the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 13 Oct 2021
| 13 Oct 2021
Behavior of KCl sorbent traps and KCl trapping solutions used for atmospheric mercury speciation: stability and specificity
Jan Gačnik
Igor Živković
Sergio Ribeiro Guevara
Radojko Jaćimović
Jože Kotnik
Gianmarco De Feo
Matthew A. Dexter
Warren T. Corns
Milena Horvat
Atmospheric mercury speciation is of paramount importance for understanding the behavior of mercury once it is emitted into the atmosphere as gaseous elemental mercury (GEM), gaseous oxidized mercury (GOM) and particulate-bound mercury (PBM). GOM and PBM can also be formed in the atmosphere; their sampling is the most problematic step in the atmospheric mercury speciation. GOM sampling with speciation traps composed of KCl sorbent materials and KCl trapping solutions are commonly used sampling methods, although the research conducted with them at ambient air concentrations is limited. The results of the specificity test demonstrated that the KCl sorbent traps are highly specific when using new traps, while their specificity drops dramatically when they are reused. The results of the stability test indicated that the highest Hg2+ losses (up to 5.5 % of Hg2+ loss) occur when low amounts of Hg2+ (< 1 ng) are loaded, due to a reduction of Hg2+ to Hg0. KCl trapping solutions have also been considered as a selective trapping media for GOM in atmospheric samples. A dimensionless Henry law constant was experimentally derived and was used to calculate the solubility of elemental Hg in KCl solution. The degree of GEM oxidation was established by purging elemental Hg calibration gas into a KCl solution and determining the GOM trapped using aqueous-phase propylation liquid–liquid extraction and gas chromatography–atomic fluorescence spectrometry (GC-AFS) measurement. A positive GOM bias was observed due to the solubility and oxidation of GEM in KCl trapping solutions, strongly suggesting that this approach is unsuitable for atmospheric mercury speciation measurements.
Please read the corrigendum first before continuing.
-
Notice on corrigendum
The requested paper has a corresponding corrigendum published. Please read the corrigendum first before downloading the article.
-
Article
(418 KB)
- Corrigendum
-
Supplement
(305 KB)
-
The requested paper has a corresponding corrigendum published. Please read the corrigendum first before downloading the article.
- Article
(418 KB) - Full-text XML
- Corrigendum
-
Supplement
(305 KB) - BibTeX
- EndNote





Since the 19th century, human activities have led to a 450 % increase in the concentration of mercury in the atmosphere (United Nations Environment Programme, 2019). In order to achieve comparability of atmospheric mercury speciation worldwide, the analytical methodology needs to be well understood in the terms of metrology and the conversion processes that may occur during the analysis. Uncertainties and lack of knowledge already appear in the sampling phase of the analytical procedure (Jaffe et al., 2014; Gustin et al., 2013).
First, the mercury species must be collected from the air and accumulated in a medium suitable for further analysis. Because some species of Hg are present in the atmosphere at very low concentrations, a highly selective pre-concentration step is required to accumulate sufficient quantity of the species for analysis. The collection of total gaseous mercury (TGM) is achieved by drawing air through different types of quartz traps filled with gold materials at a known and fixed flow rate. Types of gold materials include coiled gold wire, gold nanostructures and high specific-surface-area substrate coated with gold (U.S. EPA, 1999). The decision to use a particular gold material depends primarily on the mass of mercury to be collected. For gaseous oxidized mercury (GOM), the main sampling and preconcentration methods are KCl-coated denuders (Bu et al., 2018), KCl impinging solutions (impingers, adaptations of the Ontario Hydro method (ASTM International, 2016) and KCl sorbent traps (U.S. EPA, 2017; Prestbo and Bloom, 1995). Cation-exchange (CEM) or nylon membranes collect reactive mercury (RM – sum of GOM and PBM) but can also be used for GOM sampling if polytetrafluoroethylene (PTFE) membranes (PBM collection) are placed upstream (Bu et al., 2018; Huang et al., 2013; Gustin et al., 2021). Various types of filters (quartz-fiber, cellulose-acetate, glass-fiber and Teflon filters) are most commonly used for particulate-bound mercury (PBM) sampling (Zhang et al., 2019).
It is generally accepted that all forms of gaseous mercury are collected on gold traps and the measurement represents TGM (Dumarey, 1985; Shafawi et al., 1999). However, there is disagreement concerning whether the applied sampling protocols are optimal for GOM (Gustin and Jaffe, 2010).
Most problems with sampling of atmospheric mercury are related to GOM and PBM. Accurate quantification of PBM has proven to be one of the most difficult tasks of atmospheric mercury speciation. Problems include (i) meteorological conditions, adsorption, nucleation, gas–particle partitioning and other physical/chemical processes; (ii) ultra-low concentrations of PBM; (iii) formation of artifacts during sampling; and (iv) loss of PBM during longer sampling periods (Wang et al., 2013). Recently, research has focused on GOM sampling and its dependence on ambient conditions. Various studies of KCl-coated denuders have demonstrated that there is a clear dependence of collection efficiency on relative humidity (McClure et al., 2014; Huang and Gustin, 2015) and on the presence of ozone (McClure et al., 2014; Lyman et al., 2010). The collection efficiency has been found to be inversely dependent on ozone concentration and relative humidity, dropping as low as 13 % in some cases (Wang et al., 2013; McClure et al., 2014; Huang and Gustin, 2015). Experiments using water vapor spikes found that not only was the efficiency for GOM collection on KCl-coated denuders very low under high-humidity conditions, but also gaseous elemental mercury (GEM) concentrations were increased. This observation made it clear that under conditions of high humidity, GOM can be converted to GEM (or detected as such) (Huang and Gustin, 2015). Underestimated GOM and overestimated GEM due to processes occurring during denuder sampling have been confirmed by Lyman et al. (2016). In addition to denuders, the effectiveness of CEM and nylon membranes at different relative humidity levels was also studied. Humidity introduced a positive GOM bias for CEM while lowering the collection efficiency of nylon membranes. Nylon membrane passivation occurred with ozone exposure (Huang and Gustin, 2015). Gustin et al. (2019) compared different measurement methods for atmospheric Hg speciation. The results demonstrated that GOM and RM, measured with nylon membranes, were approximately 50 % lower than GOM and RM measured with CEM (Gustin et al., 2019). In addition to CEM and nylon membranes, various materials have been tested as membrane materials. CEM and polyethersulfone membrane (PES) have been shown to be the most quantitative sorbents. Nylon was best for identifying GOM compounds by thermal desorption. Other materials such as anion-exchange membranes, polycarbonate and polypropylene materials have obtained unsatisfactory results (Dunham-Cheatham et al., 2020). A three-membrane system (CEM + nylon + PTFE membranes) was recently applied with an attempt to distinguish between PBM and GOM. It was demonstrated that PTFE membranes retain mostly PBM, while CEM and nylon membranes retain RM if used without an upstream-placed PTFE membrane or GOM if used with an upstream-placed PTFE membrane (Gustin et al., 2021).
Various authors have studied the behavior of membranes and denuders as GOM/RM sampling methods, but less work has been done on the behavior of KCl sorbent traps and KCl trapping solutions for mercury atmospheric measurements. Initial experiments on KCl sorbent trap valuation were performed by the authors of the mercury speciation adsorption method (MESA) used for flue gas sampling (Prestbo and Bloom, 1995). The authors determined the species stability of KCl sorbent traps during storage, matrix effects, breakthrough and artifact formation (Prestbo and Bloom, 1995). Although several KCl sorbent trap behavior studies have been performed for flue gas sampling (EPRI, 2015), no studies have been conducted on their use for atmospheric mercury speciation measurements. KCl trapping solutions are commonly used for Hg speciation in flue gas (ASTM International, 2016), but the selectivity and suitability for GOM in atmospheric mercury measurements have not been previously investigated. Therefore, the aim of this work was to focus on gathering information on the behavior of KCl sorbent traps and KCl trapping solutions under different sampling conditions that would complement the aforementioned work and improve knowledge of the processes that may occur during atmospheric Hg speciation.
All chemicals and instruments that were utilized in the following experiments are listed in the Supplement (Tables S1 and S2).
