present in the chamber and their formation is enhanced in the presence of water vapour, which is a common phenomenon in Teflon chambers (Pitts et al., 1984). We also found that reversing the flows and sampling the air into the CIMS after passing through the TDI-1 or TDI-2 (at 475 K) resulted in removal of the HNO3. Sampling through TDI-1 also led to loss of HONO.
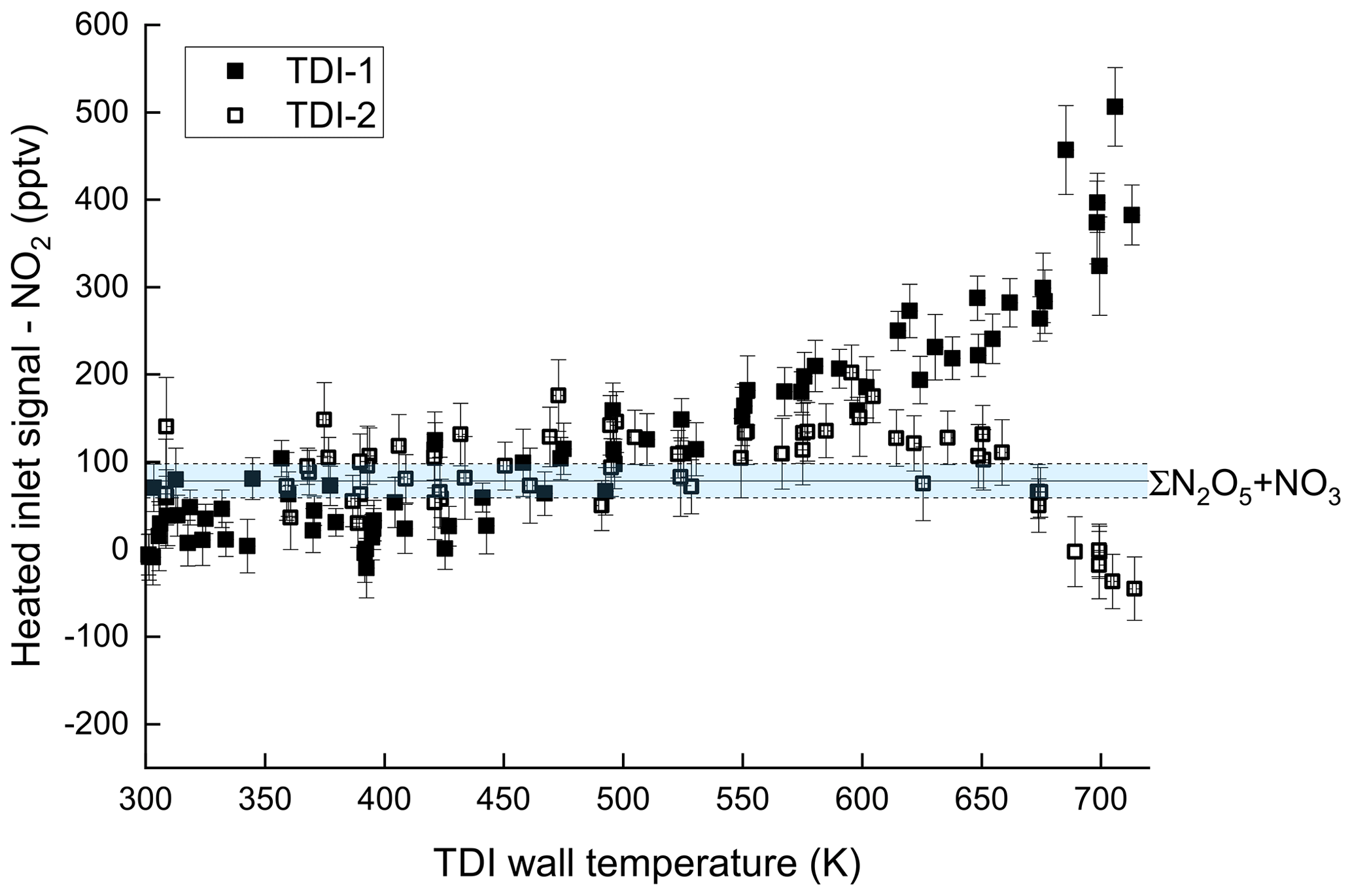
Figure 4Temperature-dependent NO2 detection when sampling 2.75 ppbv NO2 and 146 ppbv O3 in 23 SLPM dry synthetic air from SCHARK through TDI-1 and TDI-2. The signal from the NO2 cavity (no TDI) has been subtracted from both datasets. The mixing ratios of N2O5 + NO3 (with associated uncertainty) are indicated by the blue area.
Various TD-CRDS and TD-LIF instruments report the detection of HNO3 as NO2 following thermal dissociation at temperatures around 700 K (Day et al., 2002; Wild et al., 2014; Thieser et al., 2016). The sensitivity of the present set-up to HNO3 was investigated by sampling nitric acid from a calibrated permeation source (Friedrich et al., 2020) via TDI-1 and TDI-2 simultaneously. In these experiments, 22 ppbv HNO3 (with 780 pptv NO2 impurity) in dry synthetic air was delivered to the TDIs along with 350 ppbv O3. The HNO3 mixing ratio was derived using a known permeation rate (Friedrich et al., 2020) and dilution factor. Figure 5 shows the temperature-dependent conversion efficiency of HNO3 to NO2 in the presence of ozone (squares) with TDI-1 (black solid symbols) and TDI-2 (open symbols). Conversion of HNO3 to NO2 starts at ∼ 550 K and increases with rising temperature. At 680 K, the conversion efficiency is 23 % for TDI-1 and 8 % for TDI-2. No significant conversion of HNO3 to NO2 was observed when ozone was absent (blue data points) and was drastically reduced when the synthetic air was humidified to RH = 55 % at room temperature (red data points). The effect of water vapour is consistent with previous observations on the effect of humidity (Sobanski et al., 2016; Thieser et al., 2016; Friedrich et al., 2020).
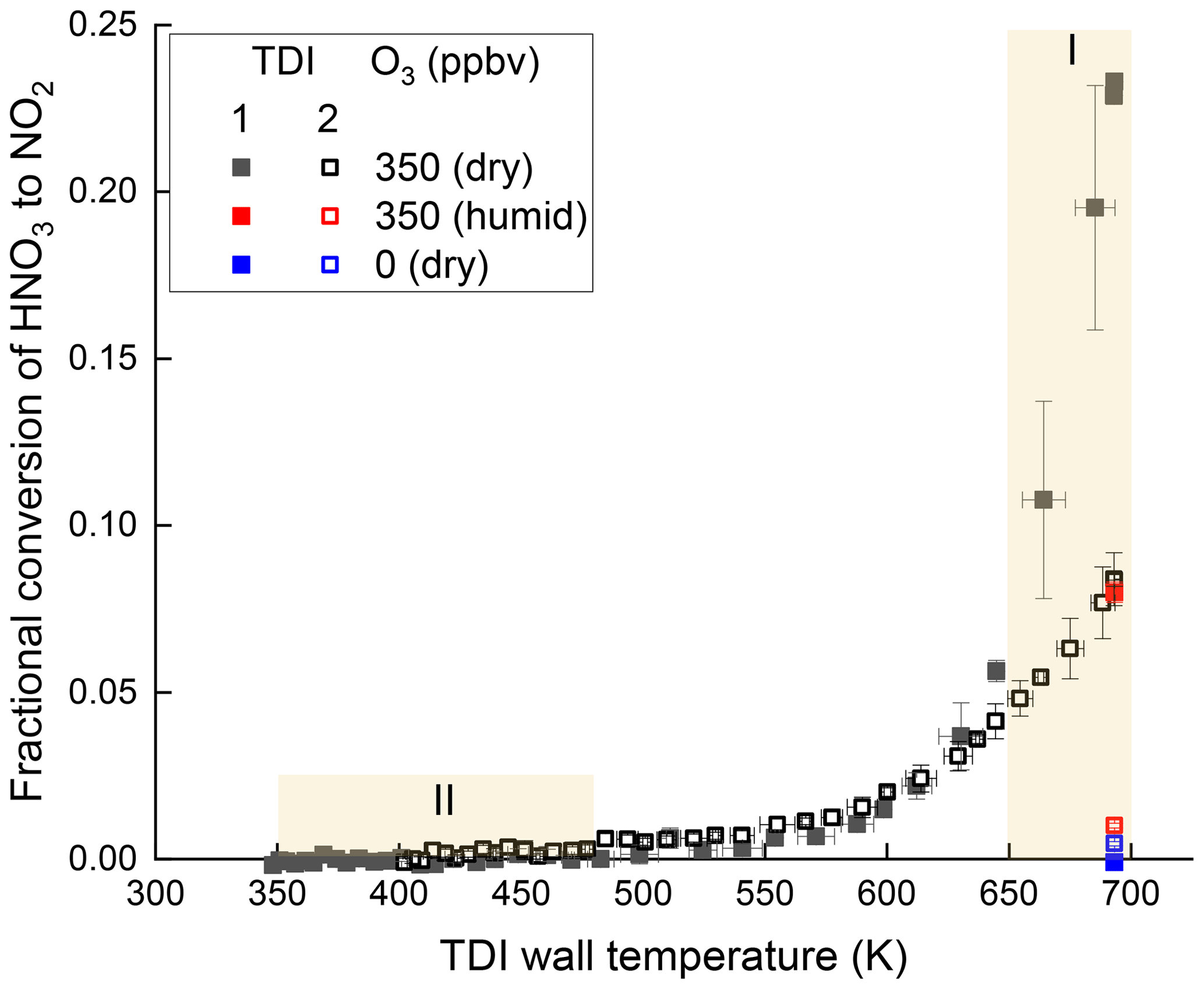
Figure 5Thermograms of nitric acid (22 ppbv) in either dry (black) or humidified air (RH = 55 % at 23 ∘C, red) obtained with TDI-1 (closed squares) and TDI-2 (open squares). The O3 mixing ratio was either zero or 350 ppbv in the non-humidified experiment and 350 ppbv in the humidified experiment. The error bars denote standard deviation (1σ, 1 min) of the signal.
The decomposition of HNO3 to NO2 thus only occurs in the presence of ozone under dry conditions, and its rate increases greatly at T > 650 K. This is consistent with the observations in Fig. 3b for TDI-1 and represents a likely explanation for the increase in signal when sampling from SCHARK to investigate the NO3 + isoprene system. The apparently more efficient (∼ factor three) conversion of HNO3 to NO2 in TDI-1 than in TDI-2 is explained by the loss of NO2 at high temperatures in TDI-2 through the reaction with O atoms (see Sect. 3.2). In TDI-1 this is prevented by the removal of O atoms by the glass beads (e.g. via scavenging or surface-catalysed recombination to O2).
The ozone-assisted conversion of HNO3 to NO2 cannot be explained by known gas-phase processes as the reaction between HNO3 and O(3P) (Reaction R12) has a low rate coefficient (k13 < 3 × 10−17 cm3 molecule−1s−1 at 298 K; Burkholder et al., 2016) and results mainly in the formation of OH and NO3 (Reaction R12). The more efficient conversion of HNO3 to NO2 in TDI-1 (with glass beads) compared to TDI-2 indicates that a surface-catalysed process involving either ozone or O(3P) is involved (Reaction R13).
Assuming that, in a Langmuir–Hinshelwood type process, the first step in the surface-catalysed reaction is physical adsorption of HNO3 to the surface, the strong reduction in conversion of HNO3 to NO2 under humid conditions is explained by the competitive adsorption of HNO3 and H2O, the latter favoured by its much larger concentrations. That is, H2O drives HNO3 from the surface and thus protects it from surface reactions.
