the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 06 Sep 2023
| 06 Sep 2023
An extraction method for nitrogen isotope measurement of ammonium in a low-concentration environment
Alexis Lamothe
Patrick Ginot
Lison Soussaintjean
Elsa Gautier
Pete D. Akers
Nicolas Caillon
Joseph Erbland
Ammonia (NH3) participates in the nucleation and growth of aerosols and thus plays a major role in atmospheric transparency, pollution, health, and climate-related issues. Understanding its emission sources through nitrogen stable isotopes is therefore a major focus of current work to mitigate the adverse effects of aerosol formation. Since ice cores can preserve the past chemical composition of the atmosphere for centuries, they are a top tool of choice for understanding past NH3 emissions through ammonium (NH, the form of NH3 archived in ice. However, the remote or high-altitude sites where glaciers and ice sheets are typically localized have relatively low fluxes of atmospheric NH deposition, which makes ice core samples very sensitive to laboratory NH3 contamination. As a result, accurate techniques for identifying and tracking NH3 emissions through concentration and isotopic measurements are highly sought to constrain uncertainties in NH3 emission inventories and atmospheric reactivity unknowns. Here, we describe a solid-phase extraction method for NH samples of low concentration that limits external contamination and produces precise isotopic results. By limiting NH3atm exposure with a scavenging fume hood and concentrating the targeted NH through ion exchange resin, we successfully achieve isotopic analysis of 50 nmol NH samples with a 0.6 ‰ standard deviation. This extraction method is applied to an alpine glacier ice core from Col du Dôme, Mont Blanc, where we successfully demonstrate the analytical approach through the analysis of two replicate 8 m water equivalent ice cores representing 4 years of accumulation with a reproducibility of ±2.1 ‰. Applying this methodology to other ice cores in alpine and polar environments will open new opportunities for understanding past changes in NH3 emissions and atmospheric chemistry.
- Article
(4649 KB) - Full-text XML
-
Supplement
(306 KB) - BibTeX
- EndNote





Ammonia (NH3) is an important compound of the nitrogen cycle that has many direct and indirect effects on public health and the environment. Not only does NH3 react with sulfate and nitrate to form secondary aerosols that adversely affect human health through air pollution (Lelieveld and Pöschl, 2017), but it also impacts soil acidification (Velthof et al., 2011) and changes radiative forcing (Szopa et al., 2021). The main sources of NH3 are assumed to be livestock waste and fertilizers, but considerable proportions are emitted by biomass burning, industry (Sutton et al., 2013), and road traffic through three-way catalyst engines (Perrino et al., 2012; Reche et al., 2015). Despite these major impacts, quantified inventories of NH3 mass and flux in the environment are highly approximated due to a lack of a NH3 monitoring networks (Fortems-Cheiney et al., 2016) and large uncertainties in emission factors (to ±300 %, Fortems-Cheiney et al., 2016), and major discrepancies therefore exist between these inventories and satellite observations as well as their subsequent top-down inventories (e.g. Zhan et al., 2021; Fortems-Cheiney et al., 2022; Van Damme et al., 2018). These factors result in NH3 inventory uncertainties that cannot be well estimated (European Environment Agency, 2021). Tracking past changes in NH3 is even more difficult because NH3 readily reacts in the environment and is generally not preserved in natural archives. At a time when reducing NH3 emissions is an increasingly important issue both in academia (Gu et al., 2021; Erisman, 2021) and in politics (European Council, 2016), an improved method of accounting for NH3 emission sources as well as tracking their changes in the past is required.
Despite the relative scarcity of NH3 analytical possibilities, ammonium (NH can serve as a proxy for NH3 as it is a generally more stable compound formed from the protonation of NH3, and it is known to be preserved in archives such as ice cores. NH3 protonation (Reaction 1) is central in aerosol chemistry through its participation in nucleation and particle growth in the presence of acids (often H2SO4 or HNO3) (Kirkby et al., 2011; Wang et al., 2022).
Stable isotope analyses have become increasingly relevant for determining emission sources of NH3 and other environmental compounds. Because one isotope of an element (e.g. 15N) is often favoured when a compound (e.g. NH undergoes physical transformations or chemical reactions, the isotopic ratios of a compound can differ based on the specific history of its formation and transportation. In this manner, nitrogen isotope ratios (δ15N defined in Eq. 1 and reported relative to standard atmospheric N2) are a means of studying the sources and reactive history of NH and thus NH3.
Glacial ice is an excellent natural archive for NH as aerosols are continuously deposited and buried with snow on the glacier surface and then preserved within the ice over long periods of time. By drilling and sequentially melting ice cores, one can reconstruct the history of changes to these aerosols at local and global scales (e.g. Preunkert et al., 2001; Gautier et al., 2019). The nitrogen isotopic ratios of NH (δ15N(NH preserved in ice cores should provide a valuable insight into past NH variability in sourcing and formation, but such a record has not yet been described in the scientific literature.
NH extraction for nitrogen isotopic analysis outside of ice core research has a long history of investigation. Analysis through an isotopic ratio mass spectrometer (IRMS) requires the conversion of NH to a gaseous species, and Rittenberg was among the first to present an extraction method using Rittenberg oxidation to produce N2 (Sprinson and Rittenberg, 1949). The subsequent development of elemental analyser combustion (Burke et al., 1990) provided an alternative method to produce N2, but both these methods require large sample sizes (µmol) unsuitable for the very low concentrations of NH in ice core samples (µmol kg−1 range). Although later developments in distillation and diffusion techniques offered additional options, substantial analytical limitations remained. NH3 distillation (Preston et al., 1996) is time-consuming and technically demanding, and it has been reported as being sensitive to external contamination (Liu et al., 2014), while the diffusion method (O'Deen and Porter, 1980) presents the same drawbacks, in addition to being unreliable at low concentrations.
To overcome these analytical obstacles, a methodology based on ion exchange resin offers more promising results by concentrating NH from large quantities of ice in a manner similar to that successfully used for analysing the nitrogen isotopes of nitrate in ice cores (Silva et al., 2000; Frey et al., 2009; Erbland et al., 2013). The ion exchange resin can be used in two ways. First, NH concentrated on the resin can be followed by immediate combustion to produce N2 (Lehmann et al., 2001), but the required sample size for this approach (100 µmol) makes it unsuitable for many ice core studies. A second technique consists of eluting the resin-concentrated NH, and the resulting aqueous NH sample can be first converted into N2O and then into N2 for the IRMS measurement (Zhang et al., 2007). This conversion is based on NH oxidation to NO by a hypobromite solution, with NO then reduced to N2O by a sodium azide acetic acid solution. This second method has been applied to aerosol samples (Kawashima et al., 2021) but has not yet been applied to ice core analyses, likely due to challenges related to the smaller NH concentrations in ice.
This paper presents an analytical method for the collection and analysis of NH in small sample sizes (< 50 nmol) suitable for ice cores, which is then applied to ice from the Col du Dôme (CDD) glacier on the Mont Blanc massif (4250 m a.s.l., 45.8421∘ N, 6.8474∘ E). This NH analysis is of particular interest because Col du Dôme ice cores have been previously found to accurately preserve atmospheric composition on European and local scales (Preunkert et al., 2001, 2003; Maupetit et al., 1995). For our proposed method, contamination control and sample collection were meticulously tested to determine best practices to preserve sample purity. Reproducibility and repeatability of this method are presented for 8 m water equivalent (m w.e.) of a Col du Dôme ice core, which shows the new potential that this method offers for understanding NH sources, transport, and reactivity.
This section gathers the details to perform an analysis presented in the paper for an ice core sample assuming that the concentration profile is already determined. Figure 1 summarizes the main steps – i.e. sample preparation, NH resin concentration and elution, isotopic measurement, and blank correction – of δ15N(NH measurement in a 10 cm diameter ice core with, for each step, the tests that were performed to develop the methodology and the final methodology. All terms are precisely defined in the following sections.
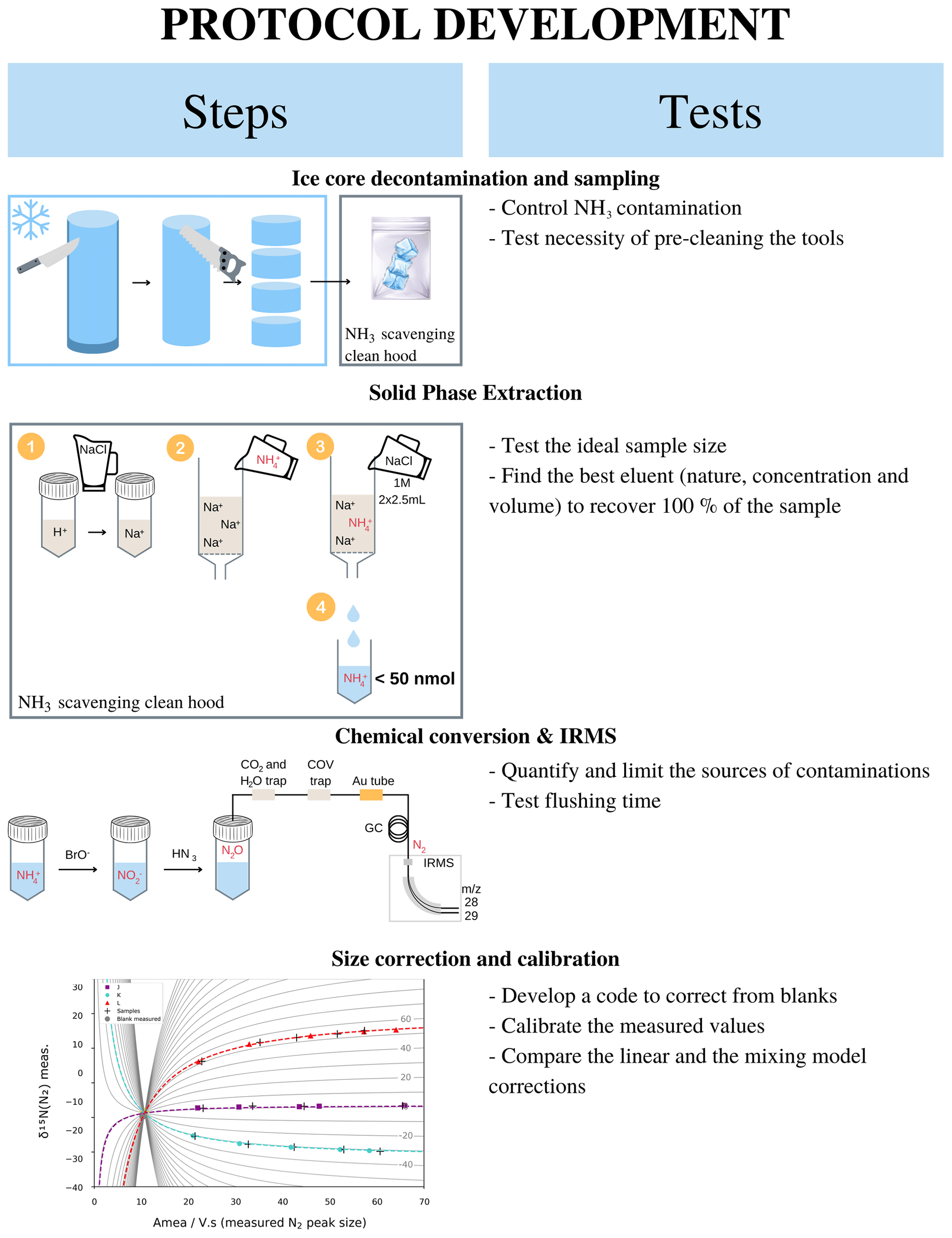
Figure 1Graphical summary of the steps involved in our proposed ice core NH isotopic analysis and the tests undertaken at each step to refine the protocol.
-
Sample preparation.
In a cold room, the first step is to cut the samples. For this, it is necessary to use clean tools that have been decontaminated with MQ water (18.2 MΩ cm) ice. The ice core is decontaminated by removing the outermost 2 mm of the external faces. The core is then sampled according to the desired sample size; in our case, with a 50 nmol target, sampling every 25 cm was sufficient. Decontaminated ice samples are sealed in clean plastic bags (Whirl-Pak) and then left to melt within the bags under the NH3-scavenging clean hood.
-
Concentration and elution step.
