the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 06 Nov 2024
| 06 Nov 2024
Alternate materials for the capture and quantification of gaseous oxidized mercury in the atmosphere
Livia Lown
Sarrah M. Dunham-Cheatham
Seth N. Lyman
Mae S. Gustin
Methodologies for identifying atmospheric oxidized mercury (HgII) compounds, including particulate-bound HgII () and gaseous oxidized mercury (), by mass spectrometry are currently under development. This method requires preconcentration of HgII for analysis due to high instrument detection limits relative to ambient HgII concentrations. The objective of this work was to identify and test materials for quantitative capture of HgII from the gas phase and to suggest potential surfaces onto which HgII can be collected, thermally desorbed, and characterized using mass spectrometry methods. From the literature, several compounds were identified as potential sorbent materials and tested in the laboratory for uptake of gaseous elemental mercury (Hg0) and (permeated from a HgBr2 salt source). Chitosan, α-Al2O3, and γ-Al2O3 demonstrated capture in ambient air laboratory tests, without sorbing Hg0 under the same conditions. When compared to cation exchange membranes (CEMs), chitosan captured a comparable quantity of , while ≤90 % of loaded was recovered from α-Al2O3 and γ-Al2O3. When deployed in the field, the capture efficiency of chitosan decreased compared to CEMs, indicating that environmental conditions impacted the sorption efficiency of this material. The poor recovery of HgII from the tested materials compared to CEMs in the field indicates that further identification and exploration of alternative sorbent materials are required to advance atmospheric mercury chemistry analysis by mass spectrometry methods.
- Article
(2442 KB) - Full-text XML
- BibTeX
- EndNote





Mercury (Hg) is a toxic global contaminant that is introduced to aquatic and terrestrial ecosystems primarily from the atmosphere (Driscoll et al., 2013). Oxidized forms (HgII), including gaseous oxidized mercury () and particulate-bound mercury (), are deposited from the atmosphere to ecosystems (Ariya et al., 2015) and may become available for transformation to and accumulation as methylmercury in food webs (Lyman et al., 2020a). A complete understanding of Hg behavior in the atmosphere is necessary to describe the fate of anthropogenic Hg pollution, assess health risks to humans and wildlife, and evaluate the effectiveness of the Minamata Convention. The mechanisms that govern the oxidation and reduction of Hg in the atmosphere are not well understood (Shah et al., 2021), and model results are uncertain because they do not consider the variety of HgII compounds that could be present (Gustin et al., 2023). For example, Shah et al. (2021) assumed that all HgII compounds volatilized from aerosols are HgCl2. This is an assumption that has not been validated.
Currently, Br− and Cl− radicals are considered to participate in elemental Hg (Hg0) oxidation. This is based on both theoretical work (Holmes et al., 2010; Horowitz et al., 2017; Song et al., 2024) and experimental observations of atmospheric Hg depletion events in the Arctic (Steffen et al., 2008), as well as observations of HgII formation at the marine boundary layer (Laurier et al., 2003). The identity of compounds in the atmosphere is currently unknown, but mass spectrometry (MS) methods capable of observing atmospheric speciation are in development (see Jones et al., 2016; Khalizov et al., 2020; Mao and Khalizov, 2021). These methods have been developed using HgBr2 and HgCl2 as model atmospheric compounds, given the role of halogen radicals in atmospheric Hg0 oxidation. However, due to differences in behavior, the use of a broad range of representative compounds is desirable in both MS method development and validation of preconcentration surfaces. MS methods will require preconcentration of ambient for detection, but the preconcentration surfaces currently available for deployment in the field have limitations that prevent their use in MS methods. Commonly used materials include potassium chloride (KCl)-coated denuders in the Tekran 2537/1130/1135 speciation system (Landis et al., 2002) and membranes deployed in the Reactive Mercury Active System (RMAS) (Luippold et al., 2020b). These membranes include cation exchange membranes (CEMs) used for quantitative HgII measurement, polytetrafluoroethylene (PTFE) membranes used to quantify (described in Luippold et al., 2020b, as PBM), and nylon membranes used for estimating Hg chemistry.
KCl denuders do not accurately measure HgII in ambient air due to ozone, humidity, and perhaps other interferences (Lyman and Jaffe, 2012; McClure et al., 2014; Huang and Gustin, 2015). PTFE membranes exposed to field conditions have also recently been found to sorb , indicating that this membrane type cannot fully separate and measurements as intended and provides measurements that are biased high (Allen et al., 2024). Although CEMs outperform KCl denuders for quantitative capture (Huang et al., 2013) and are quantitative sorbants under laboratory conditions (Miller et al., 2019; Dunham-Cheatham et al., 2020), recent work suggests that CEMs may not be fully quantitative under field conditions. For instance, Dunham-Cheatham et al. (2023) observed lower HgII concentrations collected on CEMs (discussed as RM by Dunham-Cheatham et al. (2023), which includes and as defined here) in the field relative to co-located dual-channel system (DCS) measurements. However, it is possible that the discrepancy between CEM and DCS measurements observed by Dunham-Cheatham et al. (2023) was due to the 2 L min−1 flow rate used to collect HgII on CEMs. Use of 2 L min−1 flow rates has recently been found to decrease Hg capture efficiency relative to those sampled at a 1 L min−1 flow rate (Allen et al., 2024). Increased breakthrough has been detected on downstream CEMs during field campaigns compared to CEMs exposed to for short periods in the laboratory (Allen et al., 2024, and this work), and HgII loss from CEM during long campaign periods has yet to be quantified. CEMs are not appropriate for HgII sample introduction into MS systems because when heated they generate compounds that interfere with Hg quantification by cold-vapor atomic absorption spectroscopy (Gustin et al., 2019), and research done using high HgII concentrations has suggested that exchange reactions can occur with Hg compounds on the CEM surface (Mao and Khalizov, 2021).
Thermal desorption followed by peak deconvolution of HgII compounds from nylon membranes deployed in the RMAS is currently the only method available for estimating atmospheric HgII chemistry (Huang et al., 2013; Luippold et al., 2020a; Gustin et al., 2023). This method compares thermal desorption profiles of unknown HgII compounds to reference profiles developed from HgII salts permeated onto nylon membranes to identify potential compound constituents (e.g., −O, , −N, −S, and –organic Hg compounds). The validity of thermal desorption interpretations depends on how well the desorption behavior of Hg salts represents unknown atmospheric HgII compounds. Exchange reactions involving HgBr2 and HgCl2 on CEMs and nylon membranes have also been observed at above-ambient concentrations (Mao and Khalizov, 2021), suggesting that HgII compounds desorbed from nylon membranes could be different from atmospheric HgII compounds initially captured. This may be true of any new sorptive surface and should be considered during the validation of new materials.
A DCS circumvents the need for HgII preconcentration by converting HgII to Hg0 using a thermolyzer and measuring total gaseous Hg. The HgII concentration can then be calculated by subtracting the Hg0-only fraction of atmospheric air, obtained by scrubbing the ambient sample of HgII with a CEM, from the total gaseous Hg measurement. DCSs have been successfully calibrated for measurement in the field but do not provide HgII chemistry data (Lyman et al., 2020b).