3.1.2 Region II (T=350–475 K)
In region II (350–475 K, shaded area in Fig. 3), instead of the near-zero signal expected in the absence of significant amounts of PNs or N2O5, we observe a monotonic increase in NO2 with the temperature which is a factor of ∼ 2 steeper in TDI-1 than in TDI-2. The signal in TDI-1 at 475 K, where only PNs are expected to dissociate, is ∼ 50 % of the maximum signal at 650 K. There are several potential explanations for this behaviour, which include the following: (1) the formation and detection of thermally less stable ANs (e.g. including dinitrates), which dissociate at lower temperatures than, for example, iPN; (2) the formation of non-acyl, isoprene-derived peroxy nitrates (RO2NO2) that are sufficiently long-lived to build up to appreciable concentrations in SCHARK; and (3) chemical processes taking place in the TD inlets that convert ISOP-NIT to NO2. Scenario (1) appears unlikely as several studies have shown that the O–N bond strength in various alkyl nitrates is very similar (Hao et al., 1994; Wild et al., 2014). We also note that the formation of dinitrates (in the absence of NO) only takes place when isoprene levels are very low and the first-generation nitrates formed in the NO3 + isoprene reaction can react with a further NO3. This can be ruled out for the present experiments in which the isoprene mixing ratio is always much larger than that of the first-generation nitrates formed, which in any case react much more slowly with NO3 than does isoprene. The second explanation requires that RO2 formed in the initial reaction between NO3 and isoprene react with NO2 to form RO2NO2. Given our experimental conditions, we would indeed expect that the main fate of any RO2 formed in the reaction between NO3 and isoprene is reaction with NO2, which will dominate over self-reaction or reaction with NO3, other RO2 or HO2. Non-acyl RO2NO2 are however generally highly thermally unstable, with lifetimes (at room temperature) of seconds or minutes, with respect to re-dissociation to RO2 + NO2.
For isoprene-derived RO2NO2 to contribute to the signal observed in region II would require that the RO2–NO2 bond strength be comparable to those of acyl nitrates such as PAN. The dominant 1,4-peroxy radical formed when NO3 reacts with isoprene has a nitrate group separated by two carbon atoms from the peroxy carbon. It seems unlikely that this could have a stabilizing effect on the O–N bond in RO2NO2 in the same way that an α-carbonyl group does. Indeed, chamber experiments investigating the products of the NO3 + isoprene reaction in detail (Barnes et al., 1990; Wu et al., 2021) have identified neither acyl- nor non-acyl-RO2NO2 as stable or semi-stable products formed from primary oxidation.
In support of scenario 3, Sect. 3.2 to 3.3 describe the evidence for chemical reactions leading to NO2 formation that bypass the thermodynamic barrier for direct NO2 formation but are surface-catalysed and require the presence of O3 in either SCHARK or in the inlet. These processes are peculiar to alkyl nitrates with a C=C double bond and thus have not been observed in TD inlets tested only with saturated alkyl nitrates such as the frequently used isopropyl nitrate.
3.2 The role of O3
To further investigate the conversion of ISOP-NITs to NO2 at low temperatures in the TD inlets, we generated NO3 via the room-temperature thermal decomposition of N2O5, thus ruling out chemical processes that were initiated or catalysed by O3.
In these experiments, N2O5 was transported into SCHARK by passing a flow of 0.1 SLPM dry synthetic air over a crystalline N2O5 sample cooled to −78 ∘C with further dilution in a 15 SLPM flow of zero air. The combined concentration of N2O5 + NO3 that remains after the reaction with ∼ 22 ppbv isoprene (7 sccm of 46.5 ppmv) was measured as 40.5 pptv. The use of high isoprene concentrations guarantees that the thermal decomposition of N2O5 does not contribute significantly to the thermograms. Under these conditions several parts per billion by volume of ISOP-NIT were formed. A constant flow (2 SLPM) of zero air was passed over a low-pressure Hg lamp and added between the sampling port of SCHARK and the inlets and TD inlets of the NO2, PN and AN cavities. This way the O3 mixing ratio in the TD inlets could be varied without affecting the chemistry in the chamber.
Figure 6 presents the results of one such experiment in which ISOP-NIT was sampled from SCHARK using TDI-1 and TDI-2, both initially held at 703 K with thermograms obtained by decreasing the temperature of both inlets in 25 K steps. At each temperature step (periods of 20 min), after recording the signal under O3-free conditions (black squares), a low (40–54 ppbv, green triangles), medium (97–111 ppbv, blue triangles) and high (185–219 ppbv, orange circles) mixing ratio of O3 was added in front of the TD inlets. Before cooling to the next temperature, the signal without O3 was measured again and agreed within 30–150 pptv to the value at the beginning of the corresponding period. To enable comparison with “ideal” behaviour, the thermogram of isopropyl nitrate (iPN, red circles) recorded from an experiment while flowing 162 ppbv O3 (and 3 ppbv NO2 impurity) through SCHARK is also plotted.
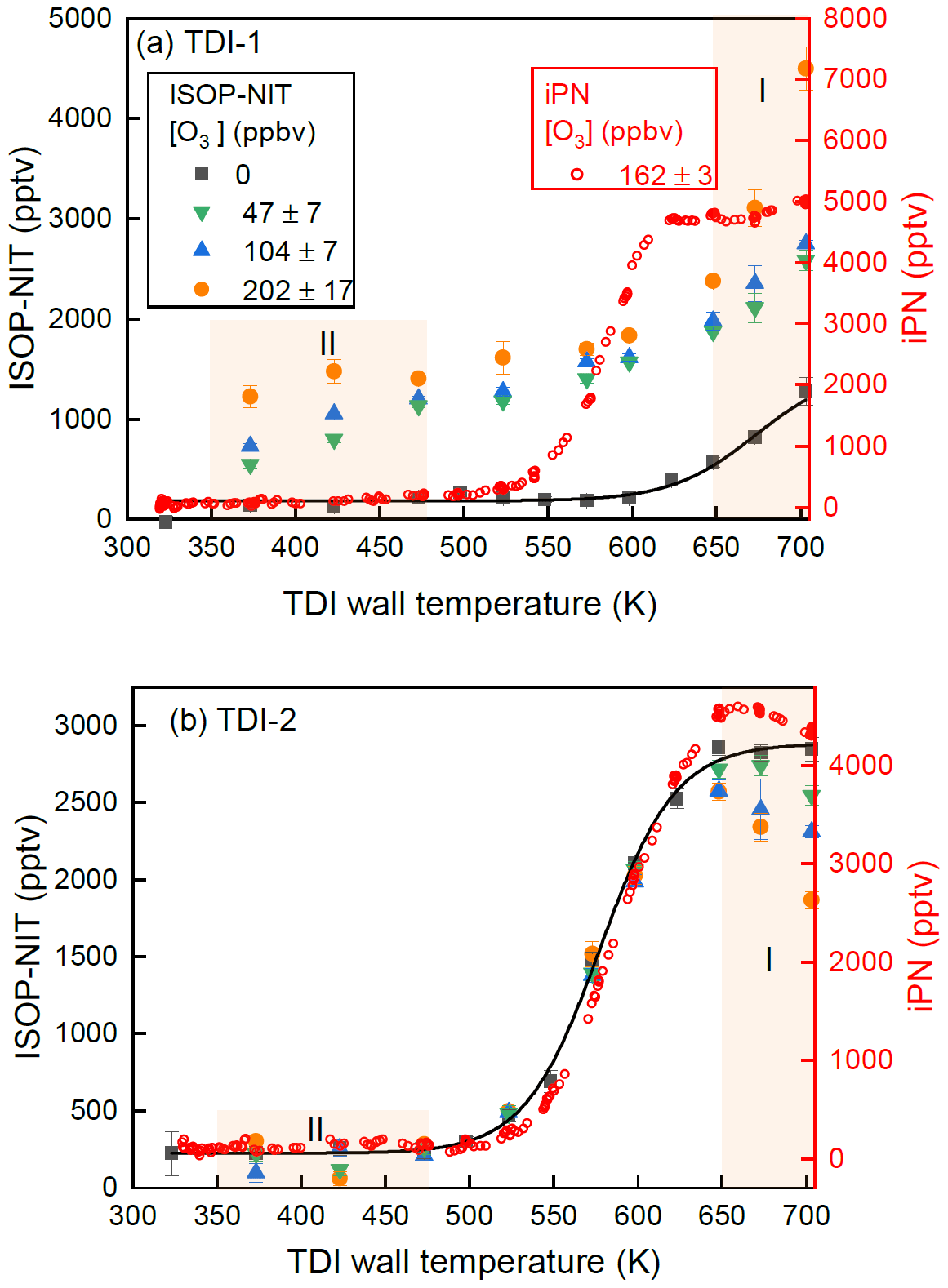
Figure 6(a) Thermograms of isoprene-derived nitrates generated by mixing N2O5 (as NO3 source) and ∼ 20 ppbv isoprene in SCHARK under dry conditions and sampling via TDI-1. The mixing ratio of O3 (added only to the inlets) was varied from 0 to 202 ppbv. A thermogram of isopropyl nitrate (iPN) in the presence of 162 ppbv O3 (and 3 ppbv NO2 impurity) sampled through the same TDI is shown for comparison. The black solid line is a Boltzmann sigmoidal fit of the ozone-free experiment. The error bars show the standard deviation of the signal (1σ). (b) Same as (a) but using TDI-2.
Figure 6a displays a thermogram obtained when sampling ISOP-NITs from SCHARK via TDI-1 (glass beads). In the presence of O3, the thermograms are very broad with substantial NO2 formation between 350 and 475 K (shaded region II) and in this sense are comparable to those displayed in Fig. 3, where NO3 was obtained from the reaction of NO2 with O3. In region I, the effect of going from ∼ 50 to ∼ 200 ppb of ozone is to increase the NO2 generated drastically. This is the opposite of that observed when sampling via TDI-2 and thus in agreement with the results of the experiment depicted in Fig. 3, where the increase in signal was assigned to detection of HNO3. For the experiments in which NO3 was generated in the room-temperature thermal dissociation of N2O5, HNO3 can arise from reactions of N2O5 with moisture on the walls and is present as impurity in the N2O5 sample. As described above, the presence of glass beads has two effects which operate in the same direction in this temperature regime: the conversion of HNO3 to NO2 is catalysed by the surface provided by the glass beads, and at the same time the loss of NO2 (via reaction with O(3P)) is reduced as O(3P) is scavenged by the glass surface.
For TDI-1, the thermograms obtained without ozone (black squares) differ greatly to those in which ozone was present. Without ozone, NO2 is not generated at temperatures lower than 550 K but its concentration increases rapidly at temperatures above ∼ 600 K with no indication of a plateau being reached. The thermograms obtained in this NO3–isoprene system using TDI-1 in the absence of ozone bear little resemblance to that of iPN. Furthermore, during periods without ozone or heat in TDI-1, compounds of lower volatility like ISOP-NITs or HNO3 appear to deposit on the glass beads and frit. This would form an explanation for a “memory effect” observed for TDI-1, whereby after exposure to HNO3 or organic nitrates during unheated periods, an increase in the NO2 signal followed by a slow decrease taking several hours is observed as soon as pure synthetic air and ozone were added to the flow through the heated inlet. An example of this phenomenon is shown in Fig. S4 in which (at peak signal) 60 ppbv of NO2 was detected just by heating TDI-1 to 703 K in the presence of O3 in synthetic air. In order to avoid bias in results caused by this effect, a cleaning procedure was adopted prior to all experiments whereby the inlet was heated to 703 K and exposed to ozone in synthetic air until a constant, low residual signal, usually between 20 and 200 pptv, was established. This memory effect seen for TDI-1 is also observed when a thermogram of ISOP-NIT (generated in a system similar to the experiment in Fig. 3) is measured by going from cold to hot temperatures (Fig. S5).
In Fig. 6b we present the results of the same experiment using TDI-2. The non-zero signal (∼ 250 pptv) at temperatures between ∼ 320 and 450 K) results from instability in the baseline. In the absence of ozone, the organic nitrates generated in the NO3-initiated oxidation of isoprene follow a well-defined thermogram (black squares and solid line) between 475 and 650 K, which is very similar to the thermogram measured for iPN (red circles). The addition of ozone does not result in the formation of NO2 in region II but does induce NO2 losses for temperatures above 650 K (region I). The fact that ISOP-NIT was not converted to NO2 at temperatures < 475 K suggests that the observation of a large signal in Fig. 3a (region II, white squares) is linked to O3-induced chemistry in SCHARK, which will be discussed in more detail in Sect. 3.3. The loss of NO2 in region I increases with increasing amounts of O3 with ∼ 35 % of the NO2 formed at 625 K lost when O3 was increased to ∼ 200 ppbv. The same behaviour is observed in the thermogram of iPN, which confirms that this process is independent of the nature of the nitrate but solely linked to thermal decomposition of O3 and subsequent reactions of O(3P) with NO2.