For each analytical batch, a minimum of two blanks, 5×3 standards (20, 40, 60, 80, and 100 nmol of the international isotopic standards IAEA-N-1, USGS 25, and USGS 26), and the samples studied must be passed through the cationic resins. The AG 50W that is originally in H+ form must be converted to the Na+ form by preparing a solution of the resin with NaCl 1 M (1:1 m m−1). Each extraction column then uses 0.6 mL of this NaCl–resin solution. To begin an NH extraction, 10 mL of 1 M NaCl solution is passed through the resin in the column and followed with five successive rinses with 10 mL of MQ water. The resin is dried by blowing 10 mL of air with a pipette, and the sample (or standard or blank) is loaded by injection at the top of the extraction column. As the sample passes gravitationally through the resin, the NH is trapped in the resin and the passed liquid discarded. Next, the now trapped NH is eluted into a vial with the injection of 2×2.5 mL of 1 M NaCl at the top of the column, allowing it to gravitationally pass through the resin. After dripping has completed, the vial containing the NH in 5 mL 1 M NaCl is sealed. Used resin can be regenerated for reuse by flushing with 10 mL of 1 M NaCl as well as 5×10 mL of MQ water and also stored in a freezer for future use.
-
Chemistry conversion and IRMS analysis.
The measurement of δ15N(NH is performed according to methods described in Zhang et al. (2007) and McIlvin and Altabet (2005). Hypobromite and the sodium arsenate solutions are prepared under the NH3-scavenging clean hood. The sodium azide acetic acid solution is flushed with He at 40 mL min−1 for 30 min. An autosampler can be used for precise and repetitive chemistry mixing. We remind readers that this method uses toxic chemicals that need to be handled respecting strict safety rules with personal protective equipment worn. After NH is converted into N2O, the latter follows an online method (Kaiser et al., 2007) to be reduced into N2 whose δ15N isotopic composition is measured with a Finnigan MAT-253 IRMS.
-
Blank correction.
The calibration and the correction from the blank are performed with the Python code available as a separate link (Lamothe et al., 2023). This correction determines the best parameters to limit the difference between a theoretical δ15N based on 15N mass balance and the measured δ15N. This allows calculating calibrated δ15N values for each standard, which can then be used to correct the sample values.
3.1 Continuous flow analysis NH concentration measurement
The NH concentration profile of an ice core is needed to plan out sampling intervals that contain the desired amount of NH and to provide geochemical interpretation. For our analyses, we determined the NH concentrations with the continuous flow analysis (CFA) PANDA platform developed at IGE in Grenoble, France (https://panda.osug.fr/, last access: 8 August 2023). This platform continuously melts and analyses ice sticks (cross-section: 34×34 mm2) with an online colorimetry module that can measure NH concentration as part of its analytical platform. The methodology for measuring NH concentration through online colorimetry is described in detail in the existing literature (Sigg et al., 1994; Röthlisberger et al., 2000; Kaufmann et al., 2008; Bigler et al., 2011). In short, soluble NH from a melted ice core reacts through the Roth ternary reaction with a reducing phthaldialdehyde (OPA) reagent and sodium sulfite buffered at pH = 9.5. The reaction rate is controlled at 75 ∘C, and the product creates a highly fluorescent signal at 430 nm (Genfa and Dasgupta, 1989). Three concentration standards at 25, 122, and 521 ppb were used for daily calibration. The overall system is suitable for ice core measurements with a limit of detection of (0.22±0.03) ppb, a linear calibration down to 1 ppm, and a repeatability <0.7 %.
3.2 Contamination control and ice core cutting
For any sample with low NH concentrations, atmospheric NH3 present during sample processing can be a substantial source of contamination. For example, NH concentrations in alpine ice cores are typically in the ng g−1 (or ppb) to µg g−1 (or ppm) range, and Preunkert et al. reported mean values in the Col du Dôme ice of 13±9 ppb in winter and 180±58 ppb in summer (Preunkert et al., 2000). To determine the potential scale of atmospheric contamination within the laboratory, MQ water samples (18.2 MΩ cm) were left open for several hours in a standard extraction fume hood. Identical experiments were carried out in a laminar flow purification hood equipped with an activated charcoal filter for NH3 scavenging (ASTM-012 filter on Iniflow-LAB1050 hood, Initioshop). NH concentrations for these contamination tests were measured through an ion chromatography device (IC) device (Thermo Fisher INTEGRION) connected to an autosampler (Thermo Fisher AS/AP), with separation on CG16-4 µm and CS16-4 µm 2 mm columns. The mobile phase is an isocratic manually mixed MSA 29 mM solution for the cations applying a 0.16 mL min−1 flow rate, resulting in a 27 min separation run with autoregenerated suppression and conductivity detection.
In addition to contamination of aqueous samples, the physical handling and processing of ice can have several potential opportunities for NH contamination. To investigate this route of contamination, we created artificial ice sticks made from cutting a block of frozen MQ pure water and replicated our typical ice core cutting and handling process. The MQ ice sticks were decontaminated by removing the outermost 2 mm of the external faces with a clean ceramic knife. To avoid confusion with other contamination, we will call this step surface decontamination. The surface-decontaminated ice sticks were then cut into individual ice samples using the saws typically used for ice core processing. We also tested the need for precleaning the tools, i.e. the ceramic knife and the two saws, before processing ice cores. Precleaning the tools consists of scratching all their faces against MQ ice for about 10 s. For this test, one MQ ice stick was surface-decontaminated using tools that had been previously precleaned with MQ ice, and another MQ ice stick was surface-decontaminated with non-precleaned tools. The resulting ice samples were sealed in clean plastic bags (Whirl-Pak) and then left to melt within the bags under the NH3-scavenging clean hood. IC measurements were performed for assessing the NH contamination levels resulting from the ice core cutting. Contamination was also a concern during the NH conversion to N2O processes, and our efforts to determine potential contamination risk for these steps will be discussed along with the conversion methodology (Sect. 3.5).
3.3 Sample size
Choosing a sample size for NH isotopic analysis is a compromise between being too large for a practical ice core study and being so small that the blank fraction's NH content – i.e. the proportion of the blank size to the sample size – substantially affects the isotopic and concentration uncertainties. To determine the ideal sample size, we targeted an initial 20–100 nmol range based on successful past experience with nitrate resin extraction and analysis (Silva et al., 2000; Erbland et al., 2013) as well as some preliminary testing. Tests were conducted using the three international NH isotopic standards IAEA-N-1 ((NH4)2SO4, δ15N = (+0.43 ± 0.07) ‰), USGS 25 ((NH4)2SO4, δ15N = (−30.41 ± 0.27) ‰), and USGS 26 ((NH4)2SO4, δ15N = () ‰), which are referred to throughout as standards J, K, and L, respectively.
Known molar quantities (20, 40, 60, 80, and 100 nmol) of each standard were passed through a cationic exchange resin, and δ15N values of each standard were determined at each molar quantity. We identified the best suitable sample size as the smallest size that has no substantial isotopic effect from the blank. In practice, this means finding the smallest molar quantity for which the blank fraction does not exceed one-third.
3.4 Elution: define the nature of the eluent, its concentration, and the volume needed
For NH capture, we used a solution of cationic resin AG 50W Bio-Rad resin (hydrogen form) in MQ water (1:1 ). To activate and clean the resin, 10 mL of a 1 M eluent is passed through the resin, followed by five rinses with 10 mL of MQ water. The choice of the eluent is discussed below. The resin was dried before and after passing samples through the resin by blowing 10 mL of air with a pipette. This step is not required for solid-phase extraction (SPE), but as it did not affect the result, we arbitrarily decided to systematically dry all resins in order to limit all the dead volumes. The volume of resin used for each NH concentration must fully capture the entire NH content of an ice sample to avoid possible isotopic fractionation issues. For this, a sufficient volume of resin must be used that considers the quantity of active sites in the resin in relation to the quantity of ions captured. All concentration and elution steps are fully carried out under the NH3-scavenging purification hood to limit external contamination. Our three isotopic NH standards were processed at 50 nmol each on three resin volumes of 0.3, 0.6, and 1.2 mL. After elution, NH was converted into N2O as described in the next section, and the isotopic and size deviations were determined on the IRMS. Note that it is not possible to directly measure the concentration of NH in the elution solution due to the high concentration of the ionic eluent; therefore, concentration measurements after elution are also done by reading the sample size in the IRMS.
We also ran a series of tests to determine which eluent compound and concentration was most effective in eluting the small volumes of ice-core-sourced NH trapped in our resin columns. To elute an ion from an ionic resin, one should take into account the counterion selectivity compared to the studied cation selectivity (here, NH with a selectivity listed as 1.95). For our Bio-Rad AG 50W resin, H+ and Na+ have a relative selectivity of 1.0 and 1.5, respectively. Consequently, we tested the easily sourced NaCl and HCl as possible eluents. KOH was also tested as an alkaline eluent to explore impacts from pH variations on our method. To determine the best eluent for NH trapped on the cationic resin, 50 nmol of NH trapped on the resin was eluted with 2×2.5 mL of 0.1 M NaCl, HCl, or KOH. This eluted NH was then converted into N2O and the yield compared to a 50 nmol NH that was not passed through the cationic resin but instead directly converted to N2O. After setting aside the choice of KOH as the eluent (see details in Sect. 4.3), a second test was performed between NaCl and HCl by increasing their concentration to 1 M.
Elution efficiency was also tested by collecting elution fractions whose sizes were measured by the IRMS. Starting with an eluent concentration of 0.1 M, we attempted to extract the isotopic international standards previously mentioned in a 30–100 nmol size range with a volume of 5 mL. Samples and eluent are gravitationally passed through the resin. After elution is completed, the resin is dried again to be consistent with sample loading as mentioned above, the NH sample is sealed with a septum in a glass vial, and the column can be reprocessed for future use. We found that one bed of resin can be used at least 25 times without affecting its effectiveness.
3.5 NH conversion and isotopic measurement
The measurement of δ15N(NH is performed according to the methods described by Zhang et al. (2007) and McIlvin and Altabet (2005). In detail, 0.4 mL of hypobromite solution is added to the NH-containing vial closed with a septum, and the reaction is run for 30 min under agitation. The hypobromite solution is prepared in a container rinsed with MQ water, into which 20 mL of MQ water is added, followed by 1 mL of a bromate bromide solution (0.6 g of NaBrO3+5 g of NaBr in 250 mL of MQ water) and then 1.05 mL of 6 M hydrochloric acid, immediately after which the reaction is left for 5 min in the dark before the final addition of 20 mL of 10 M sodium hydroxide solution. Then, the excess hypobromite from its reaction with NH is quenched by adding 0.1 mL of an arsenide solution (5.1 g of NaAsO2 in 100 mL MQ water). The solution is left to stir for 1 min. In the same vial, 1 mL of azide acetic acid solution (1:1 ) previously flushed with helium is added and the reduction is run for 30 min before the final addition of 1 mL NaOH 10 M to quench the excess reagent.
In order to reach the nanomole range size target needed for ice core studies, contamination controls must be performed. For the NH oxidation, the hypobromite solution preparation can suffer from atmospheric NH3 contamination, but this contamination is limited by preparation under the previously mentioned NH3-scavenging hood. On the other hand, sodium azide, used as a reagent during the reduction step, is a highly reactive solid which can lead to the formation of nitrite with its oxidation. We first tested the nitrite contamination of the NaN3 reagent by using a Griess colorimetric dosage (e.g. Röthlisberger et al., 2000). It has been reported that flushing the reagent solution with helium for 10 min will limit possible nitrite contamination, so we tested helium flushes of 0, 10, and 30 min to determine blank sizes of the overall method.
In the NH isotopic analysis, samples, and blanks undergo the identical analytical process as a suite of standards. Our three international NH standards J, K, and L were then used to correct for any isotopic effects resulting from processing and analysis. This was used as part of the development of the method for fractionation considerations and also, once the method is validated, for sample calibration. Measurements of δ15N(NH were performed on a Finnigan MAT-253 isotope ratio mass spectrometer (IRMS) following the protocol detailed by Kaiser et al. (2007). Briefly, N2O is decomposed into N2 and O2 that are then separated on a gas column prior to analysis by the IRMS.
3.6 Isotopic corrections
Since some degree of background contamination is inevitable, a blank is used to quantify the level of this contamination. As the blank size is not null, the sample NH mixes with the blank NH before its conversion into NO. Additionally, only one atom of N in N2O comes from NO, while the second one originates from the azide reagent (Zhang et al., 2007):
where Ameasured, Ablank, Aazide, and Asample respectively represent the measured amount, the blank amount from contamination, the chemistry reaction blank amount from azide, and the sample (or standard) amount. Equation (2) can immediately be expressed as follows since (Fig. S1 in the Supplement).