2.1 Production of 197Hg radiotracer
To allow experiments using low Hg amounts (under 1 ng), radioactive 197Hg tracer was used as it has been demonstrated to be advantageous in cases where contamination and detection limit are problematic (Ribeiro Guevara et al., 2004; Koron et al., 2012; Ribeiro Guevara and Horvat, 2013). Mercury labeled with radioactive 197Hg was used for all KCl sorbent trap experiments. Mercury enriched to 51.58 % in 196Hg isotope (only 0.15 % of 196Hg isotope is naturally present) was used to produce 197Hg ( d) via a neutron capture reaction (n,γ). A total of 2 mL of a 2 % HNO3 () solution of enriched mercury was sealed into quartz ampoules. Quartz ampoules were then irradiated in the central channel of the TRIGA Mark II (250 kW) reactor core channel (JSI, Ljubljana, Slovenia). High neutron flux (approximately 1013 cm−2 s−1 at thermal power of 250 kW) in the center of the reactor core caused the nuclear neutron capture reaction in solution to produce 197Hg during irradiation. Prior to irradiation, Hg concentration of the solution was determined by cold vapor atomic absorption spectrometry (CV AAS). The Hg concentration measured (93.3 µg mL−1 of Hg) was the reference for Hg amounts that were used in all experiments. HgX2 (X = Cl−, Br−) solutions and gases were used in the presented work; to clarify, all Hg-related concentrations that will be presented in the paper will refer to Hg concentrations and not to HgX2 concentrations if not explicitly stated otherwise. After irradiation, the Hg solution was transferred from the irradiated vial and diluted to appropriate Hg concentrations for subsequent experiments.
2.2 Determining 197Hg using an HPGe detector
The activity of radiotraced Hg in solutions was measured by means of a well-type HPGe detector (model GCW6023/S, Canberra Industries Inc., Meriden, CT, USA), while in gold traps and non-liquid samples the activity was measured using a coaxial-type HPGe detector (model 7229P, Canberra Industries Inc., Meriden, CT, USA). All activity measurements were relative to standards obtained from the irradiated solution in each experimental run, considering the Hg concentration as described in the previous paragraph. The 197Hg activity was determined by evaluating γ-ray and X-ray emissions; experimental samples were measured in the same geometry as the standards. To obtain standards for well-type detector, triplicates of a Hg radiolabeled solution (8 mL, 2 % HNO3 ()) were transferred into glass vials. The standard solution was always diluted so that the activity was similar to the activity of the measured sample. The 197Hg activity of the standards in the vials was measured using a well-type detector. Standards for the coaxial-type detector were obtained by 197Hg2+ → 197Hg0 reduction, performed in an impinger with a SnCl2 solution (100 mL, 2 % SnCl2 () and 0.5 % HCl ()), which was purged for 10 min with N2 carrier gas (purity 4.7, flow rate of 1 L min−1). Purged 197Hg0 was transferred to a gold trap by the carrier gas to obtain a measurement standard. The absence of Hg0 breakthrough was confirmed by placing an additional gold trap downstream the main gold trap. Similar to liquid samples, standards for the coaxial-type gamma detector were made in triplicate. Any time that new gold traps were prepared, new triplicate standards were also prepared following the same procedure. Gold traps were prepared in quartz tubes (170 mm long, 6 mm inner diameter) by placing 15 mm in length of the absorbing material, which was fixed in place by quartz wool. Absorbing material was prepared by dissolving 1 g of HAuCl4 ⋅ xH2O (gold chloride hydrate) in 10 mL of Milli-Q water where 10 g of Al2O3 (corundum, 0.60–0.85 mm grain size) was added. The solution was then evaporated in an automatic rotary evaporator, and the remaining material was heated to 500 ∘C for 4 h in an argon atmosphere. In order to reuse gold traps, they were heated to 300 ∘C for 30 s, which released the bounded 197Hg0. Complete release of 197Hg was confirmed by evaluation of 197Hg remains in the HPGe detector.
The evaluation of the characteristic γ-ray and X-ray emissions associated with 197Hg decay (two doublet peaks: 67.0 + 68.8 and 77.3 + 78.1 keV) was performed by computing peak areas using Genie 2000 Gamma analysis software. All activities were referred to a reference time by applying an equation derived from the exponential law of radioactive decay. The specific equations that were used for the calculation of the activity and recovery are available in the Supplement (Eqs. S1 and S2) (Ribeiro Guevara and Horvat, 2013; Koron et al., 2012; Ribeiro Guevara et al., 2007).
2.3 Specificity of KCl sorbent traps and experimental design
A scheme of the experimental setup for testing KCl sorbent trap specificity is depicted in Fig. 1. Firstly, Hg2+ (ranging from 0.1 to 1 ng) was reduced to Hg0 in the impinger using SnCl2 solution (100 mL, 2 % SnCl2 () and 0.5 % HCl ()). Hg0 was then purged out with N2 carrier gas (flow rate of 1 L min−1) for 10 min, passed through various types of KCl sorbent traps (described below) and captured at the end by a gold trap.
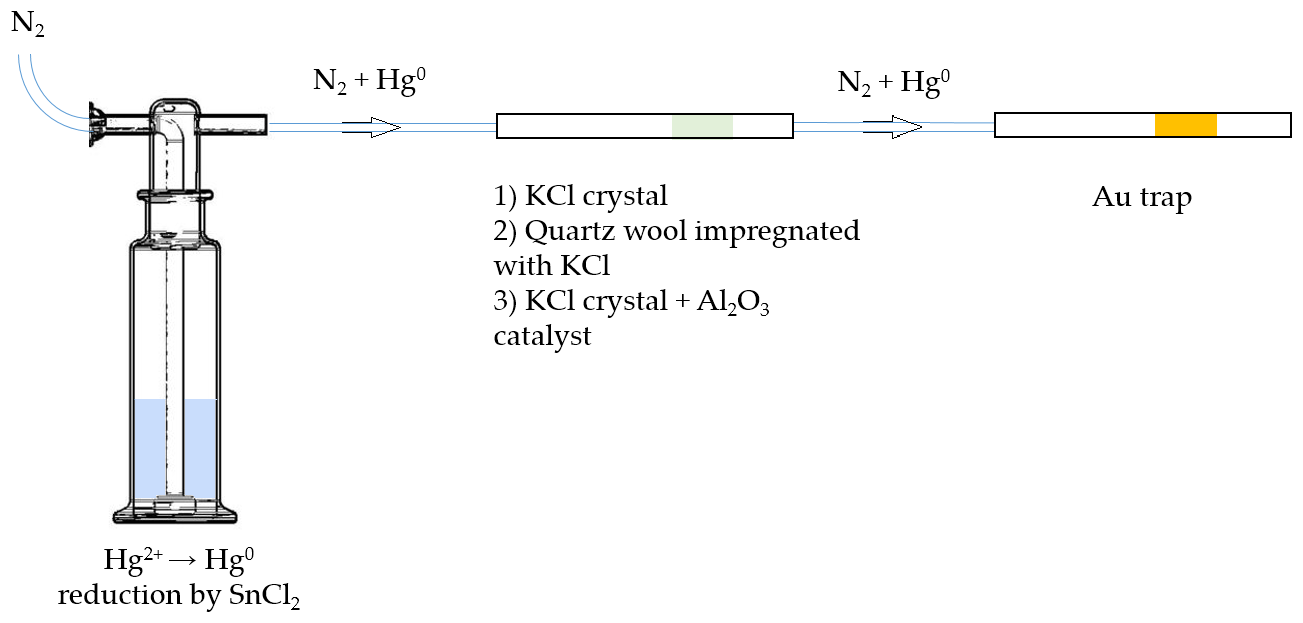
Figure 1Scheme of the experimental setup for testing the extent of undesirable Hg0 retention on three different KCl sorbent trap designs (not depicted to scale).