3.2.1 Thermal dissociation and gas-phase reactions of O3 and O(3P)
An important clue to the underlying chemical processes that lead to the conversion of ANs to NO2 at temperatures lower than those required to break the O–N bond is the fact that the thermogram of iPN (measured with TDI-1) is not significantly affected by the presence of 163 ppbv O3, whereas thermograms of ISOP-NIT, the vast majority of which are unsaturated, are greatly broadened when O3 is present.
It is well known that O(3P) reacts rapidly (via electrophilic addition) to C=C double bonds (Leonori et al., 2015), and we thus assessed the potential impact of NO2 formation via reactions of O3 or O(3P) (formed in the thermal dissociation of O3 in the TDIs) with ISOP-NIT.
Table 1Central reactions used for numerical simulation of the thermal dissociation of organic nitrates in TDI-2.

Notes: a value for 2-methyl-2-butene used. b Value for isoprene used.
The concentration of O(3P) in the TDIs depends on the concentration and rate of thermal decomposition of O3 and thus on the gas temperature as well as its rate of recombination with O2, reactions with O3, NO2, isoprene, isoprene nitrates and loss to the walls:
The contribution (in addition to the thermal dissociation of ISOP-NIT) to NO2 formation via Reactions (R19) and (R20) in TDI-2 was assessed via numerical simulation (FACSIMILE/CHEKMAT release H010; Curtis and Sweetenham, 1987). The rate coefficients for the most important reactions are listed in Table 1; the complete reaction scheme is listed in the Supplement (Sect. S9). Reaction times in heated and unheated section of TDI-2 were calculated from the temperature, internal diameter of the quartz tube and the flow rate. The rate coefficients for the gas-phase reactions of isoprene (IUPAC, 2021) and 2-methyl-2-butene (Herron and Huie, 1973) with O(3P) and O3 were used as surrogates for the reactions of ISOP-NIT for which data are not available. The temperature-dependent dissociation rate coefficient of n-propyl nitrate was used to account for NO2 from the thermal dissociation of isoprene-derived nitrates (Morin and Bedjanian, 2017). Wall loss of O(3P) in the instrument was estimated to be 90 s−1 using the method as described in Thieser et al. (2016) and implemented in the model run.
The initial conditions for the simulation were 1 ppbv ISOP-NIT, 10 ppbv isoprene and 5 ppbv NO2 at a cavity pressure of 724 hPa and a temperature of 298 K. The results obtained are shown by black curves in Fig. 7. For temperatures up to 575 K the simulated thermograms with and without O3 are almost identical, whereas at higher temperatures, the amount of NO2 exiting the inlet decreases because of its reaction with O(3P) (Reaction R17), in broad agreement with the experiments carried out using TDI-2 as shown in Fig. 3b. The model simulations show (Fig. 7) that almost no O(3P) is formed by the thermal dissociation of O3 at the lower temperatures of region II. Only at higher temperatures is a significant fraction of O3 (27 % at 703 K) converted to O(3P) with the majority subsequently lost at the inlet walls. These calculations underline the observation that the low temperature formation of NO2 from ISOP-NIT seen when using TDI-1 cannot be explained with known gas-phase chemistry.
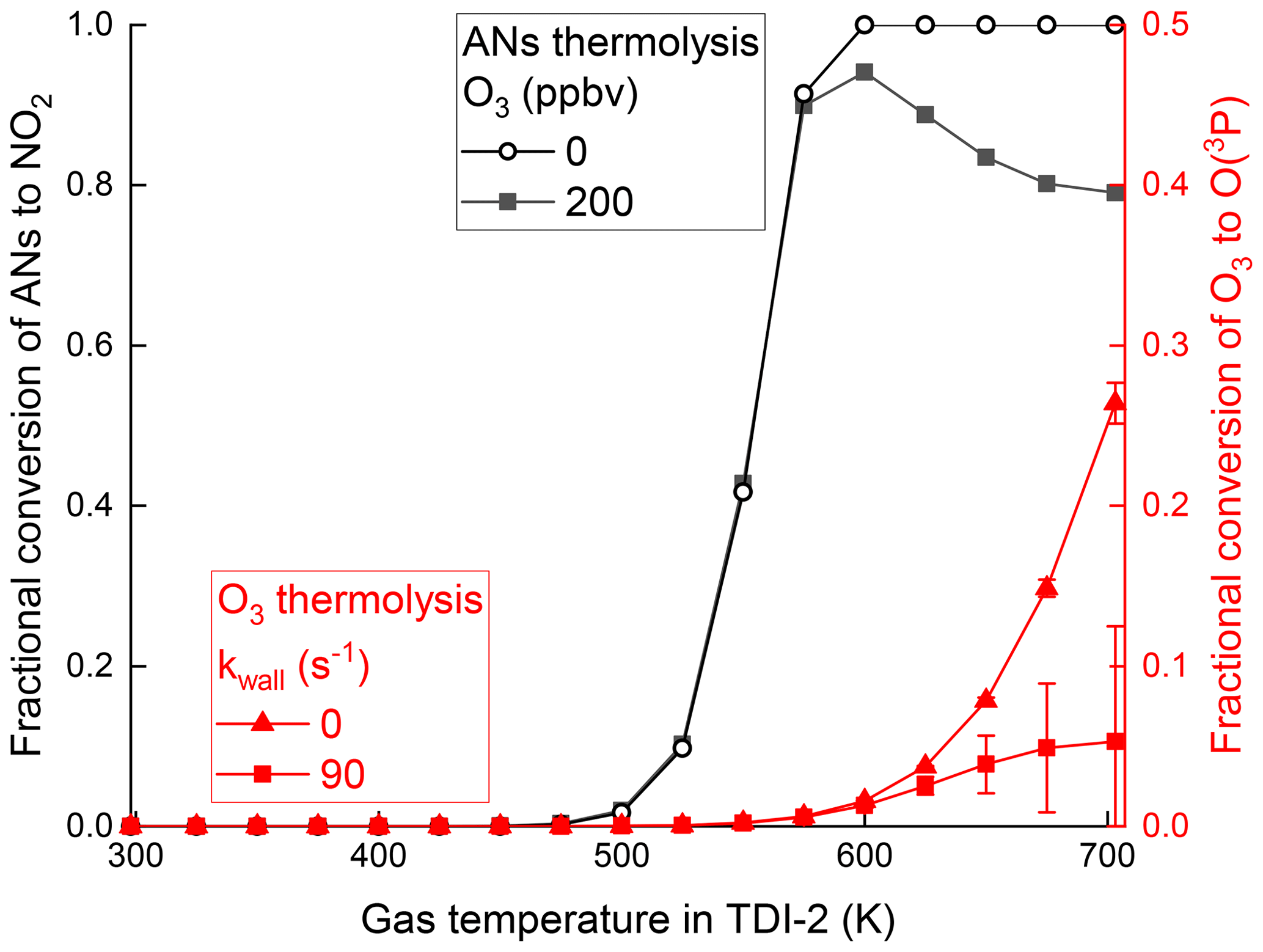
Figure 7Black lines and data points denote simulated, fractional conversion of n-propyl nitrate to NO2 for TDI-2 without and with O3 (200 ppbv). Red lines and data points denote simulated fractional conversion of O3 to O(3P) within the heated section of TDI-2 with and without wall loss of O(3P). Error bars denote the standard deviation (1σ) of the fractional conversion changing over time passed in the heated section.
In summary, the experimental observations and the numerical simulations indicate that the presence of O3 is required in the inlet for TDI-1 and in the chamber for TDI-2 to generate NO2 from isoprene-derived nitrates at temperatures less than 475 K. We have shown that the generation of NO2 from alkyl nitrates at low temperatures using TDI-1 requires that the organic nitrate has a double bond and that, while gas-phase reactions of O(3P) are responsible for the loss of NO2 at high temperatures, they are not responsible for the conversion of isoprene-derived nitrates at lower temperatures in either of the TD inlets. The presence of glass beads (large surface area) favours the formation of NO2 from ISOP-NIT at low temperatures. Altogether, these observations indicate that a surface-catalysed reaction involving ozone is the process most likely to be responsible for the conversion of ISOP-NIT to NO2 at temperatures below those required for the gas-phase thermolysis.
3.2.2 Surface-catalysed reactions with ozone
Quartz tubes contain impurities and surface defects that can provide reactive sites (RSs) at elevated temperatures, and the surface-catalysed chemistry of ozone on, for example, mineral silicates is well known (Bulanin et al., 1994; Hanisch and Crowley, 2003a, b; Usher et al., 2003). O3 can be surface-catalytically converted to O2 (Reaction R25) by the formation and loss of reactive, oxygenated surface sites (RSOs) via Reactions (R23) and (R24).
In order to test for ozone loss in our set-up, O3 in synthetic air was passed through the TDIs. The O3 mixing ratio prior to entering the inlets was measured continuously using the ozone monitor (UV absorption). The ozone exiting the TD inlets was converted to NO2 (by addition of 1 ppmv NO (Reaction R4) in a 1.5 m long PFA tubing with 0.5 in. OD and a residence time of 5.2 s) and then measured in the 409 nm cavities. Figure 8a shows the results of such an experiment for TDI-1. The concentration of ozone before entering the inlets was 20.5 ppbv (open dots), which was detected as 17.1 ppbv after passing through the inlet at 298 K (black squares, 11:15 to 11:55 LT). This represents a transition efficiency of 0.83, which matches that expected when considering the reaction time, NO concentration and the rate coefficient (k4 = 1.9 × 10−14 cm3 molecule−1 s−1, IUPAC, 2021).
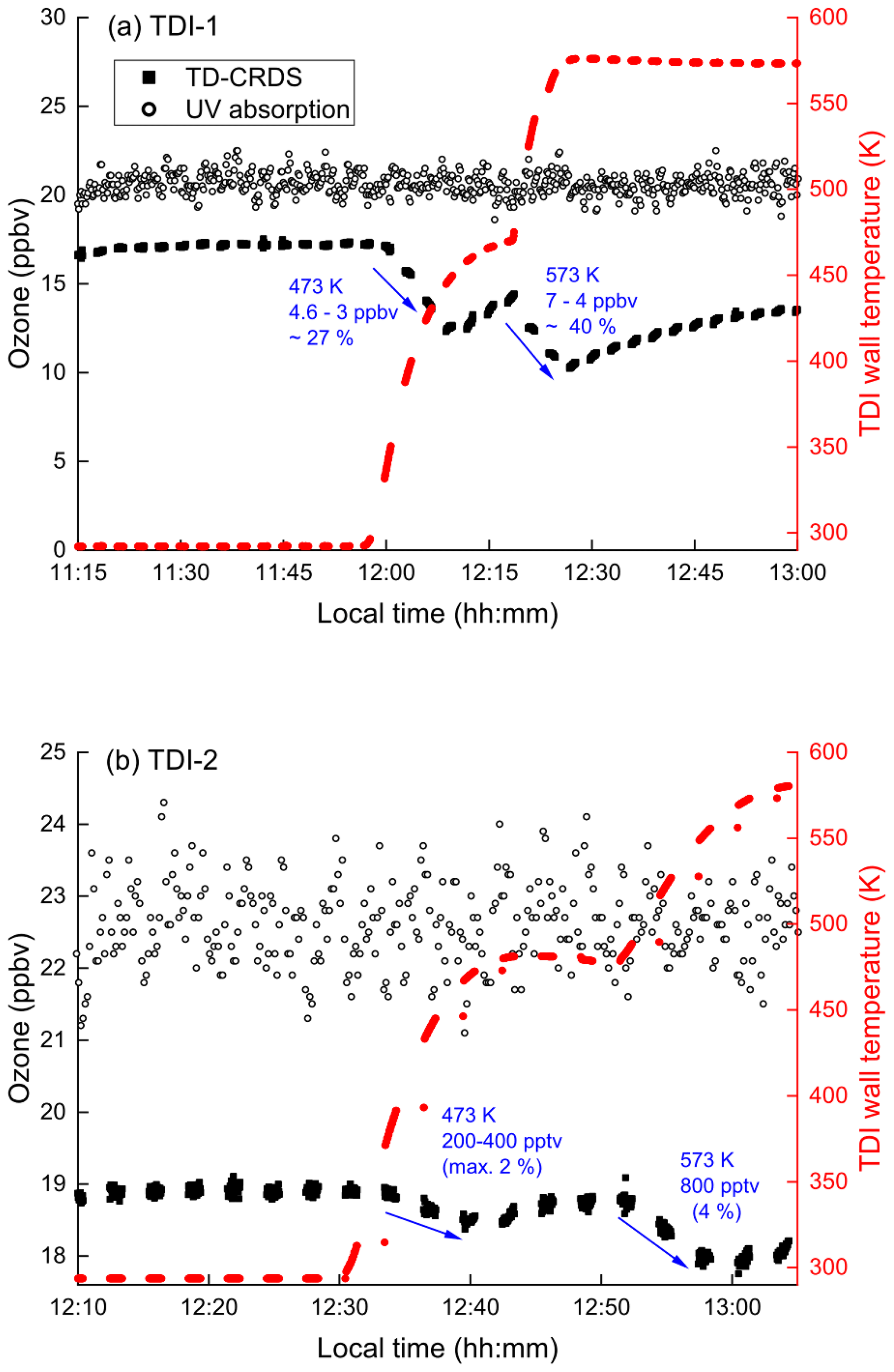
Figure 8Time series of O3 mixing ratios before and after passing though TDI-1 (a) or TDI-2 (b).