Thus, we can develop an isotopic mass balance on nitrogen in the final N2O measured on the IRMS (details in Fig. S1). We assume each step – i.e. SPE, NH oxidation by BrO−, and NO reduction by NaN3 – to be total (i.e. all the NO is consumed):
where δ15Nmeasured, δ15Nblank, δ15Nazide, and δ15Nsample respectively represent the measured 15N isotopic composition, that from the blank, that from the azide reagent, and that from the sample. In Eq. (4), three unknowns stay: Ablank, δ15Nblank, and δ15Nazide. The goal of the developed Python code is to determine this tuple, assuming it to be constant on all the samples of one batch. This code accounts for both a “size correction”, i.e. the relative contribution of the blank to the sample size, and also for any isotopic fractionation that occurs during the N2O measurement through the “gold method” conversion of N2O into N2 (Kaiser et al., 2007). To do so, we first vary the tuple of unknowns into a range of possible values, then calculate a theoretical δ15Nmeasured value, and finally correct the δ15Nmeasured values from the best tuple determined as the smallest residual difference between δ15Nscc (size-corrected and calibrated) and δ15Nmeasured. This code is performed with four main steps that are described hereafter.
-
For the international isotopic standards – IAEA-N-1, USGS 25, and USGS 26 respectively denoted J, K, and L ranging different sizes – the code calculates the δ15Nmeasured value that should theoretically be obtained and calculated with Eq. (5) based on the J, K, and L isotopic values from the literature (i.e. +0.43 ‰, −30.41 ‰, and +53.75 ‰). Ablank, δ15Nblank, and δ15Nazide vary in wide – yet realistic – intervals.
-
The second step consists of comparing the theoretically obtained δ15Nmeasured values with the empirically measured δ15Nmeasured values from the mass spectrometer. This accounts for the isotopic fractionation occurring during the N2O→N2 conversion.
-
The obtained linear relationship between the theoretical δ15Nmeasured and the observed δ15Nmeasured is used to calculate calibrated δ15N values for each standard.
-
From these calibrated standard values, Eq. (5) can be reversed to obtain δ15Nscc (size-corrected and calibrated) as shown in Eq. (6). A residual error is calculated as the standard deviation of the difference between the calculated δ15Nscc values and the international reference δ15N values. The best tuple is associated with the smallest residual error.
By comparing the calculated and measured blank isotopic compositions and sizes, one can check that the isotopic composition converges towards the blank with smaller sizes. Once the tuple is fixed, the mixing model is again reversed to correct and calibrate the samples. The Python scripts used for this correction are available as a separate link (Lamothe et al., 2023). This general correction, which considers blank size and calibration, will hereafter be referred to as the mixing model correction.
3.7 Application on a Col du Dôme ice core and reproducibility
As part of the Ice Memory programme, a 126 m ice core (CDD16-C01) was drilled in 2016 on the CDD glacier of Mont Blanc at 4250 m a.s.l. The CDD glacier is a high-accumulation glacier (from 0.5 to 2.4 m w.e. yr−1) on which a fair amount of literature has been published (e.g. Legrand et al., 2021; Preunkert et al., 2019, 2003, 2001; Guilhermet et al., 2013; Maupetit et al., 1995), largely dedicated to the dynamics of the Industrial Revolution and its anthropogenic emissions. The upper section of CDD16-C01 can be dated using seasonal variations in chemical concentrations, especially with NH (Preunkert et al., 2000). Indeed, the typically high NH concentrations found in summer reflect the vertical summer convection that transports NH from lower elevations. Winters are associated with lower NH concentration as the atmospheric boundary layer is well below the CDD site, which limits the NH supply from lower-elevation sources.
As a proof of concept, we analysed δ15N(NH using the methodology presented here for the first 8 m w.e. at a resolution of 25 cm. One-quarter of the ice core (102 mm diameter) was dedicated to δ15N(NH isotopic analyses, and this quarter was divided into two halves (named CDD16-C01-C and CDD16-C01-D) for reproducibility testing. Both of these reproducibility halves were analysed using the best-methods approach determined by the previously described methodological tests, and reproducibility was examined by analysing the two sections by different operators and with both fresh and non-fresh cationic resin.
4.1 Experimental NH sample size
The isotopic impact of NH contamination from the blank can be considered negligible when changes in NH sample size show no changes in δ15N values. Based on our test replicating sample sizes from 20 to 100 nmol, we observe that δ15N values are largely stable at sample sizes > 50 nmol (Fig. 2). Conversely, for sample sizes below 30 nmol, the NH coming from the blank becomes a substantial proportion of the total NH analysed per sample, with up to 46 % for 20 nmol samples. Unless it is possible to correct for the effects of the blank on the isotopic composition, 50 nmol is the smallest sample size that should be targeted while limiting isotopic effect from the blanks. However, the blank could also be corrected at the expense of greater isotopic value uncertainty, with the main limiting point being the acceptable level of uncertainty. This point will be further discussed in Sect. 4.5.
The tests on three resin volumes (0.3, 0.6, and 1.2 mL) reveal no fractionation linked to the resin volume choice, assuming 100 % elution of the sample. Indeed, for standards J, K, and L, the non-corrected δ15N values for the three volumes are () ‰, () ‰, and (13.0±0.1) ‰, with ± representing the repeatability variations. These variations are smaller than the chemical's method standard deviation of 0.3 ‰ (Zhang et al., 2007). We can confidently say that the resin volume we used does not affect the isotopic composition, probably because all the NH is extracted during the elution step. This aspect is further discussed in Sect. 4.3.
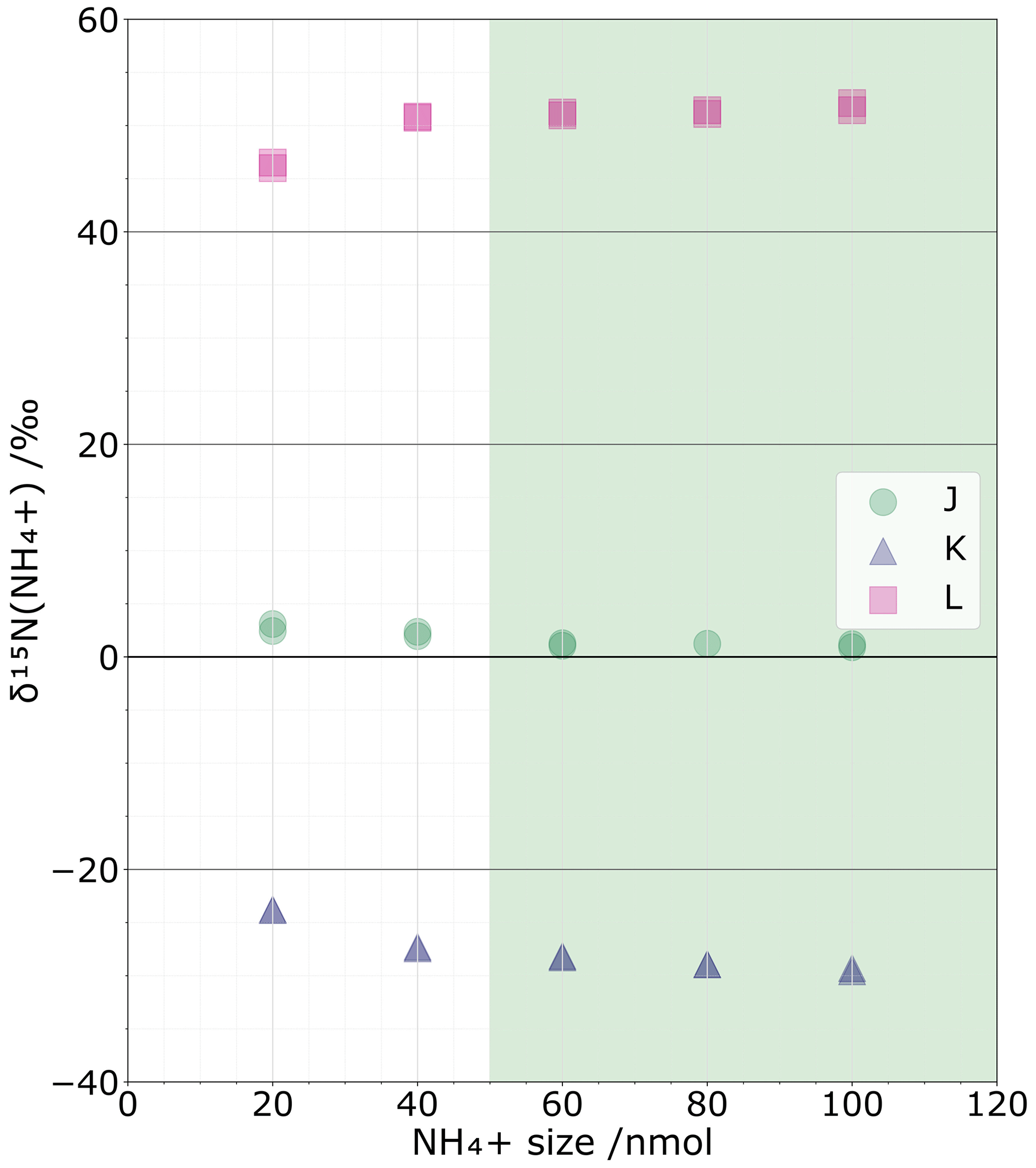
Figure 2δ15N(NH for three standards (‰) – J (red), K (green), and L (blue) – at different NH sizes (nmol) and δ15N linearity zones (faded green).
4.2 Blank control
Three sources of contamination that contribute to the blank were considered: atmospheric NH3 contamination, tool-induced contamination, and contamination during NH conversion into nitrous oxide. Regarding the first contamination source, Fig. 3 illustrates the relationship of NH concentrations in the MQ water vials kept open to the air for a certain amount of time under hoods with and without the NH3-scavenging system. Under a fume hood without the NH3-scavenging system, MQ vials are immediately contaminated. For atmospheric NH3 contamination, we found that a 5 mL MQ water sample left for 2 h in a standard fume hood reached 11 nmol NH due to atmospheric NH3 uptake. The use of the NH3-scavenging fume hood allows reducing NH3 contamination by a factor of ≈ 4, reaching ≈ 2.8 nmol NH after 3 h. While small, this NH3 contamination level is still excessively high for ice core studies. This difficulty has been solved with the replacement of the NH3 filter and its thickness that was halved to 5 cm. The previous filter was oversized, hence resulting in a pressure drop inside the laminar flow that is too high, thereby flowing in too much external air. One should therefore check the contamination levels induced by atmospheric NH3 to adjust the size of the filter initially pre-installed. The contamination is now controlled to 0.26±0.02 nmol from the atmospheric NH3.