Three different types of KCl sorbent traps were used: KCl crystal, quartz wool impregnated with KCl, and KCl crystal + Al2O3 catalyst. The latter was considered for an application that will be discussed in future work. Briefly, Al2O3 is intended to catalyze the reduction of Hg2+ to Hg0 in the next step of the analysis. All sorbent traps were prepared in quartz tubes (170 mm long, 6 mm inner diameter). In design 1, KCl crystal was 15 mm long and fixed using quartz wool. In design 2, quartz wool impregnated with KCl was 70 mm long. Quartz wool impregnated with KCl was prepared by soaking quartz wool in 1 mol L−1 KCl for 24 h, draining the excess solution and drying at 130 ∘C for 24 h. In design 3, the KCl crystal was 5 mm long and the Al2O3 catalyst part was 65 mm long. All three types of sorbent traps were tested new (no heating of the traps) and reused (traps heated to ≈ 600 ∘C three times prior to the experiment), resulting in six variations tested.
To determine the amount of Hg0 collected on the KCl sorbent traps, the traps were leached with 20 mL of 10 % HNO3 () +5 % HCl () solution and 197Hg in leachate was measured. In our research, it has previously been found that this acid mixture completely leaches Hg from KCl sorbent traps. Radiolabeled Hg0 on gold sorbent traps was also measured.
2.4 Stability of Hg2+ loading on KCl sorbent traps and experimental design
In order to test Hg2+ stability on KCl sorbent traps, the traps were exposed to ambient airflow for 30 min periods after loading with radiolabeled Hg. Potential formation of Hg0 was captured downstream of the KCl sorbent trap by an Au trap. After each 30 min exposure period, the radiolabeled Hg0 activity in the Au trap was measured. KCl sorbent traps were loaded with radiolabeled Hg2+ again (to simulate the sampling process where new Hg2+ is constantly adsorbed); the procedure was repeated 4 to 5 times. To assure that no Hg2+ breakthrough occurred and that the measured losses only comprised of Hg0, an additional KCl crystal trap was placed between the KCl sorbent trap and the Au trap to filter the potential Hg2+ breakthrough. The Hg2+ on the KCl trap filter was always below the detection limit of the gamma detector, so these results will not be presented in the results and discussion section. Since this KCl trap filter was always free of Hg, it was later checked only from time to time as a control. Figure 2 depicts a diagram of all conditions studied in the stability tests.
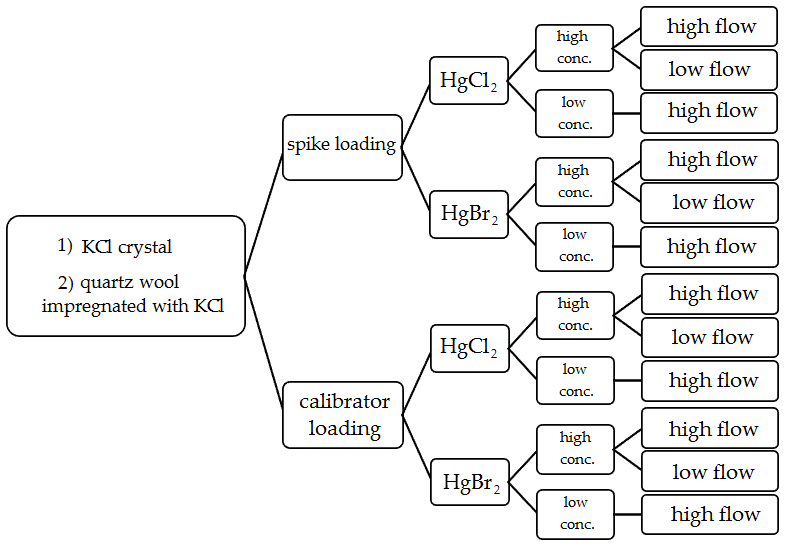
Figure 2Variations of performed experimental conditions for Hg2+ stability on KCl sorbent traps during exposure in ambient airflow.
Two types of Hg2+ loadings were tested. The first type was a direct spike of Hg2+ onto the sorbent traps. The second type of loading was performed using an Optoseven evaporative gas calibrator. This instrument enabled Hg2+ loading by evaporating the Hg2+ solution and injecting it into a carrier gas (Saxholm et al., 2020; Gačnik et al., 2021). The calibration gas was comprised of 0.07 mL min−1 of Hg2+ solution (Hg2+ concentration dependent on the concentration level tested) and 5 L min−1 of carrier gas N2. The calibration gas was formed in the evaporator at 125 ∘C. The obtained calibration gas had a Hg2+ concentration of 1178 ng m−3 for high-concentration tests and 5.90 ng m−3 for low-concentration tests. Previous studies have found that the output of the calibrator is concentration dependent; these findings were taken into account when calculating the expected calibrator output (Gačnik et al., 2021). Two Hg2+ species were tested for stability, HgCl2 and HgBr2. For Hg2+ spikes, a 4 % HCl () +3 % HNO3 () solution was used for HgCl2, and a 4 % HBr () +3 % HNO3 () solution was used for HgBr2. Compounds and their concentrations were chosen based on the composition of NIST 3177 standard reference material (Mercuric Chloride Standard Solution). Using equilibrium calculations described in the work of Gačnik et al. (2021), we confirmed that the spiking solutions contained only HgCl2 and HgBr2 without other HgxClx or HgxBrx species (Gačnik et al., 2021). In the cases where Hg2+ was loaded using the Optoseven calibrator, a 0.1 % HCl () +0.1 % HNO3 () solution was used for HgCl2 and 0.1 % HBr () +0.1 % HNO3 () for HgBr2. In addition to two loading types and two Hg species, two different KCl sorbent trap materials were tested: KCl crystal and quartz wool impregnated with KCl. Each trap material was then tested under different experimental conditions: high concentration (> 50 ng)/low concentration (< 1 ng) and high airflow (400 mL min−1)/low airflow (100 mL min−1). All variations of experimental conditions, trap types, Hg species and loading types were performed following the Fig. 2 diagram.
2.5 Solubility of elemental Hg in KCl trapping solutions and experimental design
The solubility of elemental Hg in the 1 mol L−1 KCl trapping solution was established using a method based on the Henry law constant determination. From the dimensionless Henry law constant (HLC), the amount of elemental Hg that would be collected in the KCl solution can be predicted. This approach was applied to a seawater matrix by Andersson et al. (2008).
The system consisted of an extractor stripper vessel composed of a jacketed borosilicate glass cylinder. This is represented schematically in Fig. 3. During the experiment, water at known temperature was pumped through the jacket from a temperature-controlled bath. The capacity of the extractor vessel was 1 L. At the bottom of the vessel there was an injection port connected to the argon carrier gas. The gas was bubbled inside the vessel through a glass frit on the bottom of the vessel. The gas was produced from a mass flow meter at known flow. Two holes, one on the bottom and one on the top, allow the measurement of the temperature of both the solvent and the headspace. At the top of the vessel, the pressure was measured using an absolute pressure meter.
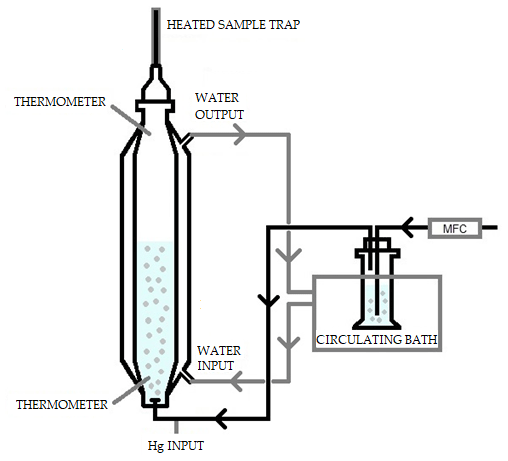
The mercury which is purged from the vessel was collected using a heated gold trap. Before and after every experiment, the recovery of the gold traps was checked to ensure that mercury collection was quantitative. The mercury extracted was compared to the mass of Hg injected as an indication of the success of the experiment and absence of oxidized mercury which would otherwise be trapped in solution. The KCl also included 11 mL L−1 of reductant solution (20 % () SnCl2 in 10 % () HCl), used to avoid oxidation.