At ≈ 12:00 LT, upon heating the inlet to 473 K the ozone exiting TDI-1 was depleted by up to 27 %, while heating to 573 K results in further loss (up to 40 %). An analogous experiment using TDI-2 (Fig. 8b) showed that while some O3 was also lost (e.g. 4 % at 573 K), this was much less than in TDI-1. As the gas-phase thermal decomposition of O3 is negligible under these conditions (0.6 % at 575 K, Fig. 7), the loss of O3 when passing through the TD inlets indicates that surface-catalysed ozone decomposition takes place (Reactions R23 and R24), especially in TDI-1 where larger surface areas are available.
We now consider the possibility that the conversion of isoprene-derived nitrates to NO2 can be catalysed by surfaces in the presence of O3. We note that previous work has shown that the heterogeneous ozonolysis of alkenes on glass or other surfaces can be more efficient than its analogous, gas-phase process (Dubowski et al., 2004; Stokes et al., 2008; Ray et al., 2013) and now consider the possibility that RSO is the mediating reactive species in TDI-1 in our experiments.
We first examine the possible contributors to ISOP-NIT formed in the reaction of NO3 with isoprene, in which the dominant initial step (in air) is a sequential 1,4 addition of NO3 and O2 to form δ- and β-peroxy radicals, e.g. O2NOCH2C(CH3)=CHCH2OO (Schwantes et al., 2015). In the presence of NO3, RO2 or HO2, the peroxy radicals react further to form “first-generation” isoprene nitrates which contain carbonyl, hydroperoxidic and alcoholic groups such as O2NOCH2C(CH3)=CHCHO, O2NOCH2C(CH3)=CHCH2OOH and O2NOCH2C(CH3)=CHCH2OH, respectively. Note that most of the known, first-generation organic nitrates retain a C=C double bond.
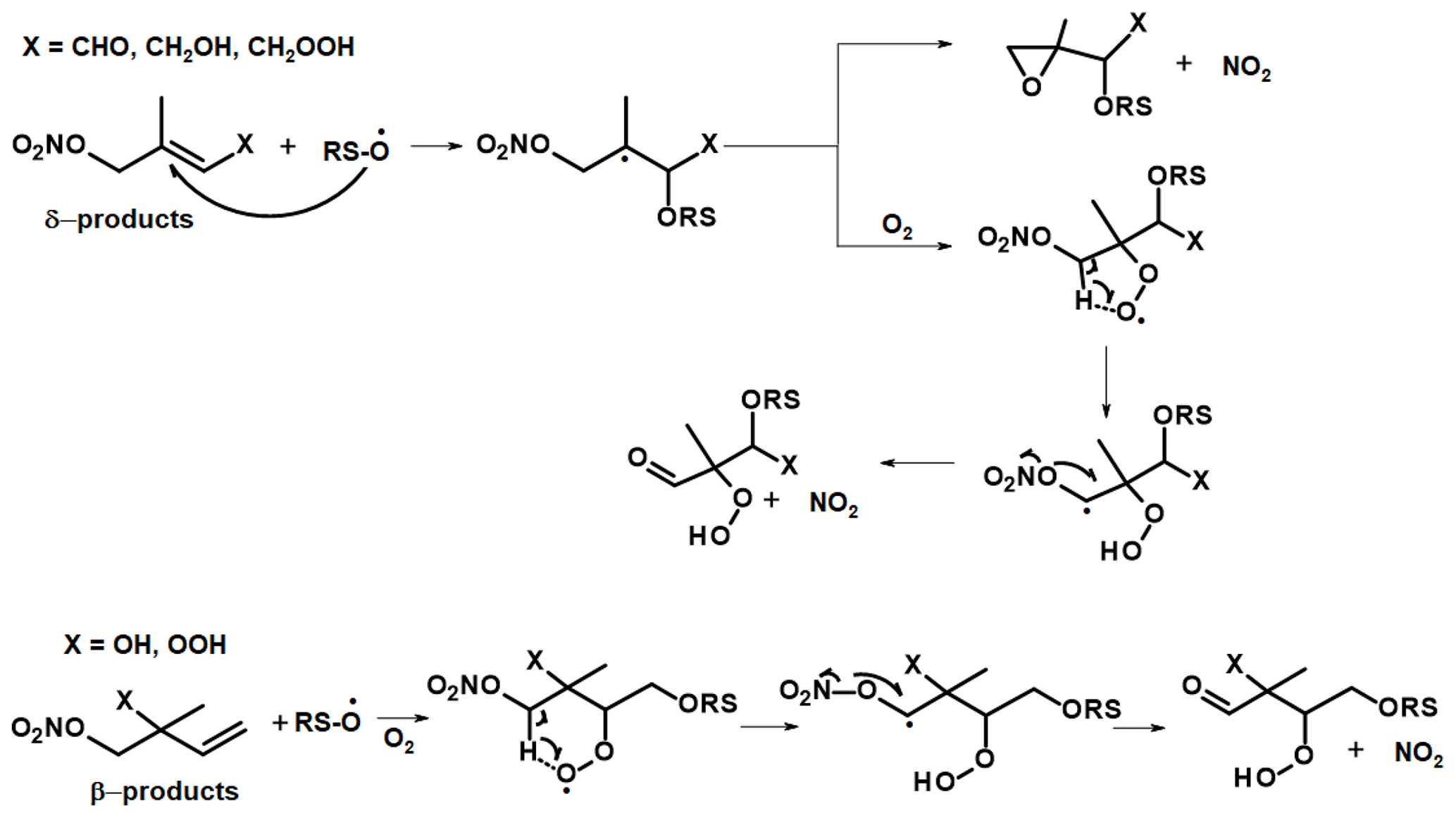
Figure 9Possible mechanism for surface-catalysed conversion of first-generation ISOP-NIT in TDI-1. RS-O represents a reactive surface site.
A hypothetical ISOP-NIT degradation scheme involving the initial attachment of RS-O to the remaining double bond is given in Fig. 9. We consider only the fate of the most stable surface adducts, i.e. tertiary radical in case of δ-products and secondary radicals in case of β-products. Both radical adducts will react with O2 to form organic peroxy radicals which may undergo H shifts (via five- or six-membered rings) resulting in formation of a radical with its unpaired electron in the direct vicinity of the nitrate functionality. Such unimolecular processes may become competitive to bimolecular reactions under atmospheric conditions (Møller et al., 2019). A possible fate of this radical is decomposition to form a carbonyl compound via NO2 elimination (Hjorth et al., 1990; Berndt and Boge, 1995; Vereecken et al., 2021). For δ-products, the tertiary product may also eliminate NO2 under the formation of an epoxide.
With TDI-2, the conversion of ISOP-NIT to NO2 at low temperatures (region I) is only observed when O3 is present in the chamber (Fig. 3a), but not when it is added only to the inlet (Fig. 6b) implying that the presence of O3 as an oxidizing agent in SCHARK is the main difference between these two reaction systems. This suggests that the ozonolysis of isoprene may play an important role. As the reaction between isoprene and O3 leads to the formation of OH and additional HO2 (Zhang et al., 2002; Malkin et al., 2010; Cox et al., 2020), the fraction of RO2 reacting with HO2 in this system is enhanced when compared to the experiments in which NO3 was generated from N2O5. The major product from the reaction between nitrated δ- and β-peroxy radicals with HO2 is hydroperoxides, such as O2NOCH2C(CH3)=CHCH2OOH (Schwantes et al., 2015). With the intention of assessing the effect of O3 on hydroperoxide yields, further model calculations were performed using the Framework for 0-D Atmospheric Modelling (F0AM) (Wolfe et al., 2016). The NO3 isoprene oxidation scheme is still the subject of current research and uncertain, which is why the Master Chemical Mechanism (MCM, version 3.3.1, http://mcm.leeds.ac.uk, last access: 7 August 2021) (Jenkin et al., 2015) and the Reduced Caltech Isoprene Mechanism Plus (RCIMP, version 5, https://data.caltech.edu/records/247, last access: 7 August 2021) (Wennberg et al., 2018; Bates and Jacob, 2019) were applied. Figure 10 shows the calculated fraction of hydroperoxides to ISOP-NIT using both models and mixing 22 ppbv isoprene with either 10.8 ppbv NO2 and 150 ppbv O3 (runs 1 and 3) or 3 ppbv N2O5 (runs 2 and 4) to achieve NO3 in a flow-through chamber with an exchange rate of 2.73 × 10−4 s−1. Generally, the MCM predicts higher hydroperoxide yields than RCIMP. A possible reason might be that MCM has higher rates of HO2 reformation from reactions of isoprene-derived alkoxy radicals than RCIMP does. For that reason, MCM only predicts an increase in the hydroperoxide yield by a factor of 1.33 when the NO3 source is changed from N2O5 to NO2 + O3 (model runs 1 and 2), whereas RCIMP predicts a factor of 2.56 (model runs 3 and 4). The calculations thus broadly support the hypothesis that unsaturated, nitrated hydroperoxides are involved in the low temperature (surface-catalysed) formation of NO2 from ISOP-NIT. Indeed, hydroperoxides not only have high affinity for surfaces but also have a rather weak O–O bond with a dissociation energy of ≈ 45 kcal mol−1 (Bach and Schlegel, 2020), and several routes to surface-catalysed elimination of NO2 appear feasible. Decomposition of isoprene-derived hydroperoxides within the sampling line has been observed for other instruments (Rivera-Rios et al., 2014). A possible degradation mechanism for an isoprene-derived hydroperoxide which is abundant from the NO3 + isoprene system (Schwantes et al., 2015) is depicted in Fig. 11. In this case, O2NOCH2C(CH3)=CHCH2OOH coordinates via hydrogen bonds to the SiO2 surface prior to cleavage of the O–O bond to form OH and an alkoxy radical. The latter may dissociate via unimolecular reactions to form closed-shell products under elimination of NO2 (Wennberg et al., 2018; Vereecken et al., 2021).