MS methods that can identify and quantify atmospheric HgII compounds could be an essential step towards describing Hg chemistry in the atmosphere, but unambiguous determination of the identity of HgII compounds via MS has not yet been achieved (see Deeds et al., 2015; Jones et al., 2016). Given the limitations of current HgII sorbents, new surfaces that can quantitatively capture HgII without compound-altering chemistry are needed to preconcentrate ambient samples to levels above MS detection limits. An ideal material for analysis of HgII compounds by MS will be inert to Hg0, capture and retain all HgII compounds with high efficiency, not promote compound-altering reactions occurring on the material surface, and release atmospherically representative Hg compounds by thermal desorption for downstream analysis.
Characteristics of promising materials include ion exchange with a porous or layered crystalline material (Manos and Kanatzidis, 2016), a high surface area, and a high melting temperature that would facilitate thermal sample recovery and analysis by MS. Materials functionalized with sulfur, such as thiol, thiosemicarbazide, sulfone, and sulfonamide groups, show promise due to their high affinity for HgII (Yu et al., 2016). Capture efficiency is increased in base materials by functionalization with active groups that interact with Hg through chemisorption, resulting in the formation of a covalent bond between the Hg atom and material (Ali et al., 2018). However, strong bonding between the material and HgII may cause the identity of the Hg compound to be lost upon collection. As a result of strong bonding, such a material may not be suitable for subsequent analysis by MS methods. Materials that capture Hg by physisorption processes (electrostatic interactions) may be desirable if Hg compounds do not undergo chemistry on the sorbent surface, as has been observed for CEMs and nylon membranes at high concentrations (Mao and Khalizov, 2021).
The objective of this work was to explore chitosan, α-Al2O3, γ-Al2O3, poly(1,4-phenylene sulfide), and perfluorosulfonic acid as candidate materials for preconcentration of atmospheric HgII that would be suitable for subsequent analysis by MS. A custom-built HgII permeation calibrator was used to load candidate materials with a known quantity of HgII for comparison to CEM. HgII capture by these materials was also compared under field conditions. It was hypothesized that chitosan, α-Al2O3, and γ-Al2O3 would quantitatively sorb HgII under ambient conditions. Chitosan sorbs HgII through both chelation and electrostatic interactions in liquid matrices via amino and hydroxyl groups (Vieira and Beppu, 2006). α-Al2O3 and γ-Al2O3, which are alumina polymorphs, were materials of interest because they are polar compounds but not acidic and may thus attract compounds without capturing Hg0 (Zheng et al., 2019). Alumina polymorphs are stable at high temperatures (Baronskiy et al., 2022), making them ideal for reuse following thermal desorption. α-Al2O3 and γ-Al2O3 differ in thermal stability and specific surface area and may thus perform differently in terms of capture efficiency and reusability.
2.1 Materials
CEMs, polyethersulfone membranes that are proprietarily treated, were purchased from Pall Corporation (0.8 µm pore size; Mustang-S, P/N MSTGS3R) as sheets and cut to 47 mm diameter disks. PTFE membranes were purchased from Sartorius Stedim Biotech (0.2 µm pore size; P/N 1180747). Chitosan (85 % deacetylated; P/N J64143.18) and α-Al2O3 (<1 µm, powder; P/N 0452572.22) were purchased from Thermo Scientific. γ-Al2O3 was obtained from Alpha Aesar as a 40 µm powder (P/N 043266.22). α-Al2O3 used in this study had a specific surface area of 2–4 m2 g−1 and thermal stability of 1200 °C, while γ-Al2O3 had a specific surface area of 100 m2 g−1 and thermal stability up to 500 °C. Perfluorosulfonic acid membrane sheets (PFSA-M) were purchased from Sigma Aldrich (trade name Aquivion E98-05, PFSA equivalent weight 980 g mol−1 SO3H, 50 µm film thickness; P/N 802697). Poly(1,4-phenylene sulfide) was also purchased from Sigma Aldrich (P/N 182354). Activated carbon was acquired from Aldrich Chemical Company (P/N 292591) as 4-14 mesh granules and crushed for use in experiments as a powder.
A set of glass tubes used to test PFSA-M were sent to SilcoTek for inert coating with deactivated silica (SilcoNert® 2000) to test if this improved HgII recovery. Based on the results of this test (discussed below), glass tubes without inert coating were used in laboratory and field tests. PTFE frits that were 10–30 µm in pore size, 2.5 mm thickness, and cut to fit 6.35 mm diameter (4 mm inner diameter) tubing were acquired from Savillex (P/N: 730-0065), as were perfluoroalkoxyalkane filter packs used to house membranes (47 mm diameter, P/N: 403-21-47-22-21-2). Optima™ HCl (A466-500), KBr (P205-500), NH2OH⋅HCl (H330-500), and SnCl2 (T142-500) were obtained from Fisher Scientific. KBrO3 was purchased from Acros Organics (268392500). All reagents were American Chemical Society-grade or higher and made with 18.2 MΩ cm type 1 water. Ultrahigh-purity argon (Linde Gas and Equipment Inc.) was used in sorption tests described in the Appendix. Gas-tight syringes were purchased from Hamilton Company (Reno, Nevada, USA). Flow rates in laboratory tests and field campaigns were controlled with critical flow orifices obtained from Teledyne API (P/N 941100).
Candidate materials that could potentially sorb without capturing Hg0 were identified from the literature and preliminary work was performed to select promising materials for further experimentation. Of the five new materials tested, only three were considered for further investigation. Preliminary work indicated poor recovery of Hg2+ from two liquid-spiked poly(1,4-phenylene sulfide) samples when analyzed using a modified EPA Method 1631 digestion (United States Environmental Protection Agency, 2002). This suggested a matrix interference, and this material was not tested further. Investigation of PFSA-M was also discontinued after poor performance in laboratory tests (discussed below). Preliminary work with poly(1,4-phenylene sulfide) is detailed in the Appendix, with additional data for chitosan, α-Al2O3, and γ-Al2O3 (Appendices A and B). An alternative digestion method (United States Environmental Protection Agency, 2001) for recovering HgII from CEMs, chitosan, α-Al2O3, and γ-Al2O3 was attempted, and these data are also available in the Appendix (Appendix C).
2.2 Laboratory loading of Hg0 and onto candidate materials
Chitosan, α-Al2O3, γ-Al2O3, and PFSA-M were tested for quantitative Hg0 and sorption in the laboratory. To test for sorption of Hg0 to candidate materials, laboratory air was drawn at 1 L min−1 through traps containing test material. A syringe was used to inject 1.2 ng Hg0 from a bell jar into the trap (n=9). As a control, activated carbon was loaded by the same method. Traps containing 30 ± 5 mg of chitosan, α-Al2O3, γ-Al2O3, or shredded PFSA-M were constructed with glass tubing containing a single PTFE frit. The glass tube (6.35 mm outer diameter) was slightly pinched at one end to prevent the frit and test material from being pulled through the trap during loading. Replicates (n=9) of each material were exposed to laboratory air for 3 min, at the end of which the Hg0 injection was made. Additional traps (n=9 per material type) that were exposed to laboratory air, but not loaded with Hg0, were used to blank-correct loaded samples. The mass of loaded Hg0 was calculated based on the Dumarey equation (Dumarey et al., 2010). Hg0 recovered from candidate materials was compared to both the calculated mass loaded and Hg0 recovered from the method control. The syringe (Hg0) tip was placed as close as possible to the materials during loading to minimize loss to the atmosphere or glass tubing.