Elemental mercury (Hg0) vapors were injected from a bell jar into the gas flow of the extractor vessel. The bell jar had a thermometer where the temperature was checked before the gas was pulled via a gastight syringe. The temperature and volume of gas are known, and therefore the mass of mercury injected could be calculated using the Dumarey equation (Dumarey et al., 2010). To make a correction on the measured mass flow and calculate the real flow, the vapor pressure must be known. The vapor pressure of 1 mol L−1 of KCl at 5 ∘C was assumed to be the same as water (Lide, 2007). The recovery of the spiked mercury into the extraction vessel was checked after the experiments. The recovery is a good indication of the success of the experiment and of the absence of oxidized mercury.
The gas coming out from the vessel was collected using heated gold traps for fixed intervals of time. These gold traps were then analyzed to determine the mass of Hg released during each time interval. For this experiment, the volume of KCl in the vessel, the pressure, the measured gas flow rate, the exact interval of each extraction time and the temperature must be precisely determined. The calculation of dimensionless HLC was performed according to the method presented by Andersson et al. (2008), which is provided in the Supplement (Eqs. S3–S12). The value of the gold trap blank was subtracted from the mass of Hg measured for every extraction. This improved the linearity of the data providing a better measurement of the dimensionless HLC. The first point of the extraction typically deviated slightly from linearity because it included the signal rise time from the point of Hg vapor injection. During the signal rise period the test solution is not under equilibrium, and therefore the first point was ignored. Additionally, the final extraction points if considered to be close to the quantification limit are ignored as the uncertainty is higher, which can affect the linear regression.
2.6 Oxidation of elemental Hg in KCl trapping solutions and experimental design
The experimental arrangement for studying the selectivity of the KCl trapping solution is depicted in Fig. 4. A PSA 10.536 elemental Hg generator was used to generate a continuous stream of calibration gas of known concentration (nominally 20 µg m−3) and flow rate. This is a considerably higher concentration of elemental Hg than ambient air concentration as the original scope of this work was to study the Hg concentration range in flue gases. The total flow generated was 5 L min−1, and both nitrogen and air (from an in-house air compressor, classified to ISO 8573-1:2010) (International Organization for Standardization, 2010) were studied. A slipstream flow of 0.5 L min−1 was pulled through the test impingers using a vacuum pump and mass flow controller arrangement. An impinger solution of 100 mL of 1 mol L−1 KCl was used as the test solution maintained at 5 ∘C in a water chiller bath. An empty impinger and an impinger of silica gel were also included to ensure that the gas going to the mass flow controller was dry. All tests were conducted over a 2 h period, during which an absolute mass of mercury of 1200 ng was passed through the impinger train. The KCl trapping solution was then analyzed by GC-AFS with aqueous-phase propylation liquid–liquid extraction to determine the oxidized Hg.
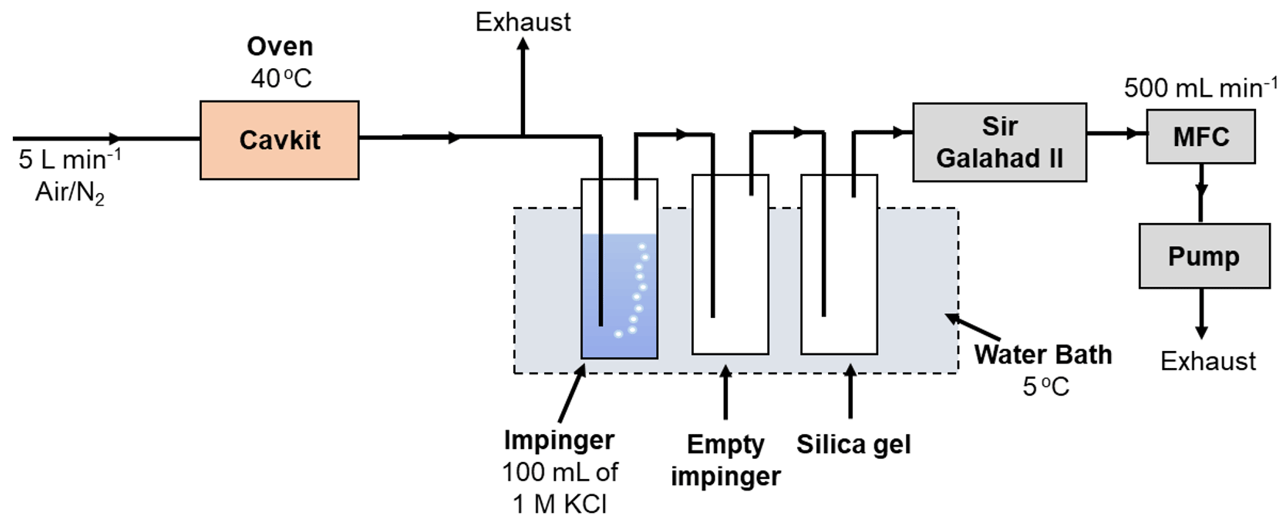
The same apparatus was used to test the stability of oxidized Hg in the KCl trapping solution. In this case a known mass of HgCl2 was added to the KCl solution. The apparatus was then run using the same conditions as described above but without elemental Hg being introduced. In this experiment, the concentration of oxidized Hg should remain the same after the 2 h period of sampling. A concentration decrease would be indicative of HgCl2 being reduced. After running the test, the KCl trapping solution was then analyzed by GC-AFS with aqueous-phase propylation liquid–liquid extraction to determine the oxidized Hg.
2.7 Determination of oxidized Hg in KCl trapping solution and experimental design
The KCl solution was propylated in the presence of an acetic acid–acetate buffer. This converts Hg2+ to dipropyl mercury. The derivatized mercury was then transferred and concentrated in an organic phase for injection into the GC-AFS instrument. To a 100 mL sample, 5 mL of buffer (0.5 M acetic acid) was added. The pH was then adjusted to 3.9. To this solution 500 µL of 2,2,4-trimethylpentane and 1 mL of the alkylation reagent were added. The solution was then shaken vigorously for 10 min and the trimethylpentane phase transferred to a GC vial. The sample was dried over anhydrous sodium sulfate and then analyzed by GC-AFS. Calibration was achieved by preparing mixed organometallic and Hg2+ standards and a blank, subjecting them to the same sample preparation. An injection volume of 2 µL was used in the splitless mode of operation with the injector at 250 ∘C. The GC temperature program used is summarized in the Supplement (Table S3). The eluent emerging from the column was thermally treated at 800 ∘C to breakdown organomercury compounds to elemental Hg before introduction to the AFS detector.
3.1 Specificity of KCl sorbent traps
The aim of this study was to determine the extent of unwanted retention of Hg0 on KCl sorbent traps. This was achieved by transferring a known amount of Hg0 in the carrier gas through a Hg2+ specific sorbent trap (where Hg0 retention is unwanted). The results of the experiment are shown in Table 1. The column “retained Hg0” represents Hg0 retained on the KCl sorbent trap. The column “mass balance” represents the sum of Hg0 on the KCl sorbent trap and Hg0 on a gold trap. All values are shown as percentages relative to the Hg amount that was purged through the system. As previously mentioned, Hg2+ breakthrough was never present since it was negligible.
Table 1Hg0 retention on various KCl sorbent trap designs, and comparison of new and reused designs. Results are presented in percentage of the initially purged Hg0 amount. Less than 1 ng of Hg was used.