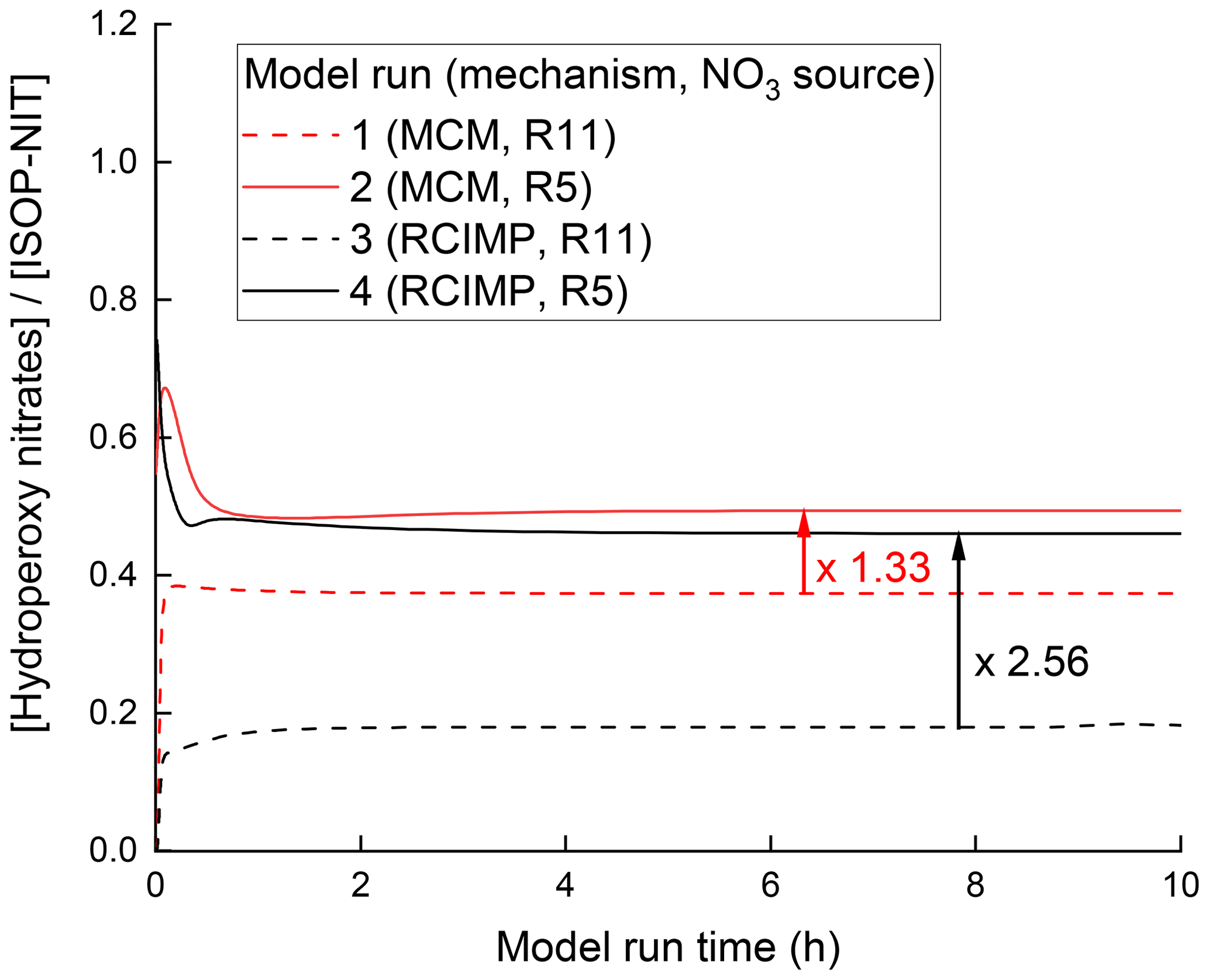
Figure 10Simulated fractional contribution of hydroperoxy nitrates to ISOP-NIT using schemes taken from the MCM (red) and from RCIMP (black). The simulations reproduce the experimental conditions in Figs. 3 and 6, with 22 ppbv of isoprene together with either 3 ppbv N2O5 as NO3 source (runs 1 and 3) or 10.8 ppbv NO2 + 150 ppbv O3 (runs 2 and 4).
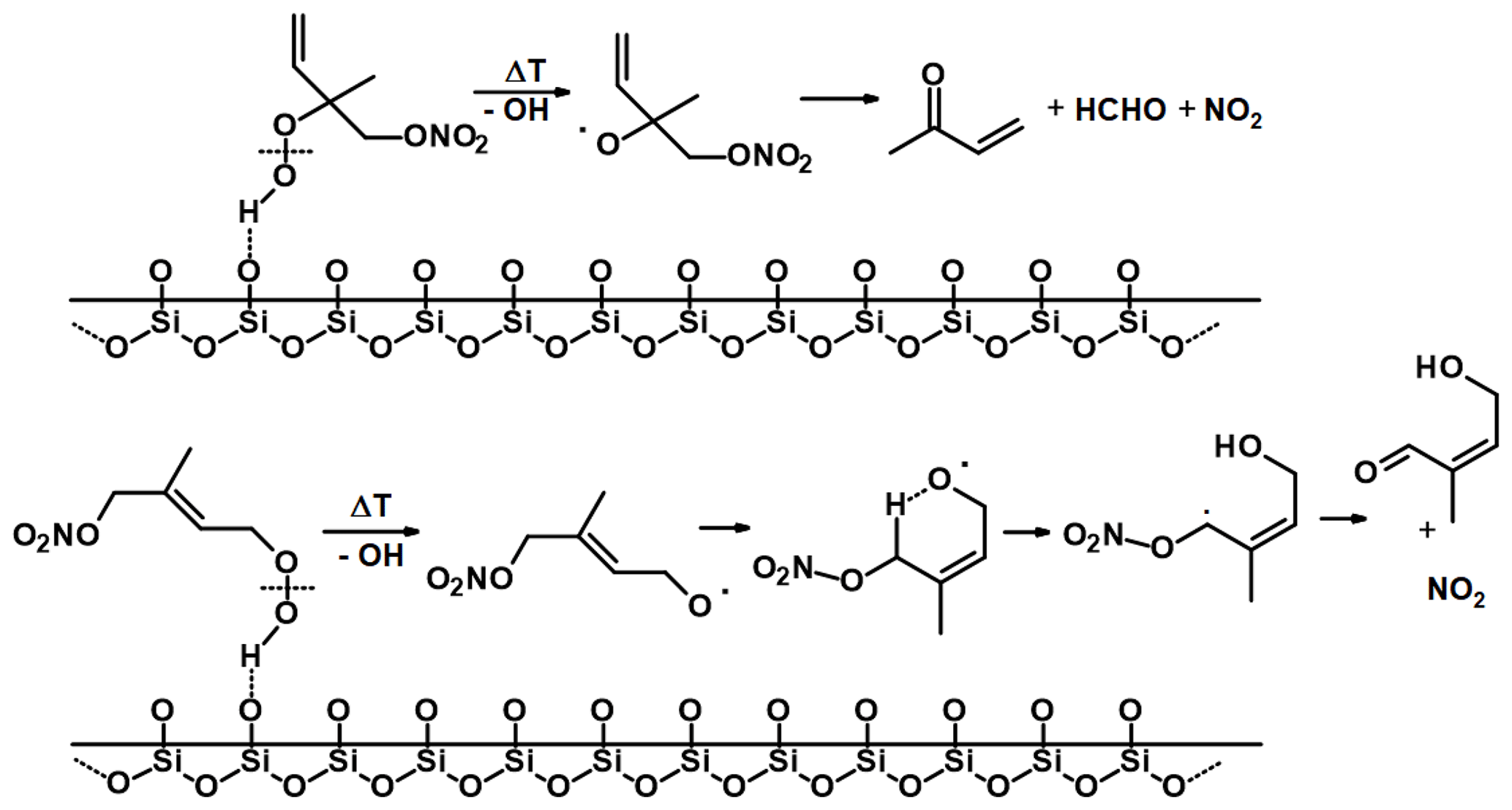
Figure 11Potential degradation pathways of isoprene-derived hydroperoxides on a heated quartz surface.
3.3 Elimination of O3 and surface-catalysed conversion of ANs at low temperatures
Our findings clearly show that the combination of heated quartz surfaces and ozone catalyse the decomposition of the unsaturated nitrates formed in the reaction between NO3 and isoprene at temperatures below 473 K. While the exemplary, surface-catalysed processes depicted in Figs. 9 and 11 fulfil the requirement of conversion of ISOP-NIT to NO2, we stress that these are purely hypothetical and we cannot state with any certainty that they are the reactions responsible for our observations. While it would be highly interesting to investigate such processes using different (e.g. surface sensitive) techniques, this is clearly beyond the scope of this paper and of the experimental capabilities of this research project. Instead, we take the more pragmatic approach and indicate potential methods to eliminate such unwanted reactions when using TD inlets.
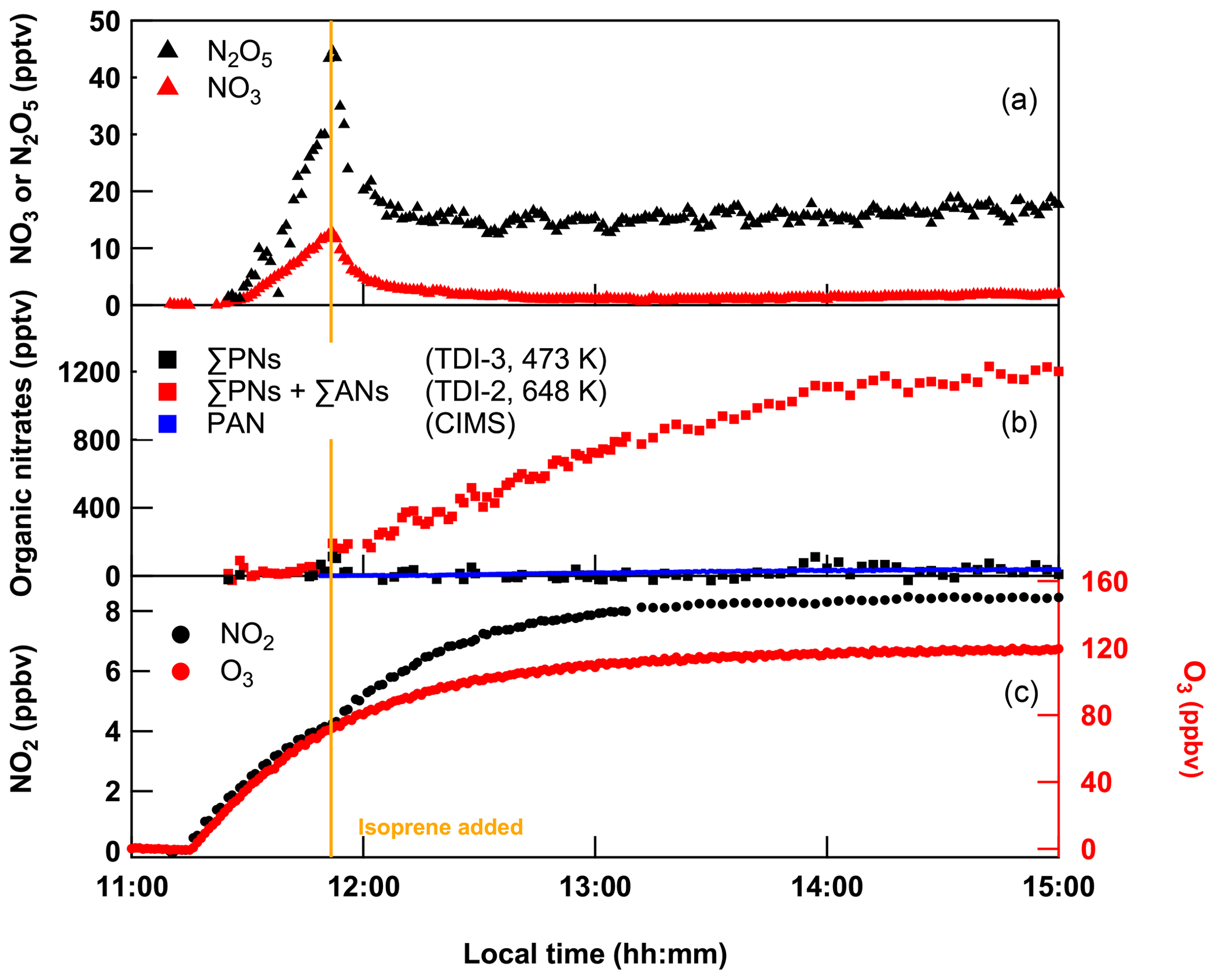
Figure 12Mixing ratios of NO2, ΣPNs with TDI-3 and ΣPNs + ΣANs with TDI-2 obtained from constant introduction of 200 sccm NO2, and 9.8 sccm isoprene in 25 SLPM (of which 5 SLPM was passed over a PenRay lamp to generate O3) dry synthetic air into SCHARK. A CIMS measurement of PAN is appended (blue squares).