Hg0 was recovered by combustion (EPA Method 7473) using a direct Hg analyzer (Nippon Instruments Corporation, MA-3000) (United States Environmental Protection Agency, 2007). This instrument was calibrated (r2≥0.999) with a primary liquid Hg2+ standard (Inorganic Ventures, MSHG-1PPM). At the beginning of each analytical run, three aliquots of a secondary standard, National Institute of Standards and Technology Standard Reference Material 1547, were analyzed to demonstrate instrument performance. Two additional aliquots of 1547 were run every 10 samples or fewer to confirm ongoing instrument performance. A recovery within ±10 % of the certified value was considered acceptable for analysis. This instrument has a detection limit of 0.10 ng Hg0.
Sorption of to candidate materials was performed with the same procedure, using a custom-built HgBr2 calibrator (Allen et al., 2024; Gačnik et al., 2024) that releases a constant stream of from a salt-based permeation source. The permeation rate is tightly controlled by maintaining constant temperature, pressure, and He flow over the permeation source. The permeation rate of the calibrator can be determined either gravimetrically or by measurement with CEMs (Lyman et al., 2016; Gačnik et al., 2024). These methods have been demonstrated to be equivalent by Elgiar et al. (2024). The CEM method was used to calculate the permeation rate of the calibrator in this study. Briefly, permeation rates are calculated by exposing CEMs to calibrator output, then digesting the CEMs by EPA Method 1631. The recovered mass of Hg is then divided by the exposure time to provide a pg s−1 permeation rate (blank-corrected). Although this calibrator can be used to calibrate measurements in other systems, it was used here to compare sorptive properties of candidate materials to CEMs by delivering consistent, measurable quantities of . Chitosan, α-Al2O3, γ-Al2O3, nylon (n=9 replicates each), and PFSA-M (n=6 replicates) were exposed to the calibrator output for the same duration to ensure equal loading of for each replicate. Triplicate blanks of each material were also exposed to laboratory air for an equivalent period of time. Initial experiments loading CEMs in this study measured a 1.76 ± 0.18 pg s−1 permeation rate (mean ± standard deviation, n=9). The permeation rate of this calibrator was reported as 2.2 ± 0.2 pg s−1 in experiments performed concurrently, discussed elsewhere (Gačnik et al., 2024). The difference in observed permeation rate between these two studies is of significance for the use of this system for calibrating HgII measurements and should be studied further before it is broadly employed by the research community. A possible explanation for the difference may be the positioning of the calibrator tip at a distance of 2 cm from the CEM during loading (Gačnik et al., 2024) versus at the filter pack inlet (5.5 cm in this work), as HgBr2 is more likely to come in contact with the filter pack when loaded at the inlet. Work by Allen et al. (2024) suggests that less than 5 % of atmospheric HgII is sorbed to the PTFE filter packs after field deployment.

Permeation, as measured by CEMs, dropped significantly to 0.42 ± 0.03 pg s−1 during experiment replication. It was suspected that the chamber containing the permeation tube overheated and shut off, cooling the HgBr2 salt. Returning the heated chamber to 50 °C restored the measured permeation rate to 1.77 ± 0.06 (data available in Table I1). Due to the change in permeation rate across replicate experiments, results are reported as a percent of recovered from CEMs rather than a percent of the expected recovery based on the perm rate that was calculated as the mass of HgII recovered from CEMs divided by exposure time (pg s−1). was quantified by following a modified version of EPA Method 1631. Briefly, BrCl solution (1.8 % KBr and 1.2 % KBrO3 in 32 %–35 % Optima™ HCl) was added to candidate materials and membranes in 1 % HCl at a ratio of 3 mL BrCl to 50 mL 1 % HCl and digested at room temperature overnight (see the SI of Dunham-Cheatham et al., 2023, for additional details). Samples were analyzed by cold-vapor atomic fluorescence spectroscopy (Tekran 2600-IVS). The instrument was calibrated (r2≥0.999) at the beginning and end of analysis. A check standard was analyzed every 10 samples and the instrument was recalibrated after a maximum of 30 samples were run. Data were considered acceptable if check standards were recovered within ±15 % of the true value, as per EPA Method 1631. Blanks for each tested material were collected and analyzed with samples during each experiment and were used to correct the analyzed value of samples. These blanks were exposed to laboratory air only (no HgBr2) during laboratory HgBr2 exposure tests or were not exposed to air during field campaigns. Mean recovery on CEM blanks (exposed to laboratory air or not) was 0.03 ± 0.01 ng per membrane (±1 standard deviation) across 28 replicates. Mean recovery of α-Al2O3, γ-Al2O3, and chitosan was 0.01 ± 0.02, 0.01 ± 0.01, and 0.02 ± 0.01 ng per target mass (30 mg), respectively (n=27 for each material).
2.3 Field comparison of candidate materials and CEM
Candidate materials were deployed for three 1-week sampling campaigns in the summer (late July through mid-September 2023) at the University of Nevada, Reno College of Agriculture, Biotechnology & Natural Resources Agricultural Experiment Station, Valley Road Greenhouse Complex (39.5375, −119.8047; 1370 m a.s.l.). This sampling location is within 100 m of Interstate 80 and is impacted by vehicle emissions and long-range transport of pollutants (Gustin et al., 2021; Luippold et al., 2020a). Environmental conditions varied between campaigns, with weekly mean temperatures falling from 25.0 ± 0.5 °C in the first campaign to 22.0 ± 3.4 and 20.8 ± 0.6 °C in the last two campaigns. Relative humidity was 20 % ± 4 %, 48 % ± 16 %, and 35 % ± 5 % for the first, second, and third campaigns, respectively. Temperature, relative humidity, solar radiation, and precipitation information was obtained from the Western Regional Climate Center (https://raws.dri.edu/, last access: 25 October 2024) measurement station located at the test site (39.53917, −119.806; 1370 m a.s.l.) and is available in the Appendix (Table D1).
Traps, constructed as described for Hg0 and sorption tests above, were deployed in inverted RMAS shields (Fig. E1). Three replicates of each material were deployed with a PTFE membrane upstream (to separate and ), and three replicate traps were deployed without an upstream PTFE membrane (providing a total HgII measurement). A CEM was deployed behind each candidate material to capture breakthrough . Filter packs containing two consecutive CEMs, both with and without an upstream PTFE membrane (n=3 for each configuration), were co-deployed with candidate materials. Critical flow orifices controlled the flow across all collection materials at 1 L min−1, and the flow rate through each trap or filter pack assembly was measured as standard flow at the beginning and end of each campaign using a volumetric airflow calibrator (BGI tetraCal). Measured masses of in each trap or filter pack were divided by the volume of air sampled, calculated as the average of flow rates measured at the beginning and end of deployment (L min−1) multiplied by the total sampling time in minutes, to calculate a concentration sampled in ambient air. All membranes and candidate materials were digested by the modified EPA Method 1631 procedure described above.
2.4 Statistical analysis
One-way ANOVA and Tukey's honestly significant difference tests (α≤0.05), comparing the recovery of HgII between material types, were performed using R (R Core Team, 2023, version 2023.06.2+561). Means, standard deviations, and t tests were calculated using Excel 2016.