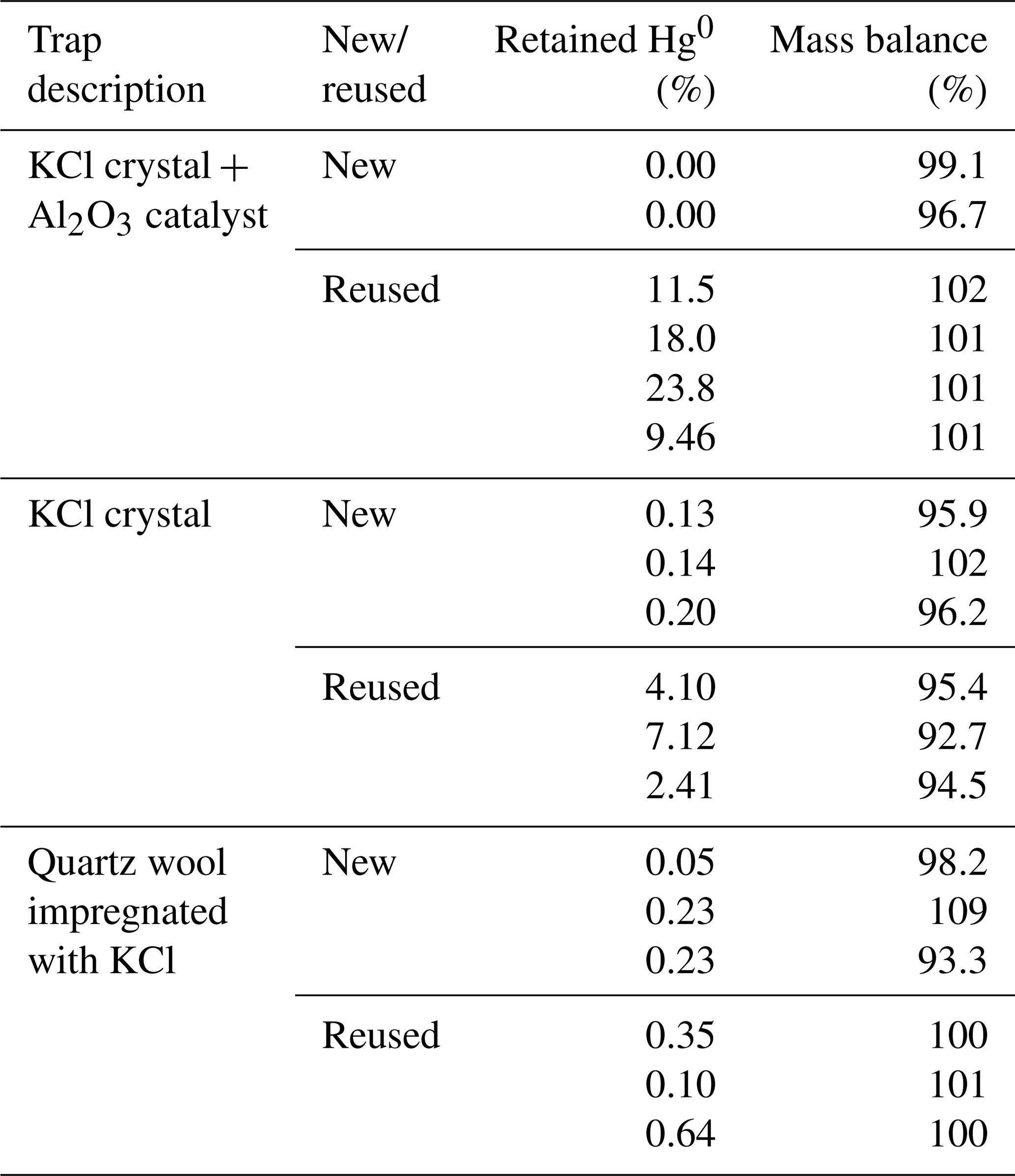
It can be observed that trap designs containing KCl crystals (KCl crystal + Al2O3 catalyst and KCl crystal traps) retained much more Hg0 when reused, which is not desirable; Hg2+ specificity is required. The amount retained also varied greatly for reused sorbent traps. Since the KCl has a melting point of 770 ∘C, this could mean that the morphology of KCl has changed at experimental temperatures (≈ 600 ∘C) approaching the melting point. The morphological change could potentially explain the increased Hg0 binding when using reused KCl sorbent traps. Traps that were unused prior to the experiment (“new” traps) always retained very small amounts of Hg0 (< 0.3 % for all designs). Mass balances were quantitative in all cases; therefore, these results can be trusted with a high level of confidence. From these findings, it can be concluded that new traps perform better than reused traps, and, therefore, to prevent the formation of artifacts only new traps should be used. It is not necessary to correct the measured values for the obtained recovery as losses are regularly lower than the variability of the recovery (standard deviation up to 4 %, losses up to 0.2 %).
3.2 Stability of Hg2+ loading on KCl sorbent traps
Exposure of the loaded sorbent trap to airflow changed the geometry of the radiolabeled Hg2+ loading. This affected the measurement as depicted in Fig. 5, resulting in biasing the results. This effect was presumed based on the fact that mass balance was always considerably above 100 %. The figure illustrates a sorbent trap that is placed over a coaxial-type gamma detector before and after exposure to airflow. The airflow shifted the distribution of the radiolabeled Hg2+ along the trap. Prolonged exposure to the airflow caused greater shifts. Due to geometrical effects, the detection system has higher detection efficiency for a radioactive source on the axis of the detector than a source at the same distance from the detector surface but far from the axis. Therefore, a distribution of the radiolabeled Hg extended to the axis of the detector generates a higher recording on the detection systems than a distribution compacted to the detector border, with the same activity. Measurement after exposure to the airflow would therefore result in greater apparent sample activity than measurement prior to exposure to airflow. Due to this observation, it was not possible to verify the overall mass balance. On the other hand, this did not affect the measurement of Hg losses (Hg0) captured on the Au trap, as Hg forms a strong amalgam with Au. Therefore, only the measured losses are presented in the tables below.
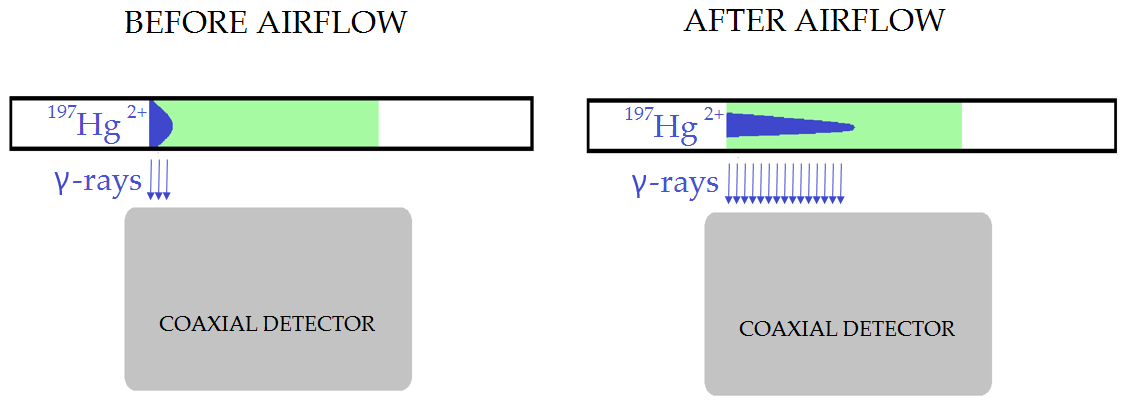
Figure 5Effect of radiotracer distribution change along the trap on the activity measurement after exposure of KCl sorbent traps to airflow (not depicted to scale).
The results of the stability tests are shown in the figures below (Figs. 6, 7, 8 and 9). Full results are available in the Supplement (Tables S4, S5, S6 and S7). Losses are presented as a percentage relative to the cumulative amount of Hg that was spiked up to that time period. Some results have four time periods (marked with an asterisk in the figures), while others have five time periods due to time constraints of each measurement day.
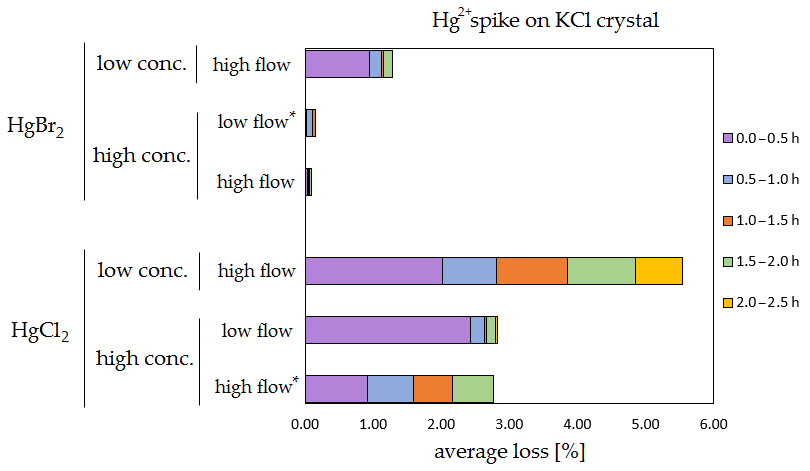
Figure 6Results of the stability test for the 197Hg2+ (radiotracer) spike on KCl crystal. Low concentrations were loaded with less than 1 ng Hg per time period, and high concentrations were loaded with more than 50 ng of Hg per time period. Low airflow experiments were performed with 100 mL min−1 airflow, while high airflow experiments were performed with 400 mL min−1 flow. Asterisks mark the results that only have the first four time periods.