In principal, as the processes that convert ISOP-NIT to NO2 are clearly surface-catalysed (involving formation of RSO), their impact can be reduced by using a surface that does not support formation of RSO. We therefore tested a third TD inlet (TDI-3), consisting of 55 cm PFA tubing (0.375 in. OD) with a 20 cm heated section. PFA tubing has been routinely used as TDI for measurement of, for example, PAN, as it is relatively unreactive to the peroxy radicals formed (Phillips et al., 2013). The C–F bond of PFA is very strong and nonpolar, which should reduce the formation of RSO as well as adsorption of ISOP-NIT to the surface. The performance of a TDI made of PFA (TDI-3) was examined by performing an experiment with SCHARK analogous to the one shown in Fig. 2. The resulting time series of NO2, ΣPNs (using TDI-3 heated to 448 K) and ΣANs (using TDI-2 heated to 648 K) are depicted in Fig. 12. At first, O3 (5 SLPM synthetic air passed over a Hg lamp) and NO2 (200 sccm of 1 ppmv) in 25 SLPM dry synthetic air were constantly introduced into SCHARK. After detectable amounts of NO3 and N2O5 had been formed, isoprene (9.8 sccm of 46.5 ppmv) was added leading (as expected) to almost quantitative depletion of NO3 and N2O5.
During the following 3 h the signal in the ΣAN cavity (TDI-2) increased to ∼ 1.2 ppbv, while the signal in the ΣPN channel (∼ 40 pptv) was close to the detection limit. A CIMS measurement obtained during this experiment validates that PAN mixing ratios are lower than 50 pptv. This result already confirms that (1) the ANs derived from NO3 + isoprene reaction are not detected at T < 448 K when PFA is used instead of quartz and (2) as expected, there is no significant generation of peroxy nitrates in these experiments which would be detectable with both ovens at T = 448 K (see Fig. S6 in the Supplement).
This result not only confirms that the previous detection of ISOP-NITs at low TD-inlet temperatures when using TDI-2 and especially TDI-1 was caused by heterogeneous reactions at the quartz surface, but also provides a solution to the problem of the separate measurement of ΣANs and ΣPNs using TD inlets.
We recall, however, that the reason for adding glass beads to the inlet was to suppress recombination reactions of NO2 with peroxy radicals by providing a surface to scavenge the latter. The use of a PFA tube rather than quartz will certainly exacerbate this effect, as the rate of loss of RO2 to PFA surfaces is expected to be lower than on quartz (Wooldridge et al., 2010). This implies that corrective procedures based on numerical simulation may be necessary in some environments as shown by Thieser et al. (2016), and this may limit the useful deployment of the method to regions where NOx levels are sufficiently low that reaction of RO2 with NO and NO2 become insignificant. In addition, similar to the observations of Sobanski et al. (2016), ClNO2 can interfere with the detection of ANs. The experiment shown in Fig. S7 reveals that ClNO2 is detected with TDI-2 at 698 K, but not with TDI-3 at 448 K.
We have shown that the detection of isoprene-derived organic nitrates via its thermal dissociation in quartz/glass inlets can (in the presence of O3) be accompanied by undesirable side reactions which broaden the thermograms and thus impede the separation of PN and AN signals by sampling through TD inlets at different temperatures as is commonly practised. While our experiments deal with the nitrates formed in the NO3-initiated oxidation of isoprene, it is very likely that similar broadening of thermograms would also occur with nitrates formed from the oxidation of other terpenoids, as some organic nitrates derived from night-time oxidation of, for example, limonene still contain a double bond (Fry et al., 2011) and/or contain hydroperoxy groups.
Specifically, we find that the presence of O3 in either the quartz TD inlet or in a Teflon simulation chamber results in the generation of NO2 from isoprene-derived nitrates in TD inlets made of quartz at temperatures less than 475 K and that this only occurs when the organic nitrate either has a double bond or a hydroperoxy group (or both). The formation of NO2 from ISOP-NIT was accelerated in the presence of glass beads, which indicates that a surface-catalysed reaction involving ozone and reactive surface sites is the process most likely to be responsible both for the conversion of ISOP-NIT to NO2 at low temperatures (375–475 K) and the conversion of HNO3 to NO2 at high temperatures (> 550 K). By avoiding the use of O3 or using a non-quartz TD inlet, we were able to show that the ISOP-NIT thermogram is entirely consistent with those of saturated alkyl nitrates.
We show that surface-catalysed reactions on quartz TD inlets involving O3 represent a potential source of bias in measurements of ΣANs and ΣPNs during field observations, especially when isoprene is abundant. For example, we previously reported results from two campaigns carried out using the TD-CRDS system described here on the same rural mountain site (Kleiner Feldberg) and season (but in different years). We found that the average, relative abundance of ΣPNs and ΣANs was quite different with ΣANs ≈ ΣPNs during the PARADE campaign in 2011 and ΣPNs > ΣANs during the NOTOMO campaign in 2015 (Sobanski et al., 2017). In 2011, the TD inlet was a quartz tube (i.e. similar to TDI-2) (Thieser et al., 2016), whereas in 2015 the inlet contained glass beads (i.e. similar to TDI-1) (Sobanski et al., 2016). With our present understanding of the role of surfaces and O3 in TD inlets, we cannot rule out that the observations during 2015 were biased to lower values for ΣANs and higher values for ΣPNs, although, as discussed by Sobanski et al. (2017), there are other, meteorological factors which would have contributed.
For the detection of PNs we have shown that (at the lower temperature required to thermally dissociate PNs to NO2) surface catalytic effects that convert ANs (or other species) to NO2 can be completely avoided by using a TD inlet made of a non-reactive material like PFA (TDI-3). In this case, O3 does not appear to have any impact.
When using a quartz TD inlet for conversion of ANs + PNs to NO2 at higher temperatures, the surface reactions that shift thermograms to lower temperatures are of less significance as, in any case, the role of the TD inlet is to convert all ANs and all PNs to NO2. However, in order to avoid detection of HNO3 or HONO other materials may be more suitable than quartz. Sapphire, commonly used in microwave discharge generated plasmas owing to its high purity and non-reactive surface, may represent a useful alternative. Under humid conditions, some of the observed interferences become negligible: HNO3 appears not to interfere with the AN measurement, and the interference of ANs to the PN measurement is reduced in TDI-2, but not in TDI-1.
This study emphasizes the importance of characterizing thermal dissociation inlets under conditions which are similar to those found in the atmosphere. We recognize that the impact of surface catalytic processes will vary from one inlet to the next (even if made from the same material), and all quartz TDIs will not necessarily exhibit the same degree of conversion of biogenic VOC-derived ANs at low temperature. For this reason, thermograms should be measured using trace gases that are abundant in the atmosphere and the effect of, for example, O3 and water vapour should be thoroughly investigated.
The data underlying the figures are available on request from the corresponding author.
The supplement related to this article is available online at: https://doi.org/10.5194/amt-14-5501-2021-supplement.
PD conducted the experiments, analysed the data and wrote the manuscript. RD was responsible for the CIMS measurements. JS designed and built SCHARK. JNC designed the experiments and together with JL contributed to the manuscript.
The authors declare that they have no conflict of interest.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The article processing charges for this open-access publication were covered by the Max Planck Society.
This paper was edited by Hendrik Fuchs and reviewed by two anonymous referees.
Bach, R. D. and Schlegel, H. B.: Bond Dissociation Energy of Peroxides Revisited, J. Phys. Chem. A, 124, 4742–4751, https://doi.org/10.1021/acs.jpca.0c02859, 2020.
Barnes, I., Bastian, V., Becker, K. H., and Tong, Z.: Kinetics and products of the reactions of NO3 with monoalkenes, dialkenes, and monoterpenes, J. Phys. Chem., 94, 2413–2419, https://doi.org/10.1021/j100369a041, 1990.
Bates, K. H. and Jacob, D. J.: A new model mechanism for atmospheric oxidation of isoprene: global effects on oxidants, nitrogen oxides, organic products, and secondary organic aerosol, Atmos. Chem. Phys., 19, 9613–9640, https://doi.org/10.5194/acp-19-9613-2019, 2019.
Beaver, M. R., Clair, J. M. St., Paulot, F., Spencer, K. M., Crounse, J. D., LaFranchi, B. W., Min, K. E., Pusede, S. E., Wooldridge, P. J., Schade, G. W., Park, C., Cohen, R. C., and Wennberg, P. O.: Importance of biogenic precursors to the budget of organic nitrates: observations of multifunctional organic nitrates by CIMS and TD-LIF during BEARPEX 2009, Atmos. Chem. Phys., 12, 5773–5785, https://doi.org/10.5194/acp-12-5773-2012, 2012.
Berndt, T. and Boge, O.: Products and Mechanism of the Reaction of NO3 with Selected Acyclic Monoalkenes, J. Atmos. Chem., 21, 275–291, https://doi.org/10.1007/Bf00696759, 1995.
Berndt, T. and Boge, O.: Gas-phase reaction of NO3 radicals with isoprene: A kinetic and mechanistic study, Int. J. Chem. Kinet., 29, 755–765, https://doi.org/10.1002/(sici)1097-4601(1997)29:10<755::aid-kin4>3.0.co;2-l, 1997.
Brownwood, B., Turdziladze, A., Hohaus, T., Wu, R., Mentel, T. F., Carlsson, P. T. M., Tsiligiannis, E., Hallquist, M., Andres, S., Hantschke, L., Reimer, D., Rohrer, F., Tillmann, R., Winter, B., Liebmann, J., Brown, S. S., Kiendler-Scharr, A., Novelli, A., Fuchs, H., and Fry, J. L.: Gas-Particle Partitioning and SOA Yields of Organonitrate Products from NO3-Initiated Oxidation of Isoprene under Varied Chemical Regimes, ACS Earth Space Chem., 5, 785–800, https://doi.org/10.1021/acsearthspacechem.0c00311, 2021.
Bulanin, K. M., Alexeev, A. V., Bystrov, D. S., Lavalley, J. C., and Tsyganenko, A. A.: IR Study of Ozone Adsorption on SiO2, J. Phys. Chem., 98, 5100–5103, https://doi.org/10.1021/j100070a026, 1994.
Burkholder, J. B., Sander, S. P., Abbatt, J., Barker, J. R., Huie, R. E., Kolb, C. E., Kurylo, M. J., Orkin, V. L., Wilmouth, D. M., and Wine, P. H.: Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies, Evaluation No. 18, JPL Publication 15-10, Jet Propulsion Laboratory, Pasadena, available at: http://jpldataeval.jpl.nasa.gov (last access: last access: 7 August 2021), 2016.
Carlton, A. G., Wiedinmyer, C., and Kroll, J. H.: A review of Secondary Organic Aerosol (SOA) formation from isoprene, Atmos. Chem. Phys., 9, 4987–5005, https://doi.org/10.5194/acp-9-4987-2009, 2009.
Cox, R. A., Ammann, M., Crowley, J. N., Herrmann, H., Jenkin, M. E., McNeill, V. F., Mellouki, A., Troe, J., and Wallington, T. J.: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume VII – Criegee intermediates, Atmos. Chem. Phys., 20, 13497–13519, https://doi.org/10.5194/acp-20-13497-2020, 2020.
Crutzen, P. J. and Lelieveld, J.: Human impacts on atmospheric chemisty, Annu. Rev. Earth Planet. Sci., 29, 17–45, https://doi.org/10.1146/annurev.earth.29.1.17, 2001.
Curtis, A. R. and Sweetenham, W. P.: Facsimile, Atomic Energy Research Establishment, Report R-12805, Harwell Laboratory, Oxfordshire, UK, 1987.
Davidson, J. A., Viggiano, A. A., Howard, C. J., Dotan, I., Fehsenfeld, F. C., Albritton, D. L., and Ferguson, E. E.: Rate Constants for Reactions of , , NO+, H3O+, , and Halide Ions with N2O5 at 300 K, J. Chem. Phys., 68, 2085–2087, https://doi.org/10.1063/1.436032, 1978.
Day, D. A., Wooldridge, P. J., Dillon, M. B., Thornton, J. A., and Cohen, R. C.: A thermal dissociation laser-induced fluorescence instrument for in situ detection of NO2, peroxy nitrates, alkyl nitrates, and HNO3, J. Geophys. Res.-Atmos., 107, ACH 4-1–ACH 4-14, https://doi.org/10.1029/2001jd000779, 2002.
Dewald, P., Liebmann, J. M., Friedrich, N., Shenolikar, J., Schuladen, J., Rohrer, F., Reimer, D., Tillmann, R., Novelli, A., Cho, C., Xu, K., Holzinger, R., Bernard, F., Zhou, L., Mellouki, W., Brown, S. S., Fuchs, H., Lelieveld, J., and Crowley, J. N.: Evolution of NO3 reactivity during the oxidation of isoprene, Atmos. Chem. Phys., 20, 10459–10475, https://doi.org/10.5194/acp-20-10459-2020, 2020.