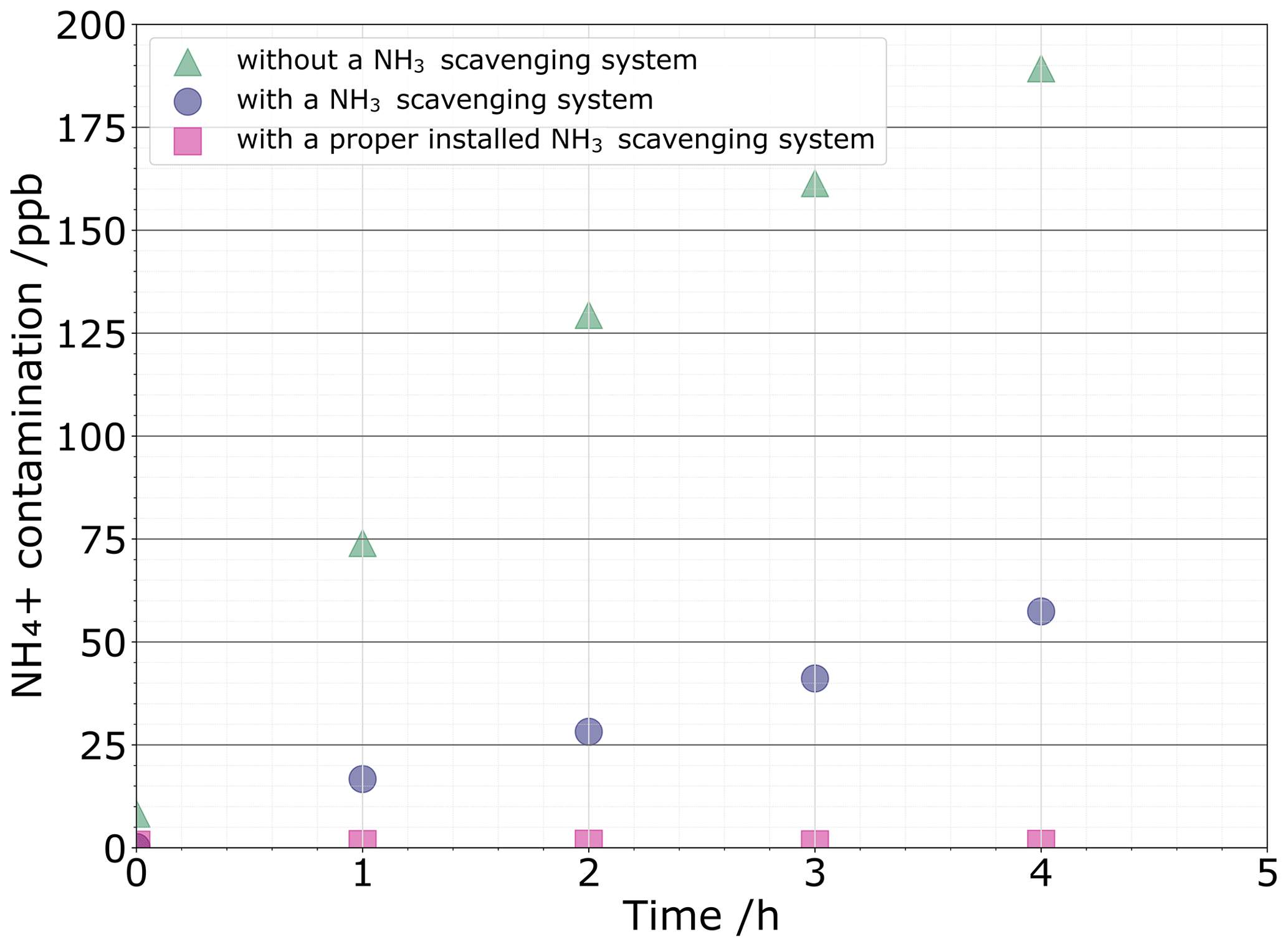
Figure 3NH contamination levels reported every hour under the standard fume hood (green triangle) and under the NH3-scavenging fume hood after installation (purple circle), as well as after changing the filter (pink square).
Tool-induced contamination proved to be negligible. For the MQ ice stick, which was decontaminated with non-precleaned tools, an NH level of 0.70 ppb is reported. This level is decreased to 0.14 ppb when using the precleaned tools. This drop of 0.56 ppb would represent only < 1 % of a typical blank size of 13 nmol. Still, we recommend precleaning as a component of good practice as it takes little time.
The most significant contamination source was found to be associated with chemistry reactions and the NaN3 acetic acid solution in particular. Although the nitrite contamination from the reducing solution (NaN3) was measured with Griess colorimetric dosage to be 27.5 ppb of NO (which represents 0.2 nmol of contamination for the 0.5 mL used as a reagent), this contamination is relatively insignificant compared to the 300 nmol NH for a standard blank test of 5 mL of MQ water that was measured for the NaN3 acetic acid solution. McIlvin and Altabet (2005) reported that this solution must be purged with He at 40 mL min−1 for 10 min. However, in our case, we found that this blank was reduced to around 20 nmol after 10 min of purging with He at 40 mL min−1, but that still remains too high for ice core analysis. We found that it was necessary to purge for at least 30 min to reach an acceptable value of 0.80 nmol. We suggest that the purging time is likely configuration-dependent, and this should be tested and set for each lab configuration.
Overall, our results suggest that contamination can be limited through meticulous planning and handling of samples. Summing all the contamination previously mentioned leads to the blank size. Relative to 50 nmol samples, the overall contamination present in the blank is still important, with an average 27 % size share relative to the sample. This requires that we develop a correction for the blank contribution through a mixing model (Sect. 4.5).
4.3 5 mL of 1 M NaCl for a complete NH elution
Eluting each 50 nmol NH sample with 0.1 M HCl, NaCl, and KOH achieved 113 %, 81 %, and 31 % sample recovery compared to the NH sample reference that was not passed through the cationic resin. The < 100 % recovery for NaCl and KOH indicates that some of the sample NH was left in the resin or otherwise lost, while the > 100 % recovery for HCl indicates that the sample was contaminated with an external source of NH. These results also highlight the important role of pH and ion affinity in selecting the eluent. Since K+ has a poorer affinity for the resin than NH, it is not unexpected that NH could be left in the resin after the elution. Additionally, for the 0.1 M KOH, the pH (13) is higher than pKa(NH, and thus the conversion of NH into NH3 is favoured. As a result, some sample NH will be lost into the atmosphere as NH3. Both explanations, although we do not know which, make the KOH elution unsuitable in our case. For the 0.1 M HCl elution, the pH = 1 and the > 100 % NH recovery suggest that the acidic conditions are fixing atmospheric NH3. In addition to this NH3 fixation, HCl was reported to affect kinetics of NH conversion into NO (McIlvin and Altabet, 2005), and therefore we reject HCl as a viable eluent.
This leaves NaCl as the best remaining eluent option, despite only 81 % recovery at 0.1 M. The conversion into N2O has not been reported due to an NaCl matrix (McIlvin and Altabet, 2005), so we assume that the low recovery of NH is due to an uncomplete elution that is improved by simply increasing the NaCl eluent concentration.
As the 81 % recovery from the 0.1 M NaCl matrix is insufficient, we next tested a 1 M NaCl solution to determine if a higher concentration could more completely push out all the NH ions. This test was comprised of 2×2.5 mL sequential elution fractions of 1 M NaCl, with each subsequent 5 mL eluent then analysed for NH concentration (Fig. 4). We found that 51.7±1.9 nmol of NH (1σ) was eluted after the first 5 mL elution. The second elution contained a very low 0.70±0.59 nmol of NH, which is unlikely to significantly alter the isotopic composition. The subsequent fractions do not contain measurable NH. Based on these results, we selected our eluent to be 2×2.5 mL of 1 M NaCl.
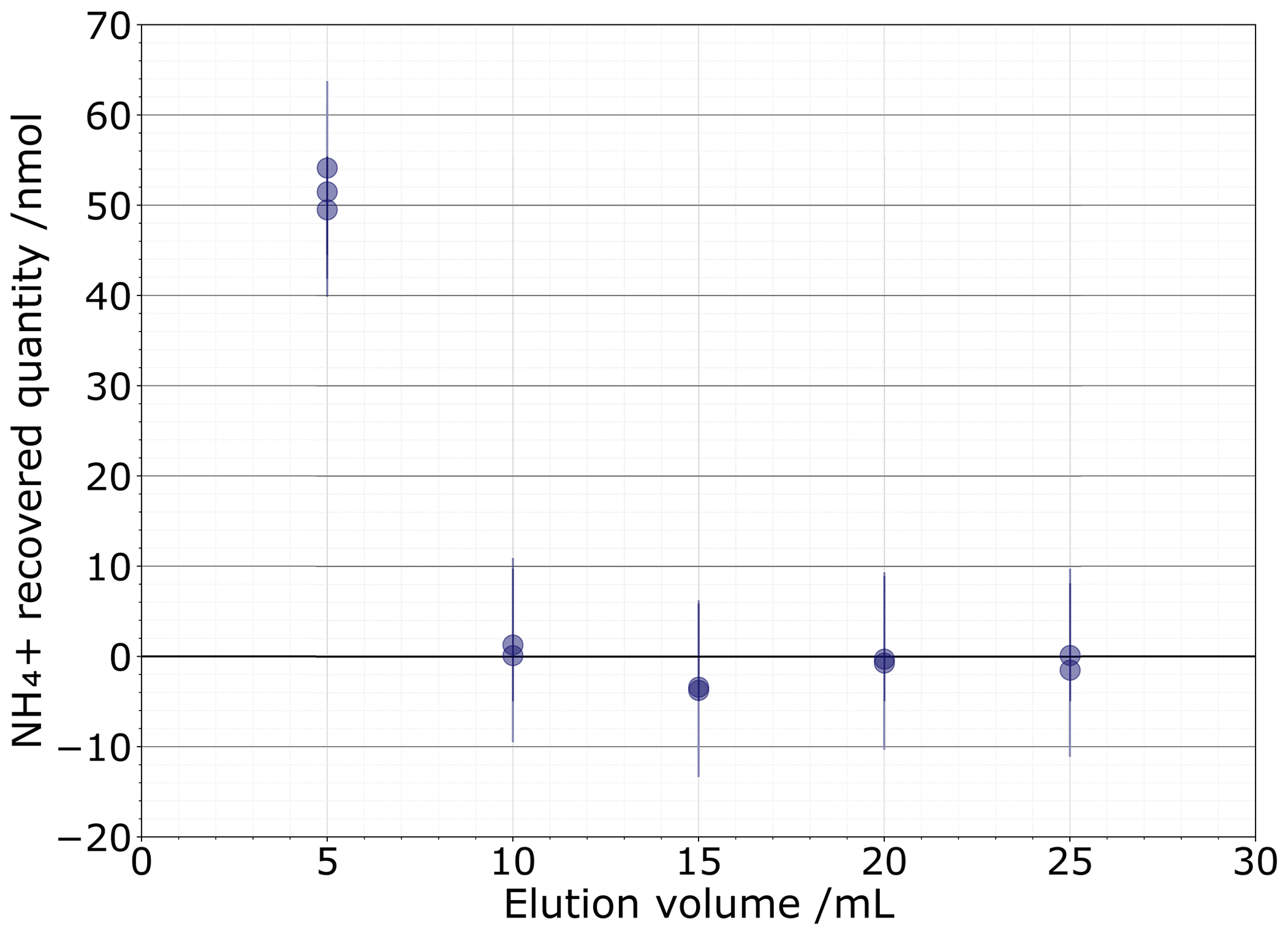
Figure 450 nmol NH recovered quantity after 5 mL elution fractions with measurement uncertainty shown by vertical bars.
4.4 Isotopic and size correction with mixing model
In contrast to a correction based on simple linear regression, which requires identical sample amounts of NH for standards and samples, our mixing model allows us to correct the method blanks and calibrate the samples. Figure 5 shows the results of a correction applied to a series of standards. For this purpose, the international standards are prepared at different sizes (20 to 100 nmol) and with processes as described above (i.e. chemical conversion and IRMS analysis). Then using the mixing model approach and assuming that each analysis shares the same blank (amount and isotopic composition) across the different standards for a given batch, the Python script calculates the shape of the mixing lines that best minimizes the difference between the modelled mixing lines and the standard points while converging toward the same origin, i.e. same blank. The best mixing line that passes through an unknown sample analysis (crosses in Fig. 5b) is then used as a calibration curve to obtain the corrected isotope composition. Figure 5a shows the IRMS linear response and variability with respect to the amount of NH, while Fig. 5b illustrates the mixing model curves (grey lines), the modelled blank, and a simulated set of unknown samples (black crosses) made from the same reference materials that are used for calibration for illustration purposes. Figure 5c shows the residual between the model mixing curves and standards. The residual δ15N of the standards is then used as a quantification of the precision of the method in the form of the standard deviation of the residual. With this approach, all standards of different sizes participate equally in the statistical correction of the method and allow covering a broad range of isotopic compositions and sample sizes in one given batch of analysis (i.e. four blanks, up to 70 samples, and 15 standards in our batch of analysis). In some cases, when the blank can be directly measured, it can be used as a post-quality-control point (grey dots of Fig. 5a and b) by comparing its size and isotope composition with the modelled one.
Comparing a calibration from standards that passed through resins with one with standards that did not results in blank sizes of 5.4 and 4.3 Vs, respectively, for the calibrations with resin-passed standards and non-resin-passed standards. The resin step increases the blank size by 20 %. An efficient mixing model is therefore needed to correct small size samples. Similarly, residual δ15N increases from 0.41 ‰ to 0.59 ‰ in this set of analysis.