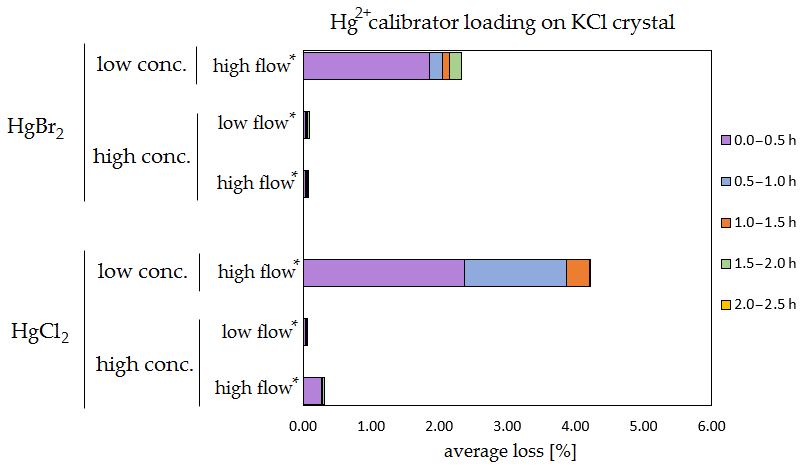
Figure 7Results of the stability test for the calibrator loading of 197Hg2+ (radiotracer) on KCl crystal. Low concentrations were loaded with less than 1 ng Hg per time period, and high concentrations were loaded with more than 50 ng of Hg per time period. Low airflow experiments were performed with 100 mL min−1 airflow, while high airflow experiments were performed with 400 mL min−1 flow. Asterisks mark the results that only have the first four time periods.

Figure 8Results of the stability test for the 197Hg2+ (radiotracer) spike on quartz wool impregnated with KCl. Low concentrations were loaded with less than 1 ng Hg per time period, and high concentrations were loaded with more than 50 ng of Hg per time period. Low airflow experiments were performed with 100 mL min−1 airflow, while high airflow experiments were performed with 400 mL min−1 flow.
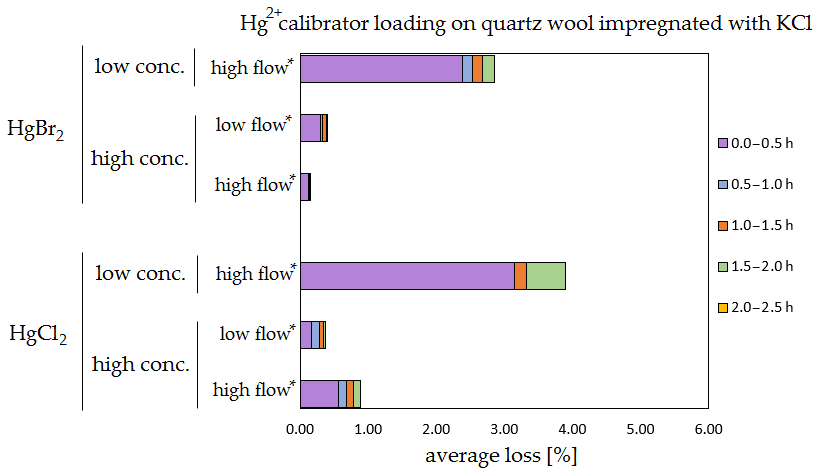
Figure 9Results of the stability test for the calibrator loading of 197Hg2+ (radiotracer) on quartz wool impregnated with KCl. Low concentrations were loaded with less than 1 ng Hg per time period, and high concentrations were loaded with more than 50 ng of Hg per time period. Low airflow experiments were performed with 100 mL min−1 airflow, while high airflow experiments were performed with 400 mL min−1 flow. Asterisks mark the results that only have the first four time periods.
The clear trend observed in the tables presented was that the relative losses were in almost all cases higher in the low-concentration experiments (5.54 % max. losses) than in high-concentration experiments (2.79 % max. losses, under 1 % in most cases). In addition, the first interval (0–0.5 h) had statistically significant maximum relative losses during the whole stability test (Kruskal–Wallis test, p < 0.001; pairwise multiple comparison procedures (Dunn's method), p < 0.05 for 0–0.5 h period against other periods). Because the variation in low/high airflow did not cause significant differences in the overall Hg2+ losses (paired t test, p = 0.471), the low airflow tests were omitted in the low-concentration stability tests. The HgCl2 HgBr2 and calibrator/spike loading variations also did not cause any significant differences in Hg2+ losses during the stability tests.
Longer sampling times are often used for low concentrations of Hg2+ (the amount of Hg2+ collected from the ambient atmospheric samples is in the order of picograms); therefore, some losses of GOM will be observed most of the time. Losses depend not only on the parameters tested in our work, but also on meteorological conditions (e.g. humidity, presence of ozone, temperature). In Hg speciation measurements, reduction of Hg2+ to Hg0 during sampling may result in a positive bias for Hg0 (GEM) and a negative bias for Hg2+ (GOM) measurement. The considerations mentioned above should be considered carefully, particularly when longer sampling times are required.
3.3 Solubility of elemental Hg in KCl trapping solutions using the dimensionless Henry law constant
Using the experimental data taken from one of the tests using 1 mol L−1 KCl at 5 ∘C, the mass of Hg released during each 2 min interval is depicted graphically in Fig. 10. The natural logarithm of the mass of the Hg released during each interval was plotted to provide the α term using the slope (see Fig. 11). The slope was established using a linear regression weighting the errors in ln (mHg(n)) using the calculated expanded uncertainty with a coverage factor k=2.

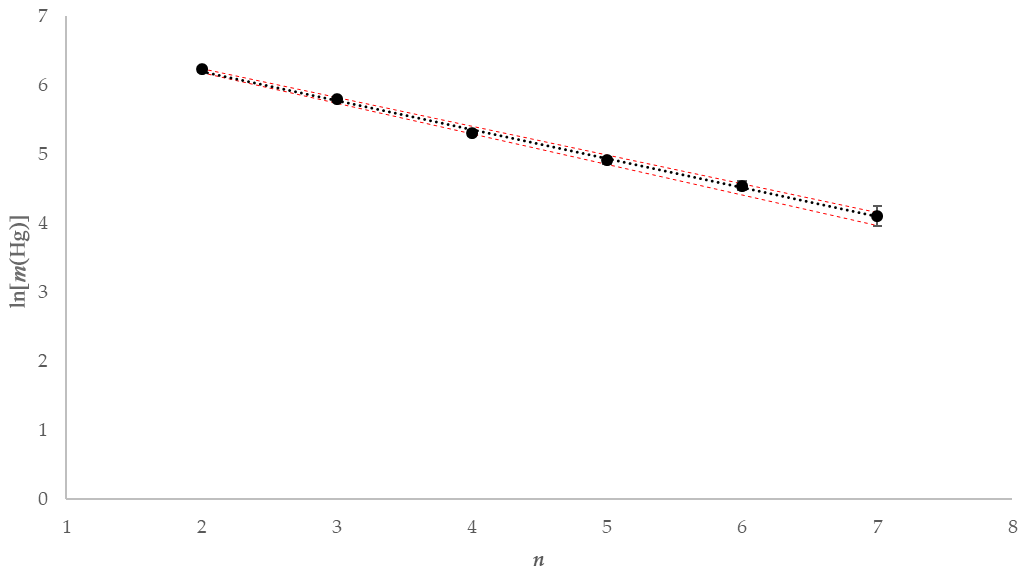
Figure 11Example ln [m(Hg)] against extraction number (n) and linear regression for KCl at 5 ∘C (red error bars ±1 uc [k=1]).