Dubowski, Y., Vieceli, J., Tobias, D. J., Gomez, A., Lin, A., Nizkorodov, S. A., McIntire, T. M., and Finlayson-Pitts, B. J.: Interaction of gas-phase ozone at 296 K with unsaturated self-assembled monolayers: A new look at an old system, J. Phys. Chem. A, 108, 10473–10485, https://doi.org/10.1021/jp046604x, 2004.
Eger, P. G., Helleis, F., Schuster, G., Phillips, G. J., Lelieveld, J., and Crowley, J. N.: Chemical ionization quadrupole mass spectrometer with an electrical discharge ion source for atmospheric trace gas measurement, Atmos. Meas. Tech., 12, 1935–1954, https://doi.org/10.5194/amt-12-1935-2019, 2019.
Finlayson-Pitts, B. J., Wingen, L. M., Sumner, A. L., Syomin, D., and Ramazan, K. A.: The heterogeneous hydrolysis of NO2 in laboratory systems and in outdoor and indoor atmospheres: An integrated mechanism, Phys. Chem. Chem. Phys., 5, 223–242, https://doi.org/10.1039/B208564J, 2003.
Friedrich, N., Tadic, I., Schuladen, J., Brooks, J., Darbyshire, E., Drewnick, F., Fischer, H., Lelieveld, J., and Crowley, J. N.: Measurement of NOx and NOy with a thermal dissociation cavity ring-down spectrometer (TD-CRDS): instrument characterisation and first deployment, Atmos. Meas. Tech., 13, 5739–5761, https://doi.org/10.5194/amt-13-5739-2020, 2020.
Fry, J. L., Kiendler-Scharr, A., Rollins, A. W., Brauers, T., Brown, S. S., Dorn, H.-P., Dubé, W. P., Fuchs, H., Mensah, A., Rohrer, F., Tillmann, R., Wahner, A., Wooldridge, P. J., and Cohen, R. C.: SOA from limonene: role of NO3 in its generation and degradation, Atmos. Chem. Phys., 11, 3879–3894, https://doi.org/10.5194/acp-11-3879-2011, 2011.
Fry, J. L., Brown, S. S., Middlebrook, A. M., Edwards, P. M., Campuzano-Jost, P., Day, D. A., Jimenez, J. L., Allen, H. M., Ryerson, T. B., Pollack, I., Graus, M., Warneke, C., de Gouw, J. A., Brock, C. A., Gilman, J., Lerner, B. M., Dubé, W. P., Liao, J., and Welti, A.: Secondary organic aerosol (SOA) yields from NO3 radical + isoprene based on nighttime aircraft power plant plume transects, Atmos. Chem. Phys., 18, 11663–11682, https://doi.org/10.5194/acp-18-11663-2018, 2018.
Guenther, A. B., Jiang, X., Heald, C. L., Sakulyanontvittaya, T., Duhl, T., Emmons, L. K., and Wang, X.: The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 5, 1471–1492, https://doi.org/10.5194/gmd-5-1471-2012, 2012.
Hamilton, J. F., Bryant, D. J., Edwards, P. M., Ouyang, B., Bannan, T. J., Mehra, A., Mayhew, A. W., Hopkins, J. R., Dunmore, R. E., Squires, F. A., Lee, J. D., Newland, M. J., Worrall, S. D., Bacak, A., Coe, H., Percival, C., Whalley, L. K., Heard, D. E., Slater, E. J., Jones, R. L., Cui, T. Q., Surratt, J. D., Reeves, C. E., Mills, G. P., Grimmond, S., Sun, Y. L., Xu, W. Q., Shi, Z. B., and Rickard, A. R.: Key Role of NO3 Radicals in the Production of Isoprene Nitrates and Nitrooxyorganosulfates in Beijing, Environ. Sci. Technol., 55, 842–853, https://doi.org/10.1021/acs.est.0c05689, 2021.
Hanisch, F. and Crowley, J. N.: Ozone decomposition on Saharan dust: an experimental investigation, Atmos. Chem. Phys., 3, 119–130, https://doi.org/10.5194/acp-3-119-2003, 2003a.
Hanisch, F. and Crowley, J. N.: Heterogeneous reactivity of NO and HNO3 on mineral dust in the presence of ozone, Phys. Chem. Chem. Phys., 5, 883–887, https://doi.org/10.1039/B211503D, 2003b.
Hao, C. S., Shepson, P. B., Drummond, J. W., and Muthuramu, K.: Gas-chromatographic detector for selective and sensitive detection of atmospheric organic nitrates, Anal. Chem., 66, 3737–3743, https://doi.org/10.1021/ac00093a032, 1994.
Herron, J. T. and Huie, R. E.: Rate Constants for the Reactions of Atomic Oxygen (O3P) with Organic Compounds in the Gas Phase, J. Phys. Chem. Ref. Data, 2, 467–518, https://doi.org/10.1063/1.3253125, 1973.
Hjorth, J., Lohse, C., Nielsen, C. J., Skov, H., and Restelli, G.: Products and Mechanisms of the Gas-Phase Reactions between NO3 and a Series of Alkenes, J. Phys. Chem., 94, 7494–7500, https://doi.org/10.1021/j100382a035, 1990.
Horowitz, L. W., Fiore, A. M., Milly, G. P., Cohen, R. C., Perring, A., Wooldridge, P. J., Hess, P. G., Emmons, L. K., and Lamarque, J.-F.: Observational constraints on the chemistry of isoprene nitrates over the eastern United States, J. Geophys. Res., 112, D12S08, https://doi.org/10.1029/2006JD007747, 2007.
IUPAC: Task Group on Atmospheric Chemical Kinetic Data Evaluation, edited by: Ammann, M., Cox, R. A., Crowley, J. N., Herrmann, H., Jenkin, M. E., McNeill, V. F., Mellouki, A., Rossi, M. J., Troe, J., and Wallington, T. J., available at: http://iupac.pole-ether.fr/index.html, last access: 23 April 2021.
Jenkin, M. E., Young, J. C., and Rickard, A. R.: The MCM v3.3.1 degradation scheme for isoprene, Atmos. Chem. Phys., 15, 11433–11459, https://doi.org/10.5194/acp-15-11433-2015, 2015.
Keehan, N. I., Brownwood, B., Marsavin, A., Day, D. A., and Fry, J. L.: A thermal-dissociation–cavity ring-down spectrometer (TD-CRDS) for the detection of organic nitrates in gas and particle phases, Atmos. Meas. Tech., 13, 6255–6269, https://doi.org/10.5194/amt-13-6255-2020, 2020.
Kirchner, F., Mayer-Figge, A., Zabel, F., and Becker, K. H.: Thermal stability of peroxynitrates, Int. J. Chem. Kinet., 31, 127–144, https://doi.org/10.1002/(SICI)1097-4601(1999)31:2<127::AID-KIN6>3.0.CO;2-L, 1999.
Kwan, A. J., Chan, A. W. H., Ng, N. L., Kjaergaard, H. G., Seinfeld, J. H., and Wennberg, P. O.: Peroxy radical chemistry and OH radical production during the NO3-initiated oxidation of isoprene, Atmos. Chem. Phys., 12, 7499–7515, https://doi.org/10.5194/acp-12-7499-2012, 2012.
Kwok, E. S. C., Aschmann, S. M., Arey, J., and Atkinson, R.: Product formation from the reaction of the NO3 radical with isoprene and rate constants for the reactions of methacrolein and methyl vinyl ketone with the NO3 radical, Int. J. Chem. Kinet., 28, 925–934, https://doi.org/10.1002/(SICI)1097-4601(1996)28:12<925::AID-KIN10>3.0.CO;2-B, 1996.
Lelieveld, J., Evans, J. S., Fnais, M., Giannadaki, D., and Pozzer, A.: The contribution of outdoor air pollution sources to premature mortality on a global scale, Nature, 525, 367–371, https://doi.org/10.1038/nature15371, 2015.
Leonori, F., Balucani, N., Nevrly, V., Bergeat, A., Falcinelli, S., Vanuzzo, G., Casavecchia, P., and Cavallotti, C.: Experimental and Theoretical Studies on the Dynamics of the O(3P) + Propene Reaction: Primary Products, Branching Ratios, and Role of Intersystem Crossing, J. Phys. Chem. C, 119, 14632–14652, https://doi.org/10.1021/jp512670y, 2015.
Liebmann, J. M., Schuster, G., Schuladen, J. B., Sobanski, N., Lelieveld, J., and Crowley, J. N.: Measurement of ambient NO3 reactivity: design, characterization and first deployment of a new instrument, Atmos. Meas. Tech., 10, 1241–1258, https://doi.org/10.5194/amt-10-1241-2017, 2017.
Malkin, T. L., Goddard, A., Heard, D. E., and Seakins, P. W.: Measurements of OH and HO2 yields from the gas phase ozonolysis of isoprene, Atmos. Chem. Phys., 10, 1441–1459, https://doi.org/10.5194/acp-10-1441-2010, 2010.
Mellouki, A., Ammann, M., Cox, R. A., Crowley, J. N., Herrmann, H., Jenkin, M. E., McNeill, V. F., Troe, J., and Wallington, T. J.: Evaluated kinetic and photochemical data for atmospheric chemistry: volume VIII – gas-phase reactions of organic species with four, or more, carbon atoms (≥ C4), Atmos. Chem. Phys., 21, 4797–4808, https://doi.org/10.5194/acp-21-4797-2021, 2021.
Møller, K. H., Bates, K. H., and Kjaergaard, H. G.: The Importance of Peroxy Radical Hydrogen-Shift Reactions in Atmospheric Isoprene Oxidation, J. Phys. Chem. A, 123, 920–932, https://doi.org/10.1021/acs.jpca.8b10432, 2019.
Morin, J. and Bedjanian, Y.: Thermal decomposition of n-propyl and n-butyl nitrates: Kinetics and products, J. Anal. Appl. Pyrolysis, 124, 576–583, https://doi.org/10.1016/j.jaap.2017.01.014, 2017.
Ng, N. L., Kwan, A. J., Surratt, J. D., Chan, A. W. H., Chhabra, P. S., Sorooshian, A., Pye, H. O. T., Crounse, J. D., Wennberg, P. O., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol (SOA) formation from reaction of isoprene with nitrate radicals (NO3), Atmos. Chem. Phys., 8, 4117–4140, https://doi.org/10.5194/acp-8-4117-2008, 2008.
Ng, N. L., Brown, S. S., Archibald, A. T., Atlas, E., Cohen, R. C., Crowley, J. N., Day, D. A., Donahue, N. M., Fry, J. L., Fuchs, H., Griffin, R. J., Guzman, M. I., Herrmann, H., Hodzic, A., Iinuma, Y., Jimenez, J. L., Kiendler-Scharr, A., Lee, B. H., Luecken, D. J., Mao, J., McLaren, R., Mutzel, A., Osthoff, H. D., Ouyang, B., Picquet-Varrault, B., Platt, U., Pye, H. O. T., Rudich, Y., Schwantes, R. H., Shiraiwa, M., Stutz, J., Thornton, J. A., Tilgner, A., Williams, B. J., and Zaveri, R. A.: Nitrate radicals and biogenic volatile organic compounds: oxidation, mechanisms, and organic aerosol, Atmos. Chem. Phys., 17, 2103–2162, https://doi.org/10.5194/acp-17-2103-2017, 2017.
Nguyen, T. B., Tyndall, G. S., Crounse, J. D., Teng, A. P., Bates, K. H., Schwantes, R. H., Coggon, M. M., Zhang, L., Feiner, P., Milller, D. O., Skog, K. M., Rivera-Rios, J. C., Dorris, M., Olson, K. F., Koss, A., Wild, R. J., Brown, S. S., Goldstein, A. H., de Gouw, J. A., Brune, W. H., Keutsch, F. N., Seinfeldcj, J. H., and Wennberg, P. O.: Atmospheric fates of Criegee intermediates in the ozonolysis of isoprene, Phys. Chem. Chem. Phys., 18, 10241–10254, https://doi.org/10.1039/c6cp00053c, 2016.