3.1 Laboratory tests for Hg0 and sorption
In this work, chitosan, α-Al2O3, and γ-Al2O3 were tested for sorption of Hg0 and HgII in the laboratory and the field. Additionally, preliminary sorption tests with poly(1,4-phenylene sulfide) and PFSA-M were performed, but these materials were abandoned when a matrix interference was identified for poly(1,4-phenylene sulfide) and PFSA-M demonstrated poor recovery. A summary of the materials tested and outcomes is available in Table 1, and the details of preliminary work are available in Appendix A and B. No quantifiable Hg0 was recovered from blanks or Hg0-loaded chitosan, α-Al2O3, or γ-Al2O3, except for one Hg0-loaded γ-Al2O3 trap that had low recovery (0.11 ng). Hg0 recovery from activated carbon was 1.3 ± 0.4 ng (mean ± standard deviation), indicating reasonably good agreement with the expected recovery of 1.1 ng based on the Dumarey equation (Fig. F1).
Chitosan traps recovered 99 % ± 36 % of the loaded compared to the CEM, while α-Al2O3, γ-Al2O3, nylon, and PFSA-M recovered less (86 % ± 15 %, 69 % ± 21 %, 81 % ± 7 %, and 26 % ± 7 %, respectively) (Fig. 1). Due to the smaller quantities of loaded following the drop in permeation, a small variation in the mass of recovered resulted in a larger percent variation. Variation in mass recovered from both the candidate material and CEM also contributed to a larger percent variation. Given the low recovery of from PFSA-M, it was not tested further. As breakthrough was not quantified for this material, it is unclear if the low recovery is due to low capture efficiency of the material itself or if the geometry of the trap (shredded membrane packed into glass tubing) permitted greater breakthrough. If this membrane material could be made porous or functionalized onto a porous substrate, it may demonstrate increased capture of due to the presence of sulfonic acid functional groups. A comparison of recoveries was made between PFSA-M loaded with in glass tubing coated with deactivated fused silica and PFSA-M in uncoated glass tubing. Although it was hypothesized that the coating would reduce sorption to the glass tubing (Jones et al., 2016), recovery was not statistically different between PFSA-M samples loaded in deactivated fused-silica-coated or uncoated tubes (p>0.05; Fig. G1). recovered on CEMs downstream of other candidate materials provided a measurement of breakthrough, calculated as a percent of the sum of recovered from the candidate material plus the CEM (n=6 replicates for chitosan, α-Al2O3, γ-Al2O3, and nylon). An average of ≤5 % was recovered downstream of candidate materials, and no quantifiable was recovered on the second-in-line CEM behind a CEM.
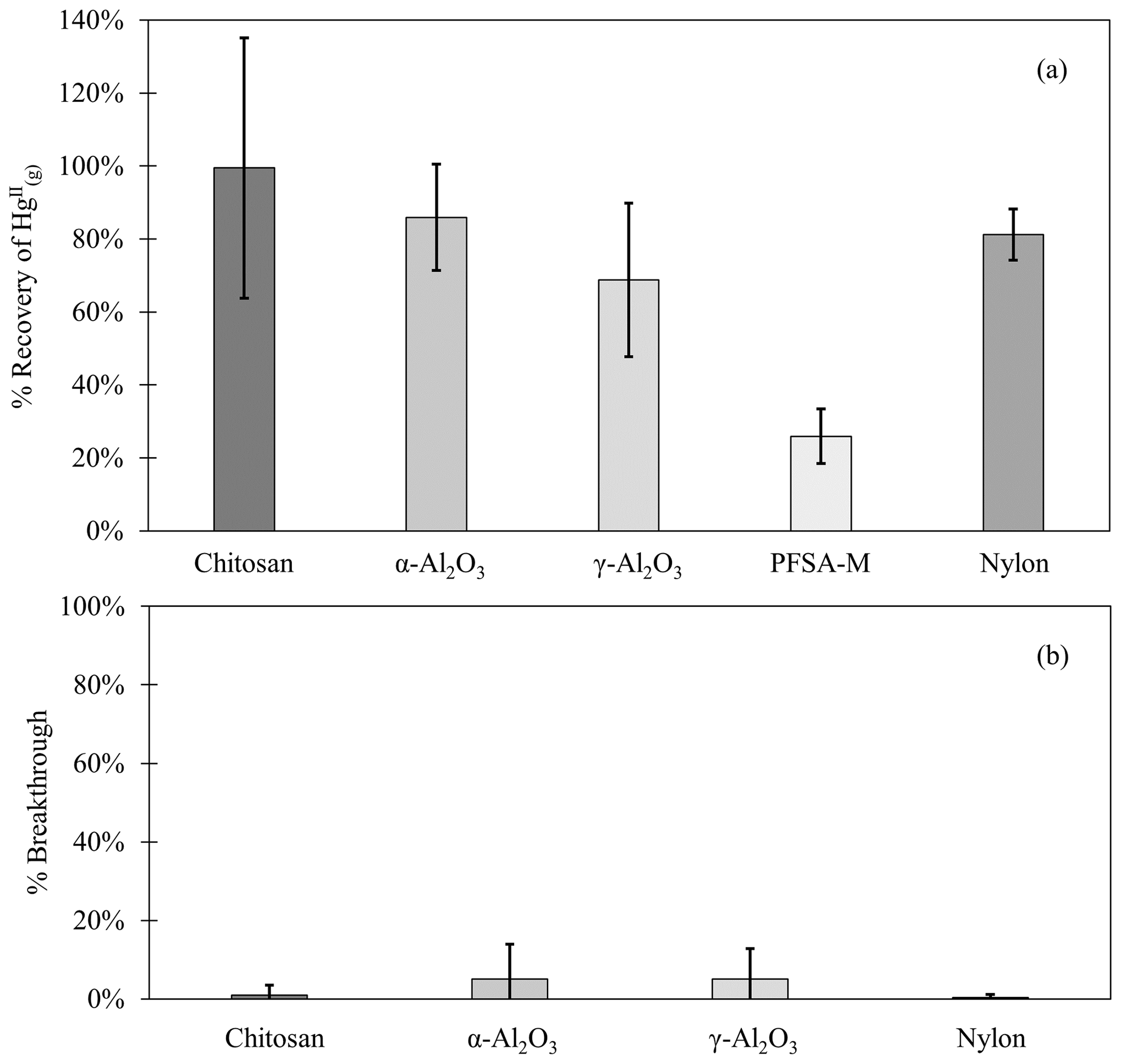
Figure 1(a) recovered from candidate materials (n=9 each for chitosan, α-Al2O3, γ-Al2O3, and nylon, and n=6 for PFSA-M) loaded with HgBr2 in laboratory air as a percentage of HgBr2 recovered on CEMs. (b) breakthrough from candidate materials (n=6 each) as a percent of HgBr2 collected on CEMs. Error bars represent ± 1 standard deviation from the mean.