As depicted in Fig. 11, plotting the logarithm of the Hg mass extracted against the extraction number gives a linear relationship that allows the calculation of the dimensionless HLC. A combined dimensionless HLC was found to be 0.1713 with an expanded uncertainty (k=2) of 0.0093 (calculation and derivation of needed equations is located in the Supplement, Eqs. S3–S12). The dimensionless HLC can be used to calculate the elemental mercury concentration collected in the KCl solution at equilibrium conditions.
3.4 Oxidation of elemental Hg in KCl trapping solution
The oxidation of elemental mercury found for the nitrogen and air matrices was 2.9 % and 3.0 % respectively. When the KCl trapping blank solution was tested, it was found to be below the GC-AFS method detection limit (1 pg). Impurities present in the KCl trapping solution appear to oxidize a small percentage of the elemental Hg vapor which was continuously introduced during the test. As a negligible difference was found when comparing air and nitrogen, it is reasonable to assume that this oxidation was not due to aerial oxidation. The results indicate that the KCl solution will also collect a small quantity of elemental Hg due to its solubility; this elemental Hg was observed in the chromatograms, as depicted in the Supplement (Fig. S1). This was not observed in standard solutions or blanks. The elemental Hg response was not quantified because the procedure used is not considered quantitative for elemental Hg, as this species does not undergo derivatization.
3.5 Retention of oxidized Hg in KCl trapping solution
In this test, bubbling the KCl solution spiked with HgCl2 with nitrogen or air for the 2 h sampling period gave a recovery of 102.3 % and 101.8 %, respectively, indicating that no HgCl2 was lost from the KCl solution. The slight increase is within the measurement uncertainty of the propylation GC-AFS.
3.6 Predicted bias calculation using KCl trapping solution at atmospheric Hg speciation concentrations
The experimental data indicate that GEM oxidation (Sect. 3.4) and GEM solubilization (Sect. 3.3) will occur in KCl trapping solutions. The combination of these two factors will produce a positive bias which can be predicted using Eq. (1):
For example, if a 1 m3 ambient air sample containing 2 ng m−3 GEM and 0.002 ng m−3 GOM (GOMnative) was sampled through a 1000 mL KCl solution, the bias can be predicted as follows: by applying the dimensionless HLC obtained from the presented work and using the equations available in the Supplement, the calculated contribution due to solubility (GEMsol) will be 0.012 ng m−3. Oxidation of 3 % of GEM was found in the KCl trapping solution. This equates to GEMoxid of 0.060 ng m−3 for this example. The apparent gas concentration in the KCl solution would be the summation of soluble GEM (0.012 ng m−3), oxidized GEM (0.060 ng m−3) and the native GOM (0.002 ng m−3), equating to 0.074 ng m−3 rather than the expected 0.002 ng m−3. From this simple calculation using Eq. (1), a GOM bias of 3500 % can be predicted due to the solubility and oxidation of GEM in the KCl solution. The bias depends on the GEM : GOM ratio – the higher the percentage of GOM relative to GEM, the lower the bias will be. For example, a similar calculation as above but with 1.980 ng m−3 GEM and 0.02 ng m−3 GOM results in 456 % bias instead of 3500 %. The calculated biases show that KCl trapping solutions are not appropriate for ambient GOM sampling, while they are still a valid choice for flue gas sampling (high GOM concentrations).
KCl sorbent traps exhibit good stability for most of the experimental conditions tested in this work. When KCl sorbent traps are used for ambient Hg concentration, the extent of GOM losses can reach up to 5.5 % (2 % to 3 % on average). When calculating the overall uncertainty of the atmospheric Hg speciation, these losses should be taken into account as the sampling uncertainty. Reuse of KCl sorbent traps resulted in large reduction in their specificity. Because the reuse of KCl-coated denuders (for ambient GOM sampling) is also common practice, a similar reduction in specificity could also occur for KCl-coated denuders. As denuders are the most commonly used means of sampling GOM, this poses a potential challenge which should be addressed.
The 197Hg radiotracer proved to be a suitable tool for studying the metrology and processes occurring during atmospheric mercury speciation. In the future, the 197Hg radiotracer could be applied for verifying other GOM sampling methods such as denuders and different membrane filters.
KCl trapping solutions cannot be considered truly selective for GOM measurements for atmospheric mercury speciation measurements. GEM will be captured in the solution due to both oxidation and solubilization producing a large bias in the GOM measurement.
Data are contained within the article (Table 1) and the Supplement (Tables S1–S7, Fig. S1).
The supplement related to this article is available online at: https://doi.org/10.5194/amt-14-6619-2021-supplement.
The conceptualization was done by JG, SRG, JK, WTC, and MH; funding was acquired by MH; the methodology was done by JG, IŽ, RJ, GDF, MAD, and SRG; the project was administered by MH; the work was supervised by JK, WTC, and MH; the validation of the analytical procedure was done by MAD, JG, RJ, and IŽ; the original draft was written by JG and WTC; the review writing and editing was done by JG, IŽ, MAD, and MH.
The contact author has declared that neither they nor their co-authors have any competing interests.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the special issue “Research results from the 14th International Conference on Mercury as a Global Pollutant (ICMGP 2019), MercOx project, and iGOSP and iCUPE projects of ERA-PLANET in support of the Minamata Convention on Mercury (ACP/AMT inter-journal SI)”. It is not associated with a conference.
Financial support from the project Integrated Global Observing Systems for Persistent Pollutants (IGOSP) funded by the European Commission in the framework of program “The European network for observing our changing planet (ERA-PLANET)”, grant agreement 689443, is also acknowledged. The authors would like to thank the TRIGA reactor staff at the Reactor Infrastructure Centre of the JSI for their availability and cooperation at all times. We would also like to thank Jarkko Makkonen and Timo Rajamäki for supplying us with the calibrator and for operational advice. The authors would like to thank Ingvar Wängberg at IVL Sweden for their initial guidance and providing detailed information regarding the extractor vessel apparatus.
This research has been supported by the European Commission, Horizon 2020 Framework Programme (ERA-PLANET (grant no. 689443)), the European Metrology Programme for Innovation and Research (grant no. 16ENV01), the Javna Agencija za Raziskovalno Dejavnost RS (grant nos. P1-0143 and PR-52044), and the Urad Republike Slovenije za Meroslovje (grant no. C3212-10-000071).
This paper was edited by Daniela Famulari and reviewed by two anonymous referees.
Andersson, M. E., Gårdfeldt, K., Wängberg, I., and Strömberg, D.: Determination of Henry's law constant for elemental mercury, Chemosphere, 73, 587–592, https://doi.org/10.1016/j.chemosphere.2008.05.067, 2008.
ASTM International: ASTM D6784-16, Standard Test Method for Elemental, Oxidized, Particle-Bound and Total Mercury in Flue Gas Generated from Coal-Fired Stationary Sources (Ontario Hydro Method), available at: http://www.astm.org (last access: 15 April 2021), 2016.
Bu, X., Zhang, H., Lv, G., Lin, H., Chen, L., Yin, X., Shen, G., Yuan, W., Zhang, W., Wang, X., and Tong, Y.: Comparison of Reactive Gaseous Mercury Collection by Different Sampling Methods in a Laboratory Test and Field Monitoring, Environ. Sci. Technol. Lett., 5, 600–607, https://doi.org/10.1021/acs.estlett.8b00439, 2018.
Dumarey, R.: Comparison of the collection and desorption efficiency of activated charcoal, silver, and gold for the determination of vapor-phase atmospheric mercury, Anal. Chem., 57, 2644–2646, 1985.
Dumarey, R., Brown, R. J. C., Corns, W. T., Brown, A. S., and Stockwell, P. B.: Elemental mercury vapour in air: The origins and validation of the “Dumarey equation” describing the mass concentration at saturation, Accredit. Qual. Assur., 15, 409–414, https://doi.org/10.1007/s00769-010-0645-1, 2010.
Dunham-Cheatham, S. M., Lyman, S., and Gustin, M. S.: Evaluation of sorption surface materials for reactive mercury compounds, Atmos. Environ., 242, 117836, https://doi.org/10.1016/j.atmosenv.2020.117836, 2020.
Electric Power Research Institute (EPRI): Guidelines for Speciated Mercury Field Measurements, Palo Alto, California, USA, 2015.