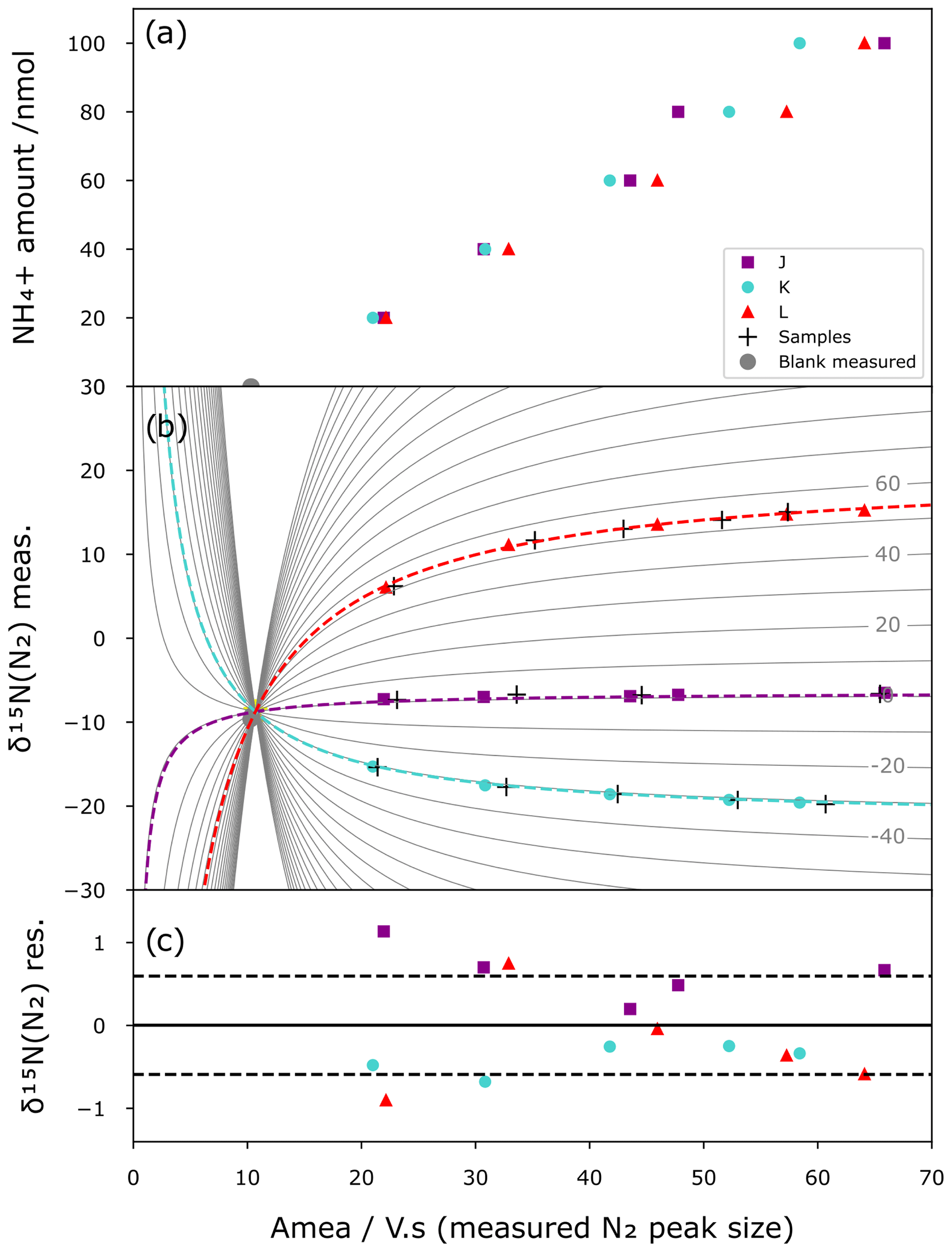
Figure 5Mixing model correction. (a) Calibration curve between standard sizes (nmol) and IRMS peak areas (Vs) for the three international isotopic standards J (green), K (blue), and L (red) as well as blanks (grey). (b) Mixing model curve fitting (dashed lines) constrained by the measured δ15N vs. the size of the standards (colour marks) and the measured blanks (grey dots). One set of standards are treated as unknown samples (black crosses) and corrected and calibrated with the mixing model (best fit of the continuous grey lines). (c) Residual δ15N from the difference between measured δ15N of the standards and calculated δ15N with the mixing model with 1σ standard deviation (dashed line).
Figure 6 compares the mixing-model-based correction with a simple linear regression correction for which we consider an average calibration with respect to standard sizes. For the set of standards that were processed as unknown samples (black crosses Fig. 5b), we quantify for both corrections the difference between the corrected values and their known isotopic standard values. For the two corrections, the general trend is an increase in divergence between corrected and known values with smaller sizes. However, the mixing-model-based correction performs better than a simple linear correction for all amounts tested. As expected, we notice a constant decline of the residual for the mixing model with respect to the NH size. This translates the increasing correction of the blank share for small sizes. For sample sizes > 40 nmol, the mixing model correction does not significantly improve the precision, showing that at this size level the blank becomes negligible, making the isotope correction insensitive to sample size, as it should be if a blank component did not exist. In the same way, one can question the necessity of using a mixing model correction at sample sizes > 40 nmol as the differences, i.e. the residual, between the linear correction and the international isotopic standard are not drastically different from each other. However, our approach demonstrates the usefulness of the mixing model correction for small size samples. We believe that this should be valuable and incorporated in the data treatment for low-concentration environments such as polar ice cores or other environments with limited sample sizes such as meteorites as well as other extraterrestrial samples. Before concluding this section, we want to emphasize that the 40 nmol threshold is laboratory-dependent and will highly depend on the blank level reached by each laboratory.
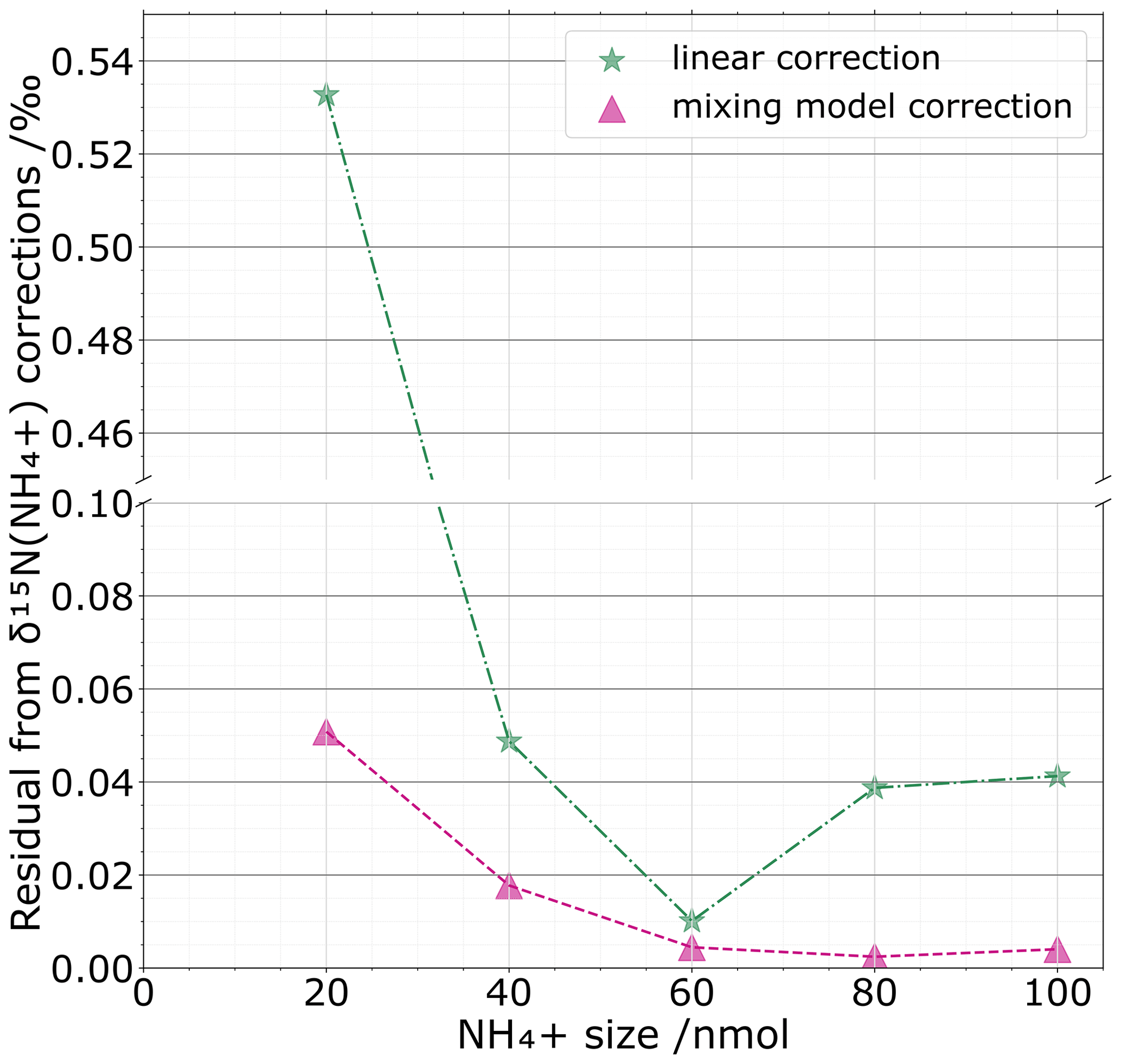
Figure 6For one set of standards processed as unknown samples and for each sample NH size (nmol), we compare the absolute value of the residuals between the linear correction (green stars) and the mixing model correction (violet triangles) with the isotopic standard values.
With the effect of the blank on the isotopic composition now under control (i.e. the residual < 0.2 ‰), samples with sizes < 50 nmol are only limited by the trade-off between size and acceptable uncertainty. The overall method we propose gives a precision of ±0.6 ‰ (1σ standard deviation) for samples greater than 20 nmol. This standard deviation is determined as the highest standard deviation for all our experiments calculated from the residuals of the mixing model. This ±0.6 ‰ value can be compared with the ±0.3 ‰ precision of the Zhang et al. (2007) NH conversion. Hence, although our proposed method has a higher standard deviation, it still offers the ability to measure δ15N(NH in low-concentration environments like ice cores with a relatively favourable standard deviation. To aptly validate the method, this standard deviation needs to be compared to ice core δ15N(NH variability.
4.5 Application of new methodology to CDD16-C01
To test the applicability of our method, a portion of the CCD ice core was cut in two parallel sections with one used for NH concentration quantification and isotope analysis, while the second was only used for isotope analysis reproducibility. The NH concentration profile measured on the continuous flow analysis system shows a well-marked seasonal pattern (Fig. 7a), in agreement with a previous study (Preunkert et al., 2000). Using this seasonal NH concentration pattern, we date the depth of 800 cm w.e. to summer 2013. Isotopic values of δ15N(NH vary between () ‰ and (5.5±1.4) ‰ across the two duplicated sections of the ice core, CDD16-C01-C and CDD16-C01-D (Fig. 7b). While the repeatability lies within ±2σ of the method precision, the reproducibility between the two parallel isotopic analyses at 2σ is ±2.1 ‰. Since the ice core's natural δ15N(NH values vary between −17.7 ‰ and 5.5 ‰, our analytical standard deviation of 0.6 ‰ and reproducibility standard deviation of 2.1 ‰ show that our overall proposed methodology has sufficiently low uncertainty for studying and interpreting ice core NH isotope compositions.