The relatively low recovery of from α-Al2O3 and γ-Al2O3, as well as the minimal breakthrough, indicated that was either lost during loading, possibly to the glass tubing, or not recovered from the material matrix by BrCl digestion. Al2O3 has been previously used to capture and thermally reduce HgII to Hg0 with high efficiency in an inert atmosphere (Gačnik et al., 2022), suggesting that EPA Method 1631 digestion is not sufficient to recover HgII sample from this matrix. Chitosan, an organic compound, is more easily decomposed, and thus, sorbed Hg is made more available for analysis by acid digestion compared to oxide crystals like α-Al2O3 and γ-Al2O3. This may explain why a higher percent recovery was observed from chitosan compared to α-Al2O3 and γ-Al2O3 and highlights the need to consider alternative digestion methods, or possibly thermal desorption, and utilize matrix-matched certified reference materials when considering new surfaces for quantitative capture.
Digestions with HF and HNO3 have been used to recover metals from refractory silicates and oxides (Zimmermann et al., 2020), and EPA Method 3052 (United States Environmental Protection Agency, 1996) is an established method for Hg. It aims to completely decompose and dissolve the sample by microwave digestion with HF and HNO3 but also offers alternative matrix-specific reagent mixtures with HCl and H2O2. This method may not be suitable for some oxides, including Al2O3 and TiO2, among others, and target analytes (including Hg) can be sequestered by undecomposed sample, leading to low recovery. Re-adsorption of Hg by residual sample matrix during digestion is also noted for activated carbon matrices in the appendix to EPA Method 1631 and could explain low recovery from α-Al2O3 and γ-Al2O3 in this study. Microwave digestion of -loaded carbon and γ-Al2O3 with HBF4 was attempted as a safer alternative to digestion with HF (Zimmermann et al., 2020), but high background Hg in analytical-grade reagents made data inconclusive. Direct Hg analyzers conveniently overcome matrix interferences by combusting the sample, but atmospheric samples will need to be preconcentrated over 2-week campaigns to collect enough Hg to exceed analytical detection limits, limiting the utility of this method for analyzing HgII trends over short timescales.
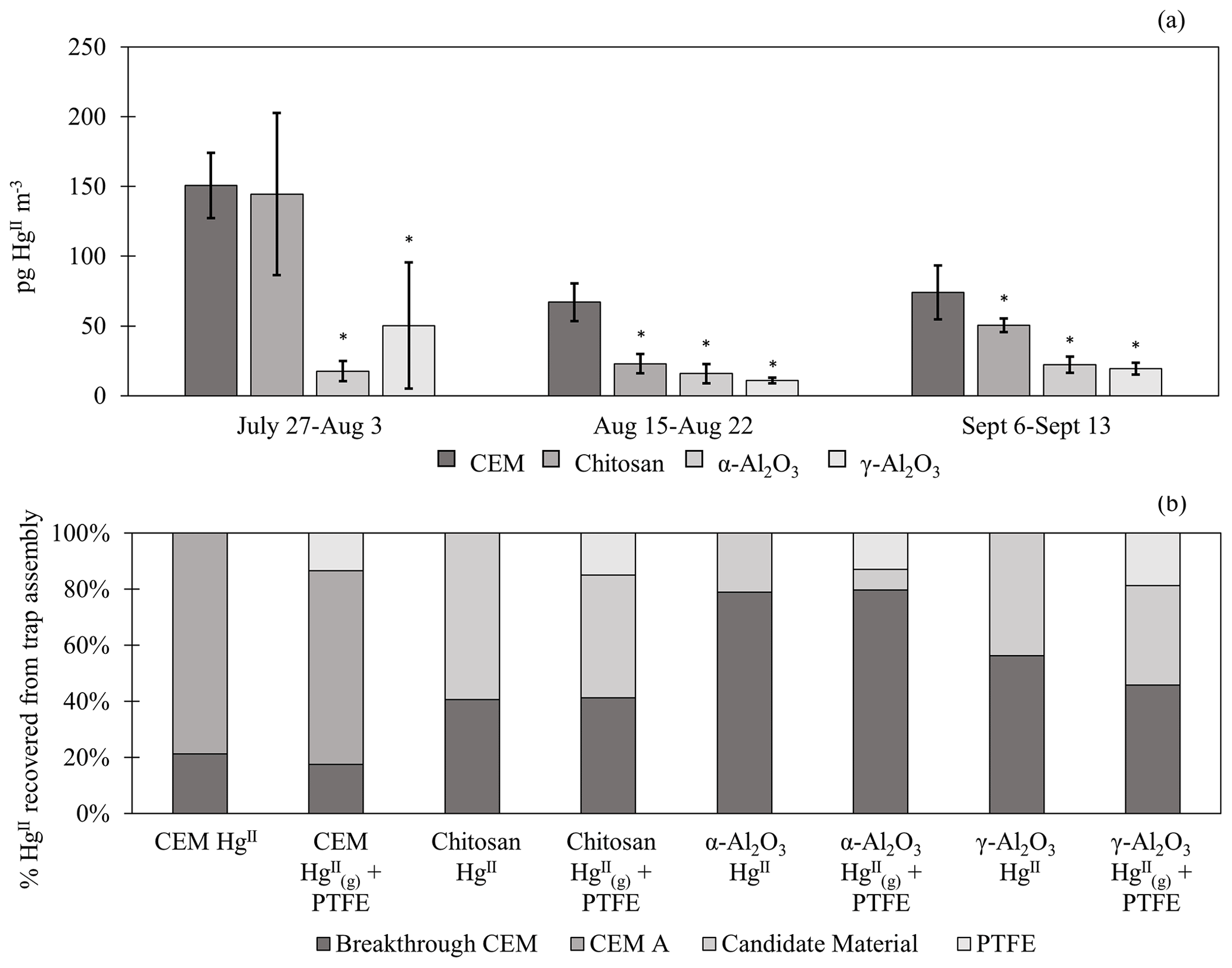
Figure 2(a) HgII recovery from PTFE + candidate materials deployed in the field for three 1-week campaigns. HgII recovery from traps with first-in-line PTFE membranes () was combined with traps without PTFE (HgII) for statistical analysis (n=6 per campaign). Data do not include recovered on breakthrough CEMs. Error bars are ± 1 standard deviation from the mean. Asterisks (∗) indicate a statistically different recovery of on candidate materials compared to CEMs (ANOVA, α≤0.05). (b) The percent of HgII recovered from each portion of the trap assembly, including from the PTFE, candidate material, first-in-line CEM (CEM A), and/or breakthrough CEM. HgII traps with no upstream PTFE are shown separately from + PTFE traps.