Gačnik, J., Živković, I., Guevara, S. R., Jaćimović, R., Kotnik, J., and Horvat, M.: Validating an evaporative calibrator for gaseous oxidized mercury, Sensors, 21, 2501, https://doi.org/10.3390/s21072501, 2021.
Gustin, M. and Jaffe, D.: Reducing the uncertainty in measurement and understanding of mercury in the atmosphere, Environ. Sci. Technol., 44, 2222–2227, https://doi.org/10.1021/es902736k, 2010.
Gustin, M. S., Huang, J., Miller, M. B., Peterson, C., Jaffe, D. A., Ambrose, J., Finley, B. D., Lyman, S. N., Call, K., Talbot, R., Feddersen, D., Mao, H., and Lindberg, S. E.: Do we understand what the mercury speciation instruments are actually measuring? Results of RAMIX, Environ. Sci. Technol., 47, 7295–7306, https://doi.org/10.1021/es3039104, 2013.
Gustin, M. S., Dunham-Cheatham, S. M., and Zhang, L.: Comparison of 4 Methos for Measurement of Reactive, Gaseous Oxiize, an Particulate Bound Mercury, Environ. Sci. Technol., 53, 14489–14495, https://doi.org/10.1021/acs.est.9b04648, 2019.
Gustin, M. S., Dunham-Cheatham, S. M., Zhang, L., Lyman, S., Choma, N., and Castro, M.: Use of Membranes and Detailed HYSPLIT Analyses to Understand Atmospheric Particulate, Gaseous Oxidized, and Reactive Mercury Chemistry, Environ. Sci. Technol., 55, 893–901, https://doi.org/10.1021/acs.est.0c07876, 2021.
Huang, J. and Gustin, M. S.: Uncertainties of gaseous oxidized mercury measurements using KCl-coated denuders, cation-exchange membranes, and nylon membranes: Humidity influences, Environ. Sci. Technol., 49, 6102–6108, https://doi.org/10.1021/acs.est.5b00098, 2015.
Huang, J., Miller, M. B., Weiss-Penzias, P., and Gustin, M. S.: Comparison of gaseous oxidized Hg measured by KCl-coated denuders, and nylon and cation exchange membranes, Environ. Sci. Technol., 47, 7307–7316, https://doi.org/10.1021/es4012349, 2013.
International Organization for Standardization: ISO 8573-1:2010 Compressed air – Part 1: Contaminants and purity classes, available at: https://www.iso.org/standard/46418.html (last access: 28 March 2021), 2010.
Jaffe, D. A., Lyman, S., Amos, H. M., Gustin, M. S., Huang, J., Selin, N. E., Levin, L., Ter Schure, A., Mason, R. P., Talbot, R., Rutter, A., Finley, B., Jaeglé, L., Shah, V., McClure, C., Ambrose, J., Gratz, L., Lindberg, S., Weiss-Penzias, P., Sheu, G. R., Feddersen, D., Horvat, M., Dastoor, A., Hynes, A. J., Mao, H., Sonke, J. E., Slemr, F., Fisher, J. A., Ebinghaus, R., Zhang, Y., and Edwards, G.: Progress on understanding atmospheric mercury hampered by uncertain measurements, Environ. Sci. Technol., 48, 7204–7206, https://doi.org/10.1021/es5026432, 2014.
Koron, N., Bratkič, A., Ribeiro Guevara, S., Vahčič, M., and Horvat, M.: Mercury methylation and reduction potentials in marine water: An improved methodology using 197Hg radiotracer, Appl. Radiat. Isot., 70, 46–50, https://doi.org/10.1016/j.apradiso.2011.07.015, 2012.
Lide, D. R.: CRC Handbook of Chemistry and Physics, 88th edn., edited by: Lide, D. R., Taylor & Francis, Bosa Roca, USA, 2007.
Lyman, S., Jones, C., O'Neil, T., Allen, T., Miller, M., Gustin, M. S., Pierce, A. M., Luke, W., Ren, X., and Kelley, P.: Automated calibration of atmospheric oxidized mercury measurements, Environ. Sci. Technol., 50, 12911–12927, https://doi.org/10.1021/acs.est.6b04211, 2016.
Lyman, S. N., Jaffe, D. A., and Gustin, M. S.: Release of mercury halides from KCl denuders in the presence of ozone, Atmos. Chem. Phys., 10, 8197–8204, https://doi.org/10.5194/acp-10-8197-2010, 2010.
McClure, C. D., Jaffe, D. A., and Edgerton, E. S.: Evaluation of the KCl denuder method for gaseous oxidized mercury using HgBr2 at an in-service AMNet site, Environ. Sci. Technol., 48, 11437–11444, https://doi.org/10.1021/es502545k, 2014.
Prestbo, E. M. and Bloom, N. S.: Mercury Speciation Adsorption (Mesa) Method for Combustion Flue Gas: Methodology, Artifacts, Intercomparison, and Atmospheric Implications, Water Air Soil Pollut., 80, 145–158, https://doi.org/10.1007/BF01189663, 1995.
Ribeiro Guevara, S. and Horvat, M.: Stability and behaviour of low level spiked inorganic mercury in natural water samples, Anal. Methods, 5, 1996–2006, https://doi.org/10.1039/c3ay26496c, 2013.
Ribeiro Guevara, S., Jereb, V., Arribére, M. A., Pérez Catán, S., and Horvat, M.: The production and use of 197Hg g radiotracer to study mercury transformation processes in environmental matrices, RMZ Mater. Geoenvironment J., 51, 1928–1931, 2004.
Ribeiro Guevara, S., Žižek, S., Repinc, U., Catán, S. P., Jaćimović, R., and Horvat, M.: Novel methodology for the study of mercury methylation and reduction in sediments and water using 197Hg radiotracer, Anal. Bioanal. Chem., 387, 2185–2197, https://doi.org/10.1007/s00216-006-1040-y, 2007.
Saxholm, S., Rajamäki, T., Hämäläinen, J., and Hildén, P.: Dynamic calibration method for reactive gases, Meas. Sci. Technol., 31, 034001, https://doi.org/10.1088/1361-6501/ab4d68, 2020.
Shafawi, A., Ebdon, L., Foulkes, M., Stockwell, P., and Corns, W.: Determination of total mercury in hydrocarbons and natural gas condensate by atomic fluorescence spectrometry, Analyst, 124, 185–189, https://doi.org/10.1039/a809679a, 1999.
United Nations Environment Programme: Global mercury assessment 2018, available at: https://www.unep.org/resources/publication/global-mercury-assessment-2018 (last access: 9 April 2021), 2019.
United States Environmental Protection Agency (U.S. EPA): IO Compendium Method IO-5: Compendium of Methods for the Determination of Inorganic Compounds in Ambient Air: Sampling and Analysis for Vapor and Particle Phase Mercury in Ambient Air Utilizing Cold Vapor Atomic Fluorescence Spectrometry (CVAFS), available at: https://www.epa.gov/esam/epa-io-inorganic-compendium-method-io-5-sampling-and-analysis -vapor-and-particle-phase-mercury (last access: 15 February 2021), 1999.
United States Environmental Protection Agency (U.S. EPA): Method 30A – Determination Of Total Vapor Phase Mercury Emissions From Stationary Sources (Instrumental Analyzer Procedure), available at: https://www.epa.gov/emc/method-30a-mercury-instrumental-procedure (last access: 20 February 2021), 2017.
Wang, S., Holsen, T. M., Huang, J., and Han, Y.-J.: Evaluation of various methods to measure particulate bound mercury and associated artifacts, Atmos. Chem. Phys. Discuss., 13, 8585–8614, https://doi.org/10.5194/acpd-13-8585-2013, 2013.
Zhang, H., Fu, X., Wang, X., and Feng, X.: Measurements and Distribution of Atmospheric Particulate-Bound Mercury: A Review, Bull. Environ. Contam. Toxicol., 103, 48–54, https://doi.org/10.1007/s00128-019-02663-5, 2019.
The requested paper has a corresponding corrigendum published. Please read the corrigendum first before downloading the article.
- Article
(418 KB) - Full-text XML
- Corrigendum
-
Supplement
(305 KB) - BibTeX
- EndNote