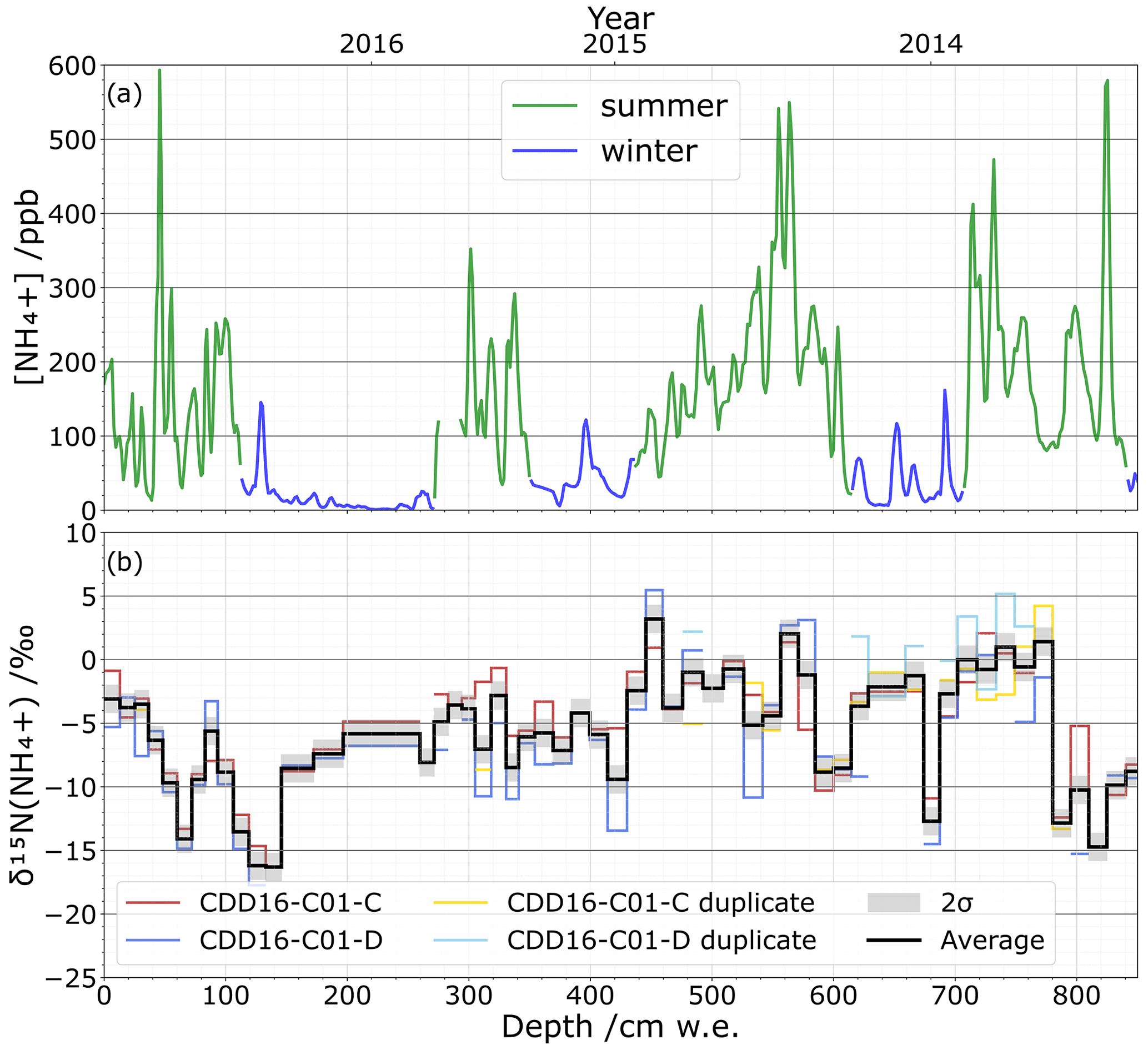
Figure 7(a) Continuous flow analysis NH concentration profile (ppb) in summer (green) and winter (blue) from 2016 to 2013 (top). (b) δ15N(NH isotopic profile (‰) with the 25 cm ice sampling resolution for the two parallel cores CDD16-C01-C (red) and CDD16-C01-D (blue) as well as their respective repeatability tests (yellow, light blue); the average value of all tests (black) is shown with the ±2σ reproducibility.
Although the environmental interpretation of this NH isotopic record is not the focus of this paper, we briefly consider the potential drivers of the observed isotopic variability. The isotopic composition of NH in an ice sample can be explained by the isotopic composition of the NH3 emission sources and/or the isotopic fractionation that may take place in the atmosphere or snow.
Because different NH3 sources often have different ranges of δ15N values, measuring δ15N(NH in ice cores offers the opportunity to investigate changes in NH3 sources as long-term trends, seasonal variations, or one-off events. For example, agricultural sources have low δ15N(NH3) values ranging from −51.8 ‰ to −40.9 ‰ and −31.7 ‰ to −18.8 ‰ in Chang et al. (2016) and Xiang et al. (2022), respectively, and emissions from human and pet waste also lead to 15N-depleted NH3 with signatures between −41.9 ‰ and −29.9 ‰ (Chang et al., 2019). On the other hand, higher δ15N values might suggest a greater contribution from combustion sources from industry (−14.6 ‰ to −11.3 ‰) or automobiles (4.5 ‰ to 8.7 ‰) (Walters et al., 2020). Ice cores from sites such as the CDD should record seasonal variations in local and/or regional NH3 sources. One study of the δ15N(NH composition of atmospheric particles in Lyon (150 km from Mont Blanc) has shown that the pollution peaks of ammonium nitrate in spring were accompanied by a strong decrease in δ15N (Favez et al., 2021), potentially explained by agricultural sources (livestock or fertilizers). However, this and other studies highlight the lack of broad understanding of isotopic variations over the rest of the year and difficulties in assigning diagnostic isotopic labelling to different emission pools (Mariappan et al., 2009; Felix et al., 2017; Favez et al., 2021).
Individual climate or environmental events may also leave distinct signals in an ice core's NH record. In the Alps, for example, Saharan dust events are an important source of NH, and a single event can deliver high amounts of NH within a brief period. We captured one such dust event in the CDD ice core at 820 cm w.e. where the NH concentration reaches its maximum value of nearly 600 ppb (Fig. 7a). Based on our chronology and supported by CFA data from terrigenous species (Mg2+, Ca2+, Al3+) as well as dust concentration, this NH peak corresponds to a dust event on 30 April 2013. While it is unclear at this time whether such Saharan dust events have isotopic impacts (Fig. 7b), our method offers a sampling resolution and isotopic precision fine enough to investigate these individual events.
The reactivity of NH in the atmosphere can also cause isotopic fractionation after NH3 has been emitted from its initial sources (Felix et al., 2017). NH3 in the presence of acids, especially sulfuric acid (H2SO4), can form a particle nucleus (Kürten, 2019). Subsequently, the condensation of weak volatile acids such as H2SO4 and nitric acid (HNO3) (Stolzenburg et al., 2020) with stabilization by ammonia or organic bases (Lehtipalo et al., 2016) leads the particle into a growth phase. Walters et al. (2019) point out that kinetic isotope fractionation can take place during nucleation, whereas equilibrium fractionation occurs during the growth phase (2019). In this regard, variations in the concentrations of the reagents could therefore modify these two fractionations. Assuming this process would not discriminate between different emission sources – i.e. the fractionation equally impacts all sources – long-term acidity trends could be investigated. Temperature can also affect atmospheric isotope fractionation (Li et al., 2012; Savard et al., 2017). The first results presented in Fig. 7b do not allow us to conclude on the kinetic or equilibrium fractionations. Concerning temperature, however, as we do not observe any major difference between the δ15N values measured in summer and winter (Fig. 7b), it seems unlikely that temperature plays a significant role in the fractionation. This aspect remains to be confirmed on a longer timescale.
Finally, isotopic fractionation is also possible after deposition within the snowpack through deprotonation of buried NH and its re-volatilization as NH3. These post-depositional processes function analogously to the better-documented post-deposition effects of nitrate in snow (e.g. Erbland et al., 2013). Strong 15N enrichment would alter the overall signal remaining in the snow as the isotopic fractionation constant NH NH3(g) is ε= 31 ‰ (Walters et al., 2019). However, these post-depositional processes are most impactful in the uppermost snow layers near the surface. Because CDD is a site of high accumulation (∼ 2.5 m w.e. yr−1) (Maupetit et al., 1995), any deposited NH will be rapidly buried to depths where post-depositional phenomena are unlikely. Furthermore, over the first 60 cm w.e. of the δ15N(NH signal (Fig. 7b), a decrease in δ15N is recorded with no change in [NH]. This decrease is opposite to the trend that should be observed with an enrichment of the snow in heavy isotopes if NH3 were to be reemitted; in addition, a depletion in [NH] should have been measured. We can therefore reject the idea that post-depositional processes have a substantial effect on NH isotopic values in the CDD snow. This question obviously remains to be investigated for sites marked by lower accumulation (e.g. East Antarctic highland).
About 25 years ago, Legrand and Mayewski stated that “the transfer functions of gaseous species that interact strongly with ice are the most complex”, and improving our analytical capabilities regarding this concept should be seen as one of the priorities of the ice core community (Legrand and Mayewski, 1997). New routes for studying NH3, one of the most abundant species in inorganic aerosols, and its neutralized form NH are therefore valuable and needed. This study presents a method for extracting NH and analysing its 15N isotopic composition in ice cores. The method hinges on three meticulous steps: a workflow under clean conditions, a cationic exchange resin with 1 M NaCl solution as the eluent to recover < 50 nmol of NH, and its conversion into a suitable gas for δ15N(NH analysis. The overall standard deviation of 0.6 ‰ and its reproducibility of 2.1 ‰ in an ice core from the Col du Dôme (Mont Blanc, France) are both much smaller than natural 15N variability. These results validate our methodological approach, and we believe that our NH analytical techniques can be helpful to reveal long-term past atmospheric changes in NH3 chemistry and emissions. Room for improvement still remains in the precision of the measurement, an issue that could be crucial for some sites where the range of δ15N(NH values would be lower than that measured at CDD. We suggest that some additional precision could be gained by pre-drying the NaCl before preparing the eluent to eliminate possible contamination with moisture and flushing the vials with helium before chemical conversion to N2O.
While NH3 emissions have long been driven by natural soil emissions, the increasing anthropogenic impact should also be detectable in δ15N(NH in ice cores both in alpine glaciers and polar ice sheets. As proposed controls and policies on NH3 emissions are debated, ice core analysis of δ15N(NH from alpine glaciers could allow an improved understanding of regional emission inventories. Looking to the future, the application of this method to Greenland ice could better constrain the changes and the origins of biomass burnings across the broader Northern Hemisphere (Rubino et al., 2016), while Antarctic NH isotopic analysis may also be useful for exploring how marine biomass is affected by sea ice changes (e.g. Thomas et al., 2019). In these and other ways, our newly refined methodology opens many new opportunities for atmospheric chemists and glaciologists to better understand how NH3 and NH have responded over time to human and natural changes in emissions, climate, and atmospheric composition.
The scripts are available here: https://doi.org/10.5281/zenodo.7728983 (Lamothe et al., 2023).
All the data mentioned in the article will be accessible in an open-access dataset still under construction, which will be named CDD16 after the Col du Dôme drilling campaign in 2016. When finalized, the link to this dataset will be found on the PANDA analytical platform website (https://panda.osug.fr/; PANDA, 2023). Until then, one can contact the authors, who will share the data.
The supplement related to this article is available online at: https://doi.org/10.5194/amt-16-4015-2023-supplement.
AL led the study, performed the laboratory measurements with the help of PG, LS, EG, and NC, and wrote the first draft with the help of PDA. All co-authors contributed to the improvement of the paper. JS and PG supervised AL. PG conducted the CDD drilling with the help of NC. JE developed the size correction model.
The contact author has declared that none of the authors has any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
We acknowledge the technical support from the F2G (French National Platform for Coring and Drilling, handled by INSU, the MIUR – Ministry of Education, University and Research) for the CDD16 drilling. We also thank Sarah Albertin for scientific advice, Selin Bagci for lab handling, and Sophie Darfeuil for CFA colorimetric module development.
This research has been supported by LabEx OSUG@2020 (“Investissements d’avenir” – ANR10 LABX56), the ANR project ANR-15-IDEX-02 (IDEX-UGA), the Ice Memory programme, and the Grenoble University Foundation (FUGA).
This paper was edited by Pierre Herckes and reviewed by two anonymous referees.
Bigler, M., Svensson, A., Kettner, E., Vallelonga, P., Nielsen, M. E., and Steffensen, J. P.: Optimization of High-Resolution Continuous Flow Analysis for Transient Climate Signals in Ice Cores, Environ. Sci. Technol., 45, 4483–4489, https://doi.org/10.1021/es200118j, 2011.
Burke, I. C., O'Deen, L. A., Mosier, A. R., and Porter, L. K.: Diffusion of Soil Extracts for Nitrogen and Nitrogen-15 Analyses by Automated Combustion/Mass Spectrometry, Soil Sci. Soc. Am. J., 54, 1190–1192, https://doi.org/10.2136/sssaj1990.03615995005400040047x, 1990.
Chang, Y., Liu, X., Deng, C., Dore, A. J., and Zhuang, G.: Source apportionment of atmospheric ammonia before, during, and after the 2014 APEC summit in Beijing using stable nitrogen isotope signatures, Atmos. Chem. Phys., 16, 11635–11647, https://doi.org/10.5194/acp-16-11635-2016, 2016.
Chang, Y., Zou, Z., Zhang, Y., Deng, C., Hu, J., Shi, Z., Dore, A. J., and Collett, J. L.: Assessing Contributions of Agricultural and Nonagricultural Emissions to Atmospheric Ammonia in a Chinese Megacity, Environ. Sci. Technol., 53, 1822–1833, https://doi.org/10.1021/acs.est.8b05984, 2019.
Erbland, J., Vicars, W. C., Savarino, J., Morin, S., Frey, M. M., Frosini, D., Vince, E., and Martins, J. M. F.: Air–snow transfer of nitrate on the East Antarctic Plateau – Part 1: Isotopic evidence for a photolytically driven dynamic equilibrium in summer, Atmos. Chem. Phys., 13, 6403–6419, https://doi.org/10.5194/acp-13-6403-2013, 2013.