3.2 HgII recovery from candidate materials in the field
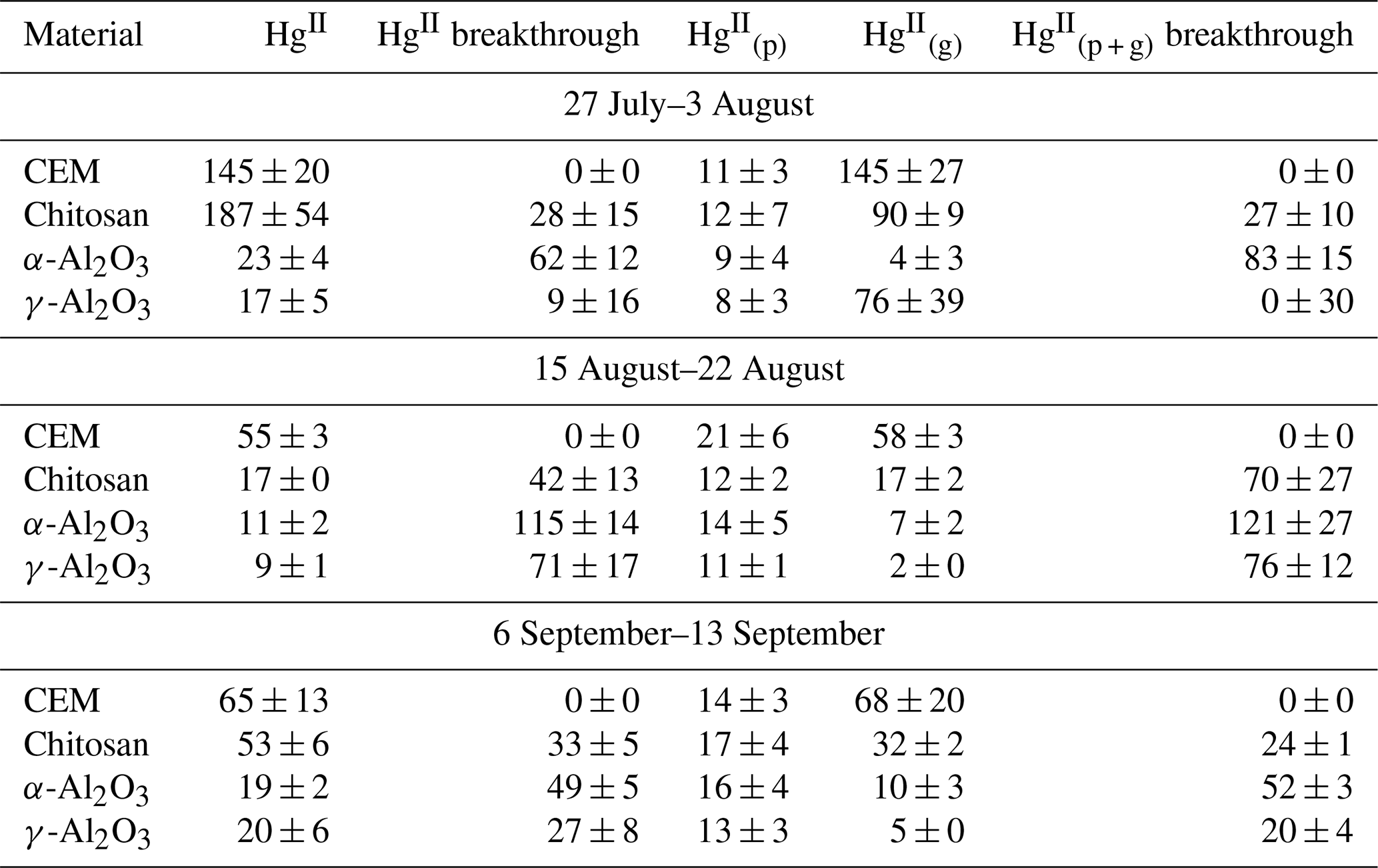
Candidate materials were deployed with or without a PTFE membrane upstream and with a CEM downstream. Of the total HgII recovered from PTFE + Al2O3 traps, 66 % was recovered from the PTFE portion of α-Al2O3 traps and 55 % of HgII was recovered from the PTFE on γ-Al2O3 traps, indicating that half or more of the HgII recovered was particulate-bound or sorbed to particles (see below). More was captured on chitosan behind PTFE compared to the equivalent α-Al2O3 and γ-Al2O3 traps (an average across all three campaigns of 7 ± 3 pg m−3 α-Al2O3 vs. 28 ± 43 pg m−3 γ-Al2O3 and 46 ± 36 pg m−3 chitosan), but not as much as compared to CEMs (91 ± 45 pg m−3). This agrees with the laboratory tests that show poor recovery from α-Al2O3 and γ-Al2O3 and relatively greater recovery from chitosan. Total HgII recovery from the entire trap assembly (PTFE membrane (if present) + candidate material or first-in-line CEM + breakthrough CEM) was not statistically different between chitosan and CEM traps during any campaign. Recovery was statistically lower for α-Al2O3 and γ-Al2O3 in the first and third campaigns and higher for α-Al2O3 during the second campaign (Fig. H1a). Field measurements included a downstream CEM that captured HgII not sorbed by candidate materials (i.e., “breakthrough”), if present. These data also suggest that either BrCl digestion was not sufficient to recover HgII from Al2O3 matrices or reduction was occurring on the material surface during the week-long sampling period and HgII was lost as Hg0.
Recent work by Allen et al. (2024) demonstrated that can be sorbed by particulates on PTFE filters, suggesting that measurements are biased high with additional sorbed. For this reason, HgII recovered on PTFE filters was added to recovered from downstream candidate materials to yield a total HgII measurement, and these data were combined with HgII measurements from candidate materials without upstream PTFE. The sum of + recovered from PTFE + CEM, respectively, has been well correlated with HgII measurements on CEMs in previous work (Gustin et al., 2019, 2023). CEMs (n=6, 3 of which included HgII recovery from PTFE + CEM) recovered the highest mass of HgII m−3 air sampled of all materials tested (Fig. 2a). More than 20 % of the HgII recovered from the entire trap (PTFE membrane, candidate material, and breakthrough CEMs) was recovered downstream of candidate materials (Fig. 2b), indicating that chitosan, α-Al2O3, and γ-Al2O3 did not quantitatively measure HgII under field conditions. There was a decrease in the HgII measured by all materials in the second and third campaigns that coincided with periods of rain and increased humidity, which is consistent with observations of HgII washout during rain events (Kaulfus et al., 2017). Of the candidate materials, chitosan performed the best during the first campaign, recovering a similar quantity of HgII as CEMs, but decreased in relative recovery during the second and third campaigns. Chitosan is highly hygroscopic (Szymańska and Winnicka, 2015), and the amino functional groups on chitosan are easily protonated at pH<6; thus, it is possible the increased humidity led to a decrease in sorption capacity due to electrostatic repulsion between protonated amino groups and (Vieira and Beppu, 2006).
CEMs outperformed chitosan, α-Al2O3, and γ-Al2O3 for measurement in the laboratory and the field, indicating they do not quantitatively capture HgII. Candidate materials did not collect Hg0. Low recoveries of from α-Al2O3 and γ-Al2O3 may be due to insufficient digestion methods, demonstrating a need to use matrix-specific methods with certified reference materials when testing alternative materials in the future. Promising materials should be tested for sorption of and Hg0 capture efficiency for a broad range of representative Hg compounds (Dunham-Cheatham et al., 2020); the potential for chemical transformation on the material surface; potential reactions between the Hg sample and other atmospheric constituents, including interferences with humidity (Huang and Gustin, 2015) and ozone (McClure et al., 2014); and performance under both laboratory and field conditions.