Orphal, J., Fellows, C. E., and Flaud, P. M.: The visible absorption spectrum of NO3 measured by high-resolution Fourier transform spectroscopy, J. Geophys. Res.-Atmos., 108, 4077, https://doi.org/10.1029/2002JD002489, 2003.
Paul, D., Furgeson, A., and Osthoff, H. D.: Measurements of total peroxy and alkyl nitrate abundances in laboratory-generated gas samples by thermal dissociation cavity ring-down spectroscopy, Rev. Sci. Instrum., 80, 114101, https://doi.org/10.1063/1.3258204, 2009.
Paulson, S. E., Orlando, J. J., Tyndall, G. S., and Calvert, J. G.: Rate Coefficients for the Reactions of O(3P) with Selected Biogenic Hydrocarbons, Int. J. Chem. Kinet., 27, 997–1008, https://doi.org/10.1002/kin.550271005, 1995.
Perring, A. E., Wisthaler, A., Graus, M., Wooldridge, P. J., Lockwood, A. L., Mielke, L. H., Shepson, P. B., Hansel, A., and Cohen, R. C.: A product study of the isoprene + NO3 reaction, Atmos. Chem. Phys., 9, 4945–4956, https://doi.org/10.5194/acp-9-4945-2009, 2009.
Peukert, S. L., Sivaramakrishnan, R., and Michael, J. V.: High temperature shock tube studies on the thermal decomposition of O3 and the reaction of dimethyl carbonate with O-Atoms, J. Phys. Chem. A, 117, 3729–3738, https://doi.org/10.1021/jp400613p, 2013.
Phillips, G. J., Pouvesle, N., Thieser, J., Schuster, G., Axinte, R., Fischer, H., Williams, J., Lelieveld, J., and Crowley, J. N.: Peroxyacetyl nitrate (PAN) and peroxyacetic acid (PAA) measurements by iodide chemical ionisation mass spectrometry: first analysis of results in the boreal forest and implications for the measurement of PAN fluxes, Atmos. Chem. Phys., 13, 1129–1139, https://doi.org/10.5194/acp-13-1129-2013, 2013.
Pitts, J. N., Sanhueza, E., Atkinson, R., Carter, W. P. L., Winer, A. M., Harris, G. W., and Plum, C. N.: An investigation of the dark formation of nitrous acid in environmental chambers, Int. J. Chem. Kinet., 16, 919–939, https://doi.org/10.1002/kin.550160712, 1984.
Ray, D., Malongwe, J. K., and Klan, P.: Rate Acceleration of the Heterogeneous Reaction of Ozone with a Model Alkene at the Air-Ice Interface at Low Temperatures, Environ. Sci. Technol., 47, 6773–6780, https://doi.org/10.1021/es304812t, 2013.
Rivera-Rios, J. C., Nguyen, T. B., Crounse, J. D., Jud, W., St Clair, J. M., Mikoviny, T., Gilman, J. B., Lerner, B. M., Kaiser, J. B., de Gouw, J., Wisthaler, A., Hansel, A., Wennberg, P. O., Seinfeld, J. H., and Keutsch, F. N.: Conversion of hydroperoxides to carbonyls in field and laboratory instrumentation: Observational bias in diagnosing pristine versus anthropogenically controlled atmospheric chemistry, Geophys. Res. Lett., 41, 8645–8651, https://doi.org/10.1002/2014gl061919, 2014.
Rollins, A. W., Kiendler-Scharr, A., Fry, J. L., Brauers, T., Brown, S. S., Dorn, H.-P., Dubé, W. P., Fuchs, H., Mensah, A., Mentel, T. F., Rohrer, F., Tillmann, R., Wegener, R., Wooldridge, P. J., and Cohen, R. C.: Isoprene oxidation by nitrate radical: alkyl nitrate and secondary organic aerosol yields, Atmos. Chem. Phys., 9, 6685–6703, https://doi.org/10.5194/acp-9-6685-2009, 2009.
Schwantes, R. H., Teng, A. P., Nguyen, T. B., Coggon, M. M., Crounse, J. D., St Clair, J. M., Zhang, X., Schilling, K. A., Seinfeld, J. H., and Wennberg, P. O.: Isoprene NO3 Oxidation Products from the RO2 + HO2 Pathway, J. Phys. Chem. A, 119, 10158–10171, https://doi.org/10.1021/acs.jpca.5b06355, 2015.
Skov, H., Hjorth, J., Lohse, C., Jensen, N. R., and Restelli, G.: Products and mechanisms of the reactions of the nitrate (NO3) with isoprene, 1,3-butadiene and 2,3-dimethyl-1,3-butadiene in air, Atmos. Environ. A, 26, 2771–2783, https://doi.org/10.1016/0960-1686(92)90015-d, 1992.
Sobanski, N., Schuladen, J., Schuster, G., Lelieveld, J., and Crowley, J. N.: A five-channel cavity ring-down spectrometer for the detection of NO2, NO3, N2O5, total peroxy nitrates and total alkyl nitrates, Atmos. Meas. Tech., 9, 5103–5118, https://doi.org/10.5194/amt-9-5103-2016, 2016.
Sobanski, N., Thieser, J., Schuladen, J., Sauvage, C., Song, W., Williams, J., Lelieveld, J., and Crowley, J. N.: Day and night-time formation of organic nitrates at a forested mountain site in south-west Germany, Atmos. Chem. Phys., 17, 4115–4130, https://doi.org/10.5194/acp-17-4115-2017, 2017.
Stokes, G. Y., Buchbinder, A. M., Gibbs-Davis, J. M., Scheidt, K. A., and Geiger, F. M.: Heterogeneous Ozone Oxidation Reactions of 1-Pentene, Cyclopentene, Cyclohexene, and a Menthenol Derivative Studied by Sum Frequency Generation, J. Phys. Chem. A, 112, 11688–11698, https://doi.org/10.1021/jp803277s, 2008.
Thieser, J., Schuster, G., Schuladen, J., Phillips, G. J., Reiffs, A., Parchatka, U., Pöhler, D., Lelieveld, J., and Crowley, J. N.: A two-channel thermal dissociation cavity ring-down spectrometer for the detection of ambient NO2, RO2NO2 and RONO2, Atmos. Meas. Tech., 9, 553–576, https://doi.org/10.5194/amt-9-553-2016, 2016.
Thornton, J. A., Wooldridge, P. J., Cohen, R. C., Martinez, M., Harder, H., Brune, W. H., Williams, E. J., Roberts, J. M., Fehsenfeld, F. C., Hall, S. R., Shetter, R. E., Wert, B. P., and Fried, A.: Ozone production rates as a function of NOx abundances and HOx production rates in the Nashville urban plume, J. Geophys. Res.-Atmos., 107, ACH 7-1–ACH 7-17, https://doi.org/10.1029/2001JD000932, 2002.
Usher, C. R., Michel, A. E., and Grassian, V. H.: Reactions on Mineral Dust, Chem. Rev., 103, 4883–4940, https://doi.org/10.1021/cr020657y, 2003.
Vandaele, A. C., Hermans, C., Simon, P. C., Carleer, M., Colin, R., Fally, S., Merienne, M. F., Jenouvrier, A., and Coquart, B.: Measurements of the NO2 absorption cross-section from 42 000 cm−1 to 10 000 cm−1 (238–1000 nm) at 220 K and 294 K, J. Quant. Spectrosc. Radiat. Transfer, 59, 171–184, https://doi.org/10.1016/S0022-4073(97)00168-4, 1998.
Vansco, M. F., Caravan, R. L., Zuraski, K., Winiberg, F. A. F., Au, K., Trongsiriwat, N., Walsh, P. J., Osborn, D. L., Percival, C. J., Khan, M. A. H., Shallcross, D. E., Taatjes, C. A., and Lester, M. I.: Experimental Evidence of Dioxole Unimolecular Decay Pathway for Isoprene-Derived Criegee Intermediates, J. Phys. Chem. A, 124, 3542–3554, https://doi.org/10.1021/acs.jpca.0c02138, 2020.
Vereecken, L., Carlsson, P. T. M., Novelli, A., Bernard, F., Brown, S. S., Cho, C., Crowley, J. N., Fuchs, H., Mellouki, W., Reimer, D., Shenolikar, J., Tillmann, R., Zhou, L., Kiendler-Scharr, A., and Wahner, A.: Theoretical and experimental study of peroxy and alkoxy radicals in the NO3-initiated oxidation of isoprene, Phys. Chem. Chem. Phys., 23, 5496–5515, https://doi.org/10.1039/D0CP06267G, 2021.
Wayne, R. P., Barnes, I., Biggs, P., Burrows, J. P., Canosamas, C. E., Hjorth, J., Lebras, G., Moortgat, G. K., Perner, D., Poulet, G., Restelli, G., and Sidebottom, H.: The Nitrate Radical – Physics, Chemistry, and the Atmosphere, Atmos. Environ., 25, 1–203, https://doi.org/10.1016/0960-1686(91)90192-A, 1991.
Wennberg, P. O., Bates, K. H., Crounse, J. D., Dodson, L. G., McVay, R. C., Mertens, L. A., Nguyen, T. B., Praske, E., Schwantes, R. H., Smarte, M. D., St Clair, J. M., Teng, A. P., Zhang, X., and Seinfeld, J. H.: Gas-Phase Reactions of Isoprene and Its Major Oxidation Products, Chem. Rev., 118, 3337–3390, https://doi.org/10.1021/acs.chemrev.7b00439, 2018.
Wild, R. J., Edwards, P. M., Dube, W. P., Baumann, K., Edgerton, E. S., Quinn, P. K., Roberts, J. M., Rollins, A. W., Veres, P. R., Warneke, C., Williams, E. J., Yuan, B., and Brown, S. S.: A measurement of total reactive nitrogen, NOy, together with NO2, NO, and O3 via cavity ring-down spectroscopy, Environ. Sci. Technol., 48, 9609–9615, https://doi.org/10.1021/es501896w, 2014.
Wolfe, G. M., Thornton, J. A., McNeill, V. F., Jaffe, D. A., Reidmiller, D., Chand, D., Smith, J., Swartzendruber, P., Flocke, F., and Zheng, W.: Influence of trans-Pacific pollution transport on acyl peroxy nitrate abundances and speciation at Mount Bachelor Observatory during INTEX-B, Atmos. Chem. Phys., 7, 5309–5325, https://doi.org/10.5194/acp-7-5309-2007, 2007.
Wolfe, G. M., Marvin, M. R., Roberts, S. J., Travis, K. R., and Liao, J.: The Framework for 0-D Atmospheric Modeling (F0AM) v3.1, Geosci. Model Dev., 9, 3309–3319, https://doi.org/10.5194/gmd-9-3309-2016, 2016.
Wooldridge, P. J., Perring, A. E., Bertram, T. H., Flocke, F. M., Roberts, J. M., Singh, H. B., Huey, L. G., Thornton, J. A., Wolfe, G. M., Murphy, J. G., Fry, J. L., Rollins, A. W., LaFranchi, B. W., and Cohen, R. C.: Total Peroxy Nitrates (ΣPNs) in the atmosphere: the Thermal Dissociation-Laser Induced Fluorescence (TD-LIF) technique and comparisons to speciated PAN measurements, Atmos. Meas. Tech., 3, 593–607, https://doi.org/10.5194/amt-3-593-2010, 2010.
Wu, R., Vereecken, L., Tsiligiannis, E., Kang, S., Albrecht, S. R., Hantschke, L., Zhao, D., Novelli, A., Fuchs, H., Tillmann, R., Hohaus, T., Carlsson, P. T. M., Shenolikar, J., Bernard, F., Crowley, J. N., Fry, J. L., Brownwood, B., Thornton, J. A., Brown, S. S., Kiendler-Scharr, A., Wahner, A., Hallquist, M., and Mentel, T. F.: Molecular composition and volatility of multi-generation products formed from isoprene oxidation by nitrate radical, Atmos. Chem. Phys., 21, 10799–10824, https://doi.org/10.5194/acp-21-10799-2021, 2021.
Zhang, D., Lei, W. F., and Zhang, R. Y.: Mechanism of OH formation from ozonolysis of isoprene: kinetics and product yields, Chem. Phys. Lett., 358, 171–179, https://doi.org/10.1016/S0009-2614(02)00260-9, 2002.