Erisman, J. W.: How ammonia feeds and pollutes the world, Science, 374, 685–686, https://doi.org/10.1126/science.abm3492, 2021.
European Council: Directive (EU) 2016/2284 of the European Parliament and of the Council of 14 December 2016 on the reduction of national emissions of certain atmospheric pollutants, amending Directive 2003/35/EC and repealing Directive 2001/81/EC (Text with EEA relevance), OJ L, 344, 1–31, http://data.europa.eu/eli/dir/2016/2284/oj (last access: 31 August 2023), 2016.
European Environment Agency: European Union emission inventory report 1990–2019 under the UNECE Convention on Long-range Transboundary Air Pollution (Air Convention), 1–158, https://www.eea.europa.eu//publications/lrtap-1990-2019 (last access: 8 August 2023), 2021.
Favez, O., Weber, S., Petit, J.-E., Alleman, L. Y., Albinet, A., Riffault, V., Chazeau, B., Amodeo, T., Salameh, D., Zhang, Y., Srivastava, D., Samaké, A., Aujay-Plouzeau, R., Papin, A., Bonnaire, N., Boullanger, C., Chatain, M., Chevrier, F., Detournay, A., Dominik-Sègue, M., Falhun, R., Garbin, C., Ghersi, V., Grignion, G., Levigoureux, G., Pontet, S., Rangognio, J., Zhang, S., Besombes, J.-L., Conil, S., Uzu, G., Savarino, J., Marchand, N., Gros, V., Marchand, C., Jaffrezo, J.-L., and Leoz-Garziandia, E.: Overview of the French Operational Network for In Situ Observation of PM Chemical Composition and Sources in Urban Environments (CARA Program), Atmosphere, 12, 207, https://doi.org/10.3390/atmos12020207, 2021.
Felix, J. D., Elliott, E. M., and Gay, D. A.: Spatial and temporal patterns of nitrogen isotopic composition of ammonia at U.S. ammonia monitoring network sites, Atmos. Environ., 150, 434–442, https://doi.org/10.1016/j.atmosenv.2016.11.039, 2017.
Fortems-Cheiney, A., Dufour, G., Hamaoui-Laguel, L., Foret, G., Siour, G., Van Damme, M., Meleux, F., Coheur, P.-F., Clerbaux, C., Clarisse, L., Favez, O., Wallasch, M., and Beekmann, M.: Unaccounted variability in NH3 agricultural sources detected by IASI contributing to European spring haze episode, Geophys. Res. Lett., 43, 5475–5482, https://doi.org/10.1002/2016GL069361, 2016.
Fortems-Cheiney, A., Dufour, G., Foret, G., Siour, G., Van Damme, M., Coheur, P.-F., Clarisse, L., Clerbaux, C., and Beekmann, M.: Understanding the Simulated Ammonia Increasing Trend from 2008 to 2015 over Europe with CHIMERE and Comparison with IASI Observations, Atmosphere, 13, 1101, https://doi.org/10.3390/atmos13071101, 2022.
Frey, M. M., Savarino, J., Morin, S., Erbland, J., and Martins, J. M. F.: Photolysis imprint in the nitrate stable isotope signal in snow and atmosphere of East Antarctica and implications for reactive nitrogen cycling, Atmos. Chem. Phys., 9, 8681–8696, https://doi.org/10.5194/acp-9-8681-2009, 2009.
Gautier, E., Savarino, J., Hoek, J., Erbland, J., Caillon, N., Hattori, S., Yoshida, N., Albalat, E., Albarede, F., and Farquhar, J.: 2600-years of stratospheric volcanism through sulfate isotopes, Nat. Commun., 10, 466, https://doi.org/10.1038/s41467-019-08357-0, 2019.
Genfa, Z. and Dasgupta, P. K.: Fluorometric measurement of aqueous ammonium ion in a flow injection system, ACS Publications, 61, 408–412, https://doi.org/10.1021/ac00180a006, 1989.
Gu, B., Zhang, L., Van Dingenen, R., Vieno, M., Van Grinsven, H. J., Zhang, X., Zhang, S., Chen, Y., Wang, S., Ren, C., Rao, S., Holland, M., Winiwarter, W., Chen, D., Xu, J., and Sutton, M. A.: Abating ammonia is more cost-effective than nitrogen oxides for mitigating PM2.5 air pollution, Science, 374, 758–762, https://doi.org/10.1126/science.abf8623, 2021.
Guilhermet, J., Preunkert, S., Voisin, D., Baduel, C., and Legrand, M.: Major 20th century changes of water-soluble humic-like substances (HULIS WS) aerosol over Europe inferred from Alpine ice cores, J. Geophys. Res.-Atmos., 118, 3869–3878, https://doi.org/10.1002/jgrd.50201, 2013.
Kaiser, J., Hastings, M. G., Houlton, B. Z., Röckmann, T., and Sigman, D. M.: Triple Oxygen Isotope Analysis of Nitrate Using the Denitrifier Method and Thermal Decomposition of N2O, Anal. Chem., 79, 599–607, https://doi.org/10.1021/ac061022s, 2007.
Kaufmann, P. R., Federer, U., Hutterli, M. A., Bigler, M., Schüpbach, S., Ruth, U., Schmitt, J., and Stocker, T. F.: An Improved Continuous Flow Analysis System for High-Resolution Field Measurements on Ice Cores, Environ. Sci. Technol., 42, 8044–8050, https://doi.org/10.1021/es8007722, 2008.
Kawashima, H., Yoshida, O., and Suto, N.: Ion-exchange resin and denitrification pretreatment for determining δ15N-NH4+, δ15N-NO3, and δ18O-NO3 values, Rapid Commun. Mass Sp., 35, e9027, https://doi.org/10.1002/rcm.9027, 2021.
Kirkby, J., Curtius, J., Almeida, J., Dunne, E., Duplissy, J., Ehrhart, S., Franchin, A., Gagné, S., Ickes, L., Kürten, A., Kupc, A., Metzger, A., Riccobono, F., Rondo, L., Schobesberger, S., Tsagkogeorgas, G., Wimmer, D., Amorim, A., Bianchi, F., Breitenlechner, M., David, A., Dommen, J., Downard, A., Ehn, M., Flagan, R. C., Haider, S., Hansel, A., Hauser, D., Jud, W., Junninen, H., Kreissl, F., Kvashin, A., Laaksonen, A., Lehtipalo, K., Lima, J., Lovejoy, E. R., Makhmutov, V., Mathot, S., Mikkilä, J., Minginette, P., Mogo, S., Nieminen, T., Onnela, A., Pereira, P., Petäjä, T., Schnitzhofer, R., Seinfeld, J. H., Sipilä, M., Stozhkov, Y., Stratmann, F., Tomé, A., Vanhanen, J., Viisanen, Y., Vrtala, A., Wagner, P. E., Walther, H., Weingartner, E., Wex, H., Winkler, P. M., Carslaw, K. S., Worsnop, D. R., Baltensperger, U., and Kulmala, M.: Role of sulphuric acid, ammonia and galactic cosmic rays in atmospheric aerosol nucleation, Nature, 476, 429–433, https://doi.org/10.1038/nature10343, 2011.
Kürten, A.: New particle formation from sulfuric acid and ammonia: nucleation and growth model based on thermodynamics derived from CLOUD measurements for a wide range of conditions, Atmos. Chem. Phys., 19, 5033–5050, https://doi.org/10.5194/acp-19-5033-2019, 2019.
Lamothe, A., Savarino, J., Ginot, P., Soussaintjean, L., Gautier, E., Akers, P. D., Caillon, N., and Erbland, J.: NH4_method_in_low_concentrated_ environment-size_correction_and_calibration_scripts, Zenodo [code], https://doi.org/10.5281/zenodo.7728983, 2023.
Legrand, M. and Mayewski, P.: Glaciochemistry of polar ice cores: A review, Rev. Geophys., 35, 219–243, https://doi.org/10.1029/96RG03527, 1997.
Legrand, M., McConnell, J. R., Preunkert, S., Chellman, N. J., and Arienzo, M. M.: Causes of enhanced bromine levels in Alpine ice cores during the 20th century: Implications for bromine in the free European troposphere, J. Geophys. Res.-Atmos., 126, e2020JD034246, https://doi.org/10.1029/2020JD034246, 2021.
Lehmann, M. F., Bernasconi, S. M., and McKenzie, J. A.: A Method for the Extraction of Ammonium from Freshwaters for Nitrogen Isotope Analysis, Anal. Chem., 73, 4717–4721, https://doi.org/10.1021/ac010212u, 2001.
Lehtipalo, K., Rondo, L., Kontkanen, J., Schobesberger, S., Jokinen, T., Sarnela, N., Kürten, A., Ehrhart, S., Franchin, A., Nieminen, T., Riccobono, F., Sipilä, M., Yli-Juuti, T., Duplissy, J., Adamov, A., Ahlm, L., Almeida, J., Amorim, A., Bianchi, F., Breitenlechner, M., Dommen, J., Downard, A. J., Dunne, E. M., Flagan, R. C., Guida, R., Hakala, J., Hansel, A., Jud, W., Kangasluoma, J., Kerminen, V.-M., Keskinen, H., Kim, J., Kirkby, J., Kupc, A., Kupiainen-Määttä, O., Laaksonen, A., Lawler, M. J., Leiminger, M., Mathot, S., Olenius, T., Ortega, I. K., Onnela, A., Petäjä, T., Praplan, A., Rissanen, M. P., Ruuskanen, T., Santos, F. D., Schallhart, S., Schnitzhofer, R., Simon, M., Smith, J. N., Tröstl, J., Tsagkogeorgas, G., Tomé, A., Vaattovaara, P., Vehkamäki, H., Vrtala, A. E., Wagner, P. E., Williamson, C., Wimmer, D., Winkler, P. M., Virtanen, A., Donahue, N. M., Carslaw, K. S., Baltensperger, U., Riipinen, I., Curtius, J., Worsnop, D. R., and Kulmala, M.: The effect of acid–base clustering and ions on the growth of atmospheric nano-particles, Nat. Commun., 7, 11594, https://doi.org/10.1038/ncomms11594, 2016.
Lelieveld, J. and Pöschl, U.: Chemists can help to solve the air-pollution health crisis, Nature, 551, 291–293, https://doi.org/10.1038/d41586-017-05906-9, 2017.
Li, L., Lollar, B. S., Li, H., Wortmann, U. G., and Lacrampe-Couloume, G.: Ammonium stability and nitrogen isotope fractionations for NH4+–NH3(aq)–NH3(gas) systems at 20–70 ∘C and pH of 2–13: Applications to habitability and nitrogen cycling in low-temperature hydrothermal systems, Geochim. Cosmochim. Acta, 84, 280–296, https://doi.org/10.1016/j.gca.2012.01.040, 2012.
Liu, D., Fang, Y., Tu, Y., and Pan, Y.: Chemical method for nitrogen isotopic analysis of ammonium at natural abundance, Anal. Chem., 86, 3787–3792, 2014.
Mariappan, S., Exner, M. E., Martin, G. E., and Spalding, R. F.: Variability of Anaerobic Animal Waste Lagoon delta15N Source Signatures, Environ. Forensics, 10, 18–25, https://doi.org/10.1080/15275920802502075, 2009.
Maupetit, F. and Delmas, R. J.: Snow chemistry of high altitude glaciers in the French Alps, Tellus B, 46, 304–324, https://doi.org/10.3402/tellusb.v46i4.15806, 1994.
Maupetit, F., Wagenbach, D., Weddeling, P., and Delmas, R. J.: Seasonal fluxes of major ions to a high altitude cold alpine glacier, Atmos. Environ., 29, 1–9, https://doi.org/10.1016/1352-2310(94)00222-7, 1995.
McIlvin, M. R. and Altabet, M. A.: Chemical Conversion of Nitrate and Nitrite to Nitrous Oxide for Nitrogen and Oxygen Isotopic Analysis in Freshwater and Seawater, Anal. Chem., 77, 5589–5595, https://doi.org/10.1021/ac050528s, 2005.
O'Deen, W. A. and Porter, L. K.: Devarda's alloy reduction of nitrate and tube diffusion of the reduced nitrogen for indophenol ammonium and nitrogen-15 determinations, Anal. Chem., 52, 1164–1166, https://doi.org/10.1021/ac50057a044, 1980.
PANDA: https://panda.osug.fr/, last access: 31 August 2023.