Cold-vapor atomic fluorescence spectroscopy was used to characterize the loss of Hg in an argon (Ar) carrier gas following injection of gaseous elemental mercury (Hg0) through a trap containing poly(1,4-phenylene sulfide) (PPS), chitosan, or a shredded perfluorosulfonic acid membrane (PFSA-M). The testing apparatus (Fig. A1) consisted of an Ar cylinder that provided carrier gas and pressure to the sample line, a PTFE sample line into which a trap containing a test material could be inserted with an upstream Hg0 injection port, a thermolyzer (>650 °C) to convert gaseous oxidized mercury () to Hg0, a gold cartridge to collect Hg0 for analysis, and a Tekran 2500 to measure Hg0. Materials were loaded with a gas-tight syringe by injecting Hg0 from a temperature-stabilized source through the injection port upstream of a trap containing a sorbent material. The quantity of Hg0 loaded was calculated based on the Dumarey equation (Dumarey et al., 2010). Sorbent traps containing 29.4 mg PPS, 29.4 mg chitosan, or a half of a 47 mm diameter PFSA-M (one replicate each) were constructed as described in the main text with 6.35 mm internal diameter uncoated glass tubing and quartz wool plugs.
To test for sorption of Hg0 to PPS, chitosan, and PFSA-M, the peak area of 0.4 ng Hg0 detected downstream of traps containing material was compared to a baseline peak area of 0.4 ng Hg0 injected through a glass trap without sorbent material. Peaks observed following an injection indicated no or partial sorption of Hg0 to the candidate material. A Student's t test (α=0.05) was used to assess if the peak area of Hg0 detected downstream of the candidate material (n=5 injections of 0.4 ng Hg0 each, relative standard deviation – RSD <10 % for PPS) was statistically different from the Hg0 peak area detected downstream of the empty trap (n=5 injections, RSD=7 %). The absence of a peak following an injection indicated complete sorption of Hg0 to the trap, while significantly lower peak areas indicated partial sorption, and peak areas equivalent to peaks detected downstream of an empty trap indicated no Hg0 sorption to the candidate material. PPS and chitosan did not sorb any Hg0. More Hg0 was recovered downstream of the PFSA-M compared to empty glass traps (RSD of injections <5 %, p<0.01). The reason for this was unclear and this test was repeated a second time with the same outcome (a total of 20 injections through each empty and PFSA-M trap over 2 d). It was concluded that PFSA-M did not sorb Hg0.
A trap containing 14 mg α-Al2O3 was tested using a similar procedure with a few minor differences. The glass tubing was pinched at one end and contained PTFE frits, rather than quartz wool plugs, to prevent material movement, and the Tekran 2500 was calibrated with Hg0 using the bell jar method (r2>0.998) to quantify sorption by α-Al2O3. The quantity of Hg0 injected was also increased from 0.4 to 1.1 ng Hg, so analysis was performed on Hg mass in the middle of the calibration range. α-Al2O3 sorbed 40 % of the injected Hg0, demonstrating that significant sorption of Hg0 is possible in an Ar atmosphere.
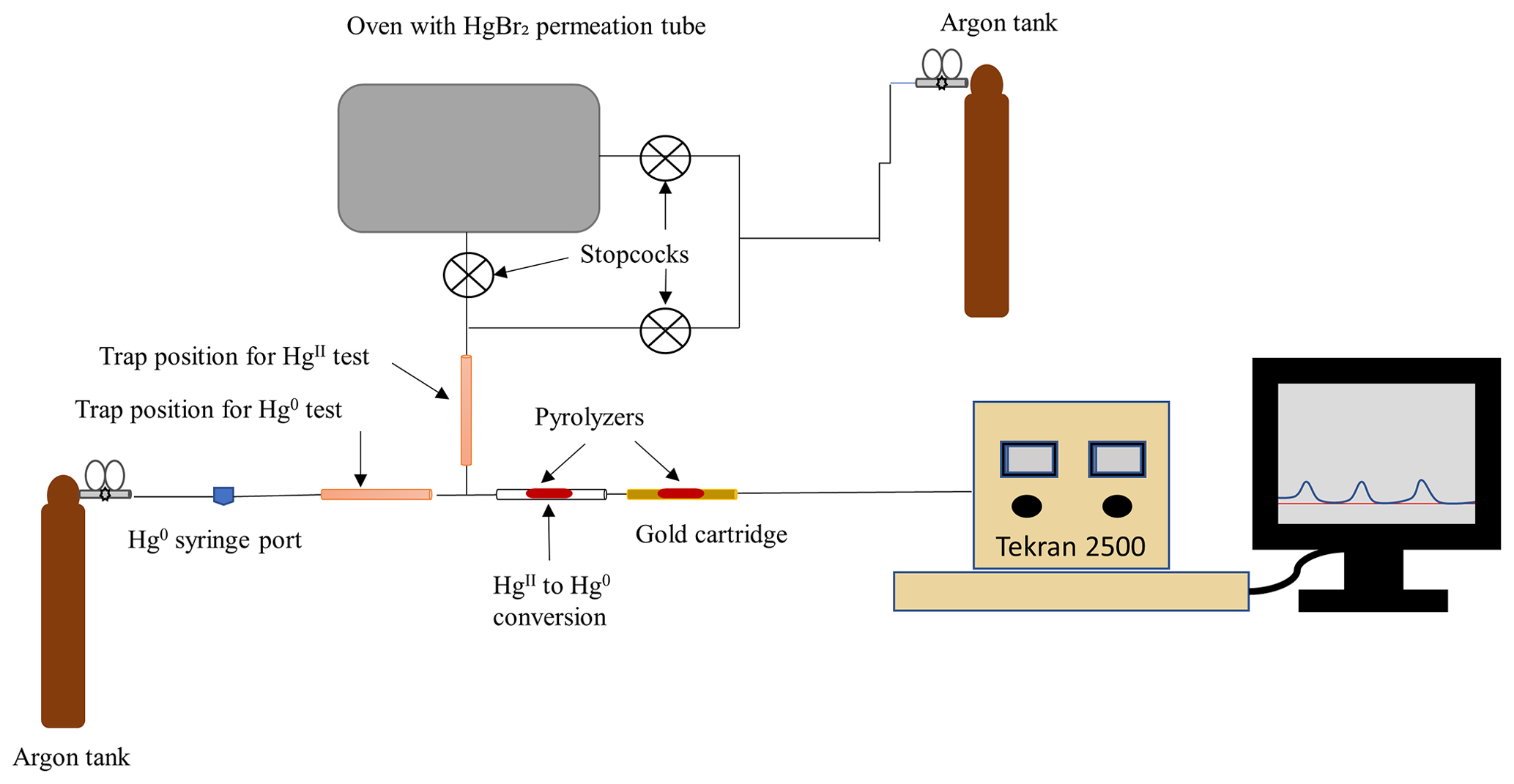
Figure A1Experimental setup for Hg0 and sorption to candidate materials in an argon atmosphere. During Hg0 sorption testing, the line containing the permeation tube was disconnected. Lines leading into the permeation source and to the Tekran 2500 were heated to a nominal 83 °C with heat tape and insulated with aluminum foil to encourage movement through the system and reduce photo-effects. Components in the figure are not to scale.