Perrino, C., Marconi, E., Tofful, L., Farao, C., Materazzi, S., and Canepari, S.: Thermal stability of inorganic and organic compounds in atmospheric particulate matter, Atmos. Environ., 54, 36–43, https://doi.org/10.1016/j.atmosenv.2012.02.078, 2012.
Preston, T., Bury, S., Présing, M., Moncoiffe, G., and Slater, C.: Isotope Dilution Analysis of Combined Nitrogen in Natural Waters: I. Ammonium, Rapid Commun. Mass Sp., 10, 958–964, 1996.
Preunkert, S., Wagenbach, D., Legrand, M., and Vincent, C.: Col du Dôme (Mt Blanc Massif, French Alps) suitability for ice-core studies in relation with past atmospheric chemistry over Europe, Tellus B, 52, 993–1012, https://doi.org/10.3402/tellusb.v52i3.17081, 2000.
Preunkert, S., Legrand, M., and Wagenbach, D.: Sulfate trends in a Col du Dôme (French Alps) ice core: A record of anthropogenic sulfate levels in the European midtroposphere over the twentieth century, J. Geophys. Res.-Atmos., 106, 31991–32004, https://doi.org/10.1029/2001JD000792, 2001.
Preunkert, S., Wagenbach, D., and Legrand, M.: A seasonally resolved alpine ice core record of nitrate: Comparison with anthropogenic inventories and estimation of preindustrial emissions of NO in Europe, J. Geophys. Res.-Atmos., 108, 4681, https://doi.org/10.1029/2003JD003475, 2003.
Preunkert, S., McConnell, J. R., Hoffmann, H., Legrand, M., Wilson, A. I., Eckhardt, S., Stohl, A., Chellman, N. J., Arienzo, M. M., and Friedrich, R.: Lead and Antimony in Basal Ice From Col du Dome (French Alps) Dated With Radiocarbon: A Record of Pollution During Antiquity, Geophys. Res. Lett., 46, 4953–4961, https://doi.org/10.1029/2019GL082641, 2019.
Reche, C., Viana, M., Karanasiou, A., Cusack, M., Alastuey, A., Artiñano, B., Revuelta, M. A., López-Mahía, P., Blanco-Heras, G., Rodríguez, S., Sánchez de la Campa, A. M., Fernández-Camacho, R., González-Castanedo, Y., Mantilla, E., Tang, Y. S., and Querol, X.: Urban NH3 levels and sources in six major Spanish cities, Chemosphere, 119, 769–777, https://doi.org/10.1016/j.chemosphere.2014.07.097, 2015.
Röthlisberger, R., Bigler, M., Hutterli, M., Sommer, S., Stauffer, B., Junghans, H. G., and Wagenbach, D.: Technique for Continuous High-Resolution Analysis of Trace Substances in Firn and Ice Cores, Environ. Sci. Technol., 34, 338–342, https://doi.org/10.1021/es9907055, 2000.
Rubino, M., D'Onofrio, A., Seki, O., and Bendle, J. A.: Ice-core records of biomass burning, The Anthropocene Review, 3, 140–162, https://doi.org/10.1177/2053019615605117, 2016.
Savard, M. M., Cole, A., Smirnoff, A., and Vet, R.: δ15N values of atmospheric N species simultaneously collected using sector-based samplers distant from sources – Isotopic inheritance and fractionation, Atmos. Environ., 162, 11–22, https://doi.org/10.1016/j.atmosenv.2017.05.010, 2017.
Sigg, A., Fuhrer, K., Anklin, M., Staffelbach, T., and Zurmuehle, D.: A continuous analysis technique for trace species in ice cores, Environ. Sci. Technol., 28, 204–209, https://doi.org/10.1021/es00051a004, 1994.
Silva, S. R., Kendall, C., Wilkison, D. H., Ziegler, A. C., Chang, C. C. Y., and Avanzino, R. J.: A new method for collection of nitrate from fresh water and the analysis of nitrogen and oxygen isotope ratios, J. Hydrol., 228, 22–36, https://doi.org/10.1016/S0022-1694(99)00205-X, 2000.
Sprinson, D. B. and Rittenberg, D. J.: The rate of utilization of ammonia for protein synthesis, Biol. Chem., 180, 707–714, 1949.
Stolzenburg, D., Simon, M., Ranjithkumar, A., Kürten, A., Lehtipalo, K., Gordon, H., Ehrhart, S., Finkenzeller, H., Pichelstorfer, L., Nieminen, T., He, X.-C., Brilke, S., Xiao, M., Amorim, A., Baalbaki, R., Baccarini, A., Beck, L., Bräkling, S., Caudillo Murillo, L., Chen, D., Chu, B., Dada, L., Dias, A., Dommen, J., Duplissy, J., El Haddad, I., Fischer, L., Gonzalez Carracedo, L., Heinritzi, M., Kim, C., Koenig, T. K., Kong, W., Lamkaddam, H., Lee, C. P., Leiminger, M., Li, Z., Makhmutov, V., Manninen, H. E., Marie, G., Marten, R., Müller, T., Nie, W., Partoll, E., Petäjä, T., Pfeifer, J., Philippov, M., Rissanen, M. P., Rörup, B., Schobesberger, S., Schuchmann, S., Shen, J., Sipilä, M., Steiner, G., Stozhkov, Y., Tauber, C., Tham, Y. J., Tomé, A., Vazquez-Pufleau, M., Wagner, A. C., Wang, M., Wang, Y., Weber, S. K., Wimmer, D., Wlasits, P. J., Wu, Y., Ye, Q., Zauner-Wieczorek, M., Baltensperger, U., Carslaw, K. S., Curtius, J., Donahue, N. M., Flagan, R. C., Hansel, A., Kulmala, M., Lelieveld, J., Volkamer, R., Kirkby, J., and Winkler, P. M.: Enhanced growth rate of atmospheric particles from sulfuric acid, Atmos. Chem. Phys., 20, 7359–7372, https://doi.org/10.5194/acp-20-7359-2020, 2020.
Sutton, M. A., Reis, S., Riddick, S. N., Dragosits, U., Nemitz, E., Theobald, M. R., Tang, Y. S., Braban, C. F., Vieno, M., Dore, A. J., Mitchell, R. F., Wanless, S., Daunt, F., Fowler, D., Blackall, T. D., Milford, C., Flechard, C. R., Loubet, B., Massad, R., Cellier, P., Personne, E., Coheur, P. F., Clarisse, L., Van Damme, M., Ngadi, Y., Clerbaux, C., Skjøth, C. A., Geels, C., Hertel, O., Wichink Kruit, R. J., Pinder, R. W., Bash, J. O., Walker, J. T., Simpson, D., Horváth, L., Misselbrook, T. H., Bleeker, A., Dentener, F., and de Vries, W.: Towards a climate-dependent paradigm of ammonia emission and deposition, Philos. T. R. Soc. B, 368, 20130166, https://doi.org/10.1098/rstb.2013.0166, 2013.
Szopa, S., Naik, V., Adhikary, B., Artaxo, P., Berntsen, T., Collins, W. D., Fuzzi, S., Gallardo, L., Kiendler-Scharr, A., Klimont, Z., Liao, H., Unger, N., and Zanis, P.: Short-Lived Climate Forcers, edited by: Masson-Delmotte, V., Zhai, P., Pirani, A., Connors, S. L., Péan, C., Berger, S., Caud, N., Chen, Y., Goldfarb, L., Gomis, M. I., Huang, M., Leitzell, K., Lonnoy, E., Matthews, J. B. R., Maycock, T. K., Waterfield, T., Yelekçi, O., Yu, R., and Zhou, B., Climate Change 2021: The Physical Science Basis. Contribution of Working Group I to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change, 817–922, https://doi.org/10.1017/9781009157896.008, 2021.
Thomas, E. R., Allen, C. S., Etourneau, J., King, A. C. F., Severi, M., Winton, V. H. L., Mueller, J., Crosta, X., and Peck, V. L.: Antarctic Sea Ice Proxies from Marine and Ice Core Archives Suitable for Reconstructing Sea Ice over the Past 2000 Years, Geosciences, 9, 506, https://doi.org/10.3390/geosciences9120506, 2019.
Van Damme, M., Clarisse, L., Whitburn, S., Hadji-Lazaro, J., Hurtmans, D., Clerbaux, C., and Coheur, P.-F.: Industrial and agricultural ammonia point sources exposed, Nature, 564, 99–103, https://doi.org/10.1038/s41586-018-0747-1, 2018.
Velthof, G., Barot, S., Bloem, J., Butterbach-Bahl, K., de Vries, W., Kros, J., Lavelle, P., Olesen, J. E., and Oenema, O.: Nitrogen as a threat to European soil quality, in: The European Nitrogen Assessment: Sources, Effects and Policy Perspectives, edited by: Sutton, M. A., Howard, C. M., Erisman, J. W., Billen, G., Bleeker, A., Grennfelt, P., van Grinsven, H., and Grizzetti, B., Cambridge University Press, Cambridge, 495–510, https://doi.org/10.1017/CBO9780511976988.024, 2011.
Walters, W. W., Chai, J., and Hastings, M. G.: Theoretical Phase Resolved Ammonia–Ammonium Nitrogen Equilibrium Isotope Exchange Fractionations: Applications for Tracking Atmospheric Ammonia Gas-to-Particle Conversion, ACS Earth and Space Chemistry, 3, 79–89, https://doi.org/10.1021/acsearthspacechem.8b00140, 2019.
Walters, W. W., Song, L., Chai, J., Fang, Y., Colombi, N., and Hastings, M. G.: Characterizing the spatiotemporal nitrogen stable isotopic composition of ammonia in vehicle plumes, Atmos. Chem. Phys., 20, 11551–11567, https://doi.org/10.5194/acp-20-11551-2020, 2020.
Wang, M., Xiao, M., Bertozzi, B., Marie, G., Rörup, B., Schulze, B., Bardakov, R., He, X.-C., Shen, J., Scholz, W., Marten, R., Dada, L., Baalbaki, R., Lopez, B., Lamkaddam, H., Manninen, H. E., Amorim, A., Ataei, F., Bogert, P., Brasseur, Z., Caudillo, L., De Menezes, L.-P., Duplissy, J., Ekman, A. M. L., Finkenzeller, H., Carracedo, L. G., Granzin, M., Guida, R., Heinritzi, M., Hofbauer, V., Höhler, K., Korhonen, K., Krechmer, J. E., Kürten, A., Lehtipalo, K., Mahfouz, N. G. A., Makhmutov, V., Massabò, D., Mathot, S., Mauldin, R. L., Mentler, B., Müller, T., Onnela, A., Petäjä, T., Philippov, M., Piedehierro, A. A., Pozzer, A., Ranjithkumar, A., Schervish, M., Schobesberger, S., Simon, M., Stozhkov, Y., Tomé, A., Umo, N. S., Vogel, F., Wagner, R., Wang, D. S., Weber, S. K., Welti, A., Wu, Y., Zauner-Wieczorek, M., Sipilä, M., Winkler, P. M., Hansel, A., Baltensperger, U., Kulmala, M., Flagan, R. C., Curtius, J., Riipinen, I., Gordon, H., Lelieveld, J., El-Haddad, I., Volkamer, R., Worsnop, D. R., Christoudias, T., Kirkby, J., Möhler, O., and Donahue, N. M.: Synergistic HNO3–H2SO4–NH3 upper tropospheric particle formation, Nature, 605, 483–489, https://doi.org/10.1038/s41586-022-04605-4, 2022.
Xiang, Y.-K., Dao, X., Gao, M., Lin, Y.-C., Cao, F., Yang, X.-Y., and Zhang, Y.-L.: Nitrogen isotope characteristics and source apportionment of atmospheric ammonium in urban cities during a haze event in Northern China Plain, Atmos. Environ., 269, 118800, https://doi.org/10.1016/j.atmosenv.2021.118800, 2022.
Zhan, X., Adalibieke, W., Cui, X., Winiwarter, W., Reis, S., Zhang, L., Bai, Z., Wang, Q., Huang, W., and Zhou, F.: Improved Estimates of Ammonia Emissions from Global Croplands, Environ. Sci. Technol., 55, 1329–1338, https://doi.org/10.1021/acs.est.0c05149, 2021.
Zhang, L., Altabet, M. A., Wu, T., and Hadas, O.: Sensitive Measurement of NHN N (δ15NH) at Natural Abundance Levels in Fresh and Saltwaters, Anal. Chem., 79, 5297–5303, https://doi.org/10.1021/ac070106d, 2007.