To test sorption to candidate materials, the experimental apparatus included a heated (nominally 90 °C) impinger containing a HgBr2 permeation tube (Fig. A1) that could provide controlled injections of through a candidate material trap (35.6 mg PPS or a half of a 47 mm diameter PFSA-M). For HgII tests, the sample line was also kept warm with heat tape (∼83 °C) and insulated with aluminum foil. The same injection procedure as described above was used to determine sorption to PPS and PFSA-M, although the empty trap contained PTFE plugs rather than quartz wool. To confirm source stability over time, peak area was observed through an empty trap before and after injections, and a t test was used to check that peak area from injections did not differ from the beginning of the test to the end of the test. Beginning and end injections were not statistically different in peak area.
Statistically different peak areas (p<0.05) were observed between an empty glass trap and both PFSA-M and PPS, indicating these materials sorbed (n=5, RSD≤10 % for injections through an empty trap; n=5, RSD<10 % for PFSA-M; and n=5, RSD=14 % for injections through the PPS trap).
An alternative digestion method was attempted to improve recovery of from α-Al2O3 and γ-Al2O3. The appendix to EPA Method 1631 is a similar digestion procedure to Method 1631, but with an additional leaching step using aqua regia (3:1 HCl:HNO3) before digestion with BrCl. A calibrator (described in the main text) was used to load materials with a known mass of , in lieu of an appropriate certified reference material. An expected 0.25 ng was loaded onto each material (30 ± 5 mg chitosan, α-Al2O3, or γ-Al2O3; n=3 traps each), which was then digested by the appendix to EPA Method 1631 and analyzed by cold-vapor atomic fluorescence. Activated carbon (30 ± 5 mg) was used downstream of chitosan, α-Al2O3, and γ-Al2O3 to measure not captured by the candidate material. A second-in-line cation exchange membrane (CEM) captured breakthrough from a first-in-line CEM. All measurements were blank-corrected with the appropriate material. Results were highly variable for α-Al2O3 (0.13 ± 0.12 ng recovered on α-Al2O3 and 0.17 ± 0.15 on breakthrough carbon), and no was recovered from γ-Al2O3, with little recovered from downstream carbon (0.03 ± 0.06 ng ). CEM reasonably recovered the expected loaded mass (0.23 ± 0.06 ng ), with no quantifiable breakthrough (Fig. C1). The expected 0.25 ng of was reasonably recovered from traps containing chitosan and breakthrough activated carbon (0.13 ± 0.06 ng recovered on chitosan, 0.1 ng recovered on downstream activated carbon), indicating that the appendix method worked to recover the mass balance from CEM, chitosan, and carbon matrices. The lack of recovery from α-Al2O3, γ-Al2O3, and downstream carbon suggests that α-Al2O3 and γ-Al2O3 may be sorbing HgII, but this digestion method is insufficient to quantify it. The appendix to EPA Method 1635 was chosen as a digestion procedure because it is intended for recalcitrant matrices, including coal; however, aqua regia has a matrix-dependent leaching efficiency (Zimmermann et al., 2020). A certified reference material matrix matched to Al2O3 may conclusively demonstrate this.
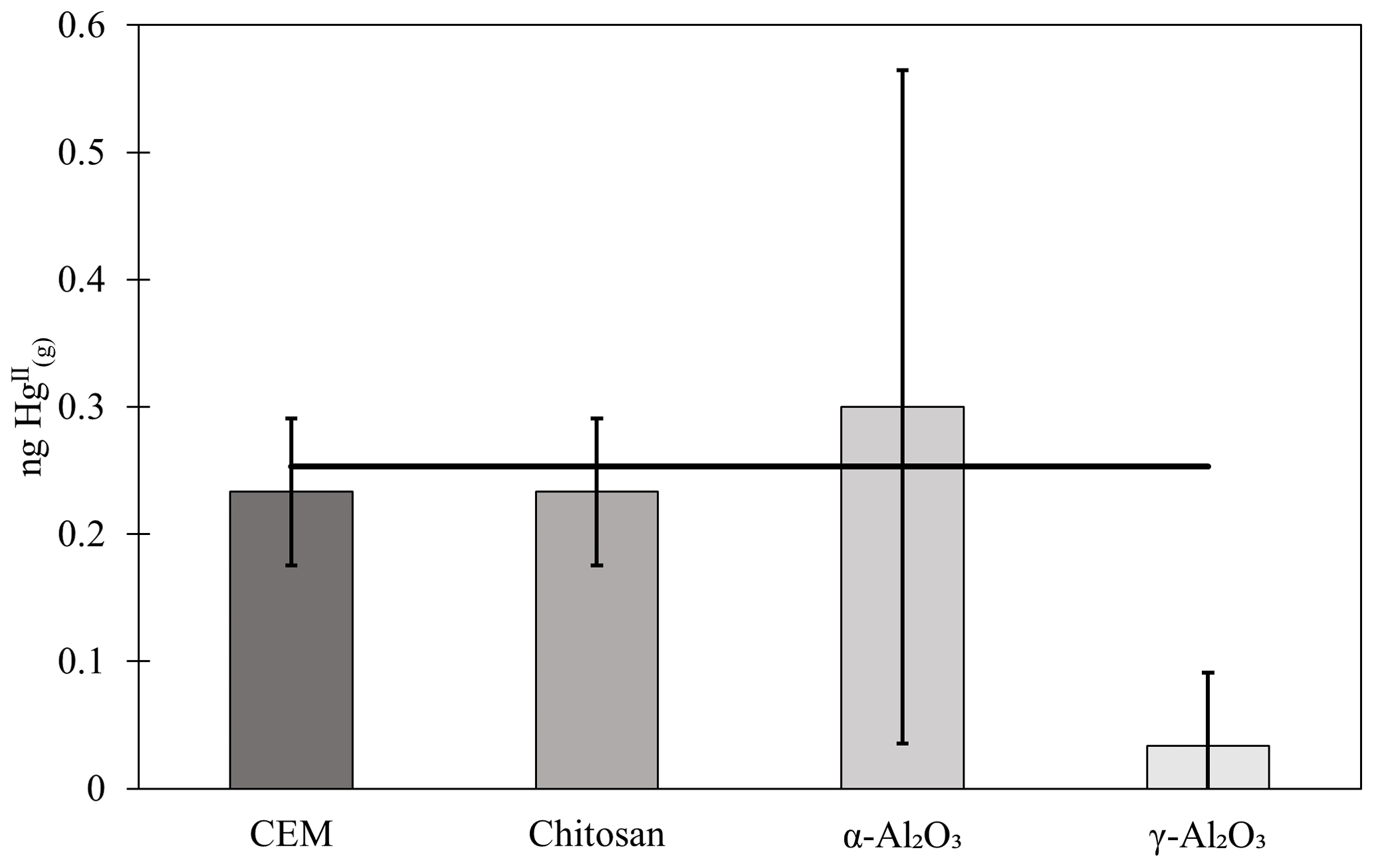
Figure C1 (ng) recovered by the appendix to EPA Method 1631 on chitosan, α-Al2O3, and γ-Al2O3 compared to an equivalent mass of loaded on CEMs. Masses include measured on downstream activated carbon. Error bars represent 1 standard deviation from the mean.
Table D1Environmental conditions during Reactive Mercury Active System (RMAS) campaigns. These data were downloaded as shown from the Western Regional Climate Center (https://raws.dri.edu/, last access: 25 October 2024). The measurement station was located at the test site (39.53917, −119.806,; 1370 m a.s.l.). Means and standard deviations (highlighted in italics) were calculated by the authors from the presented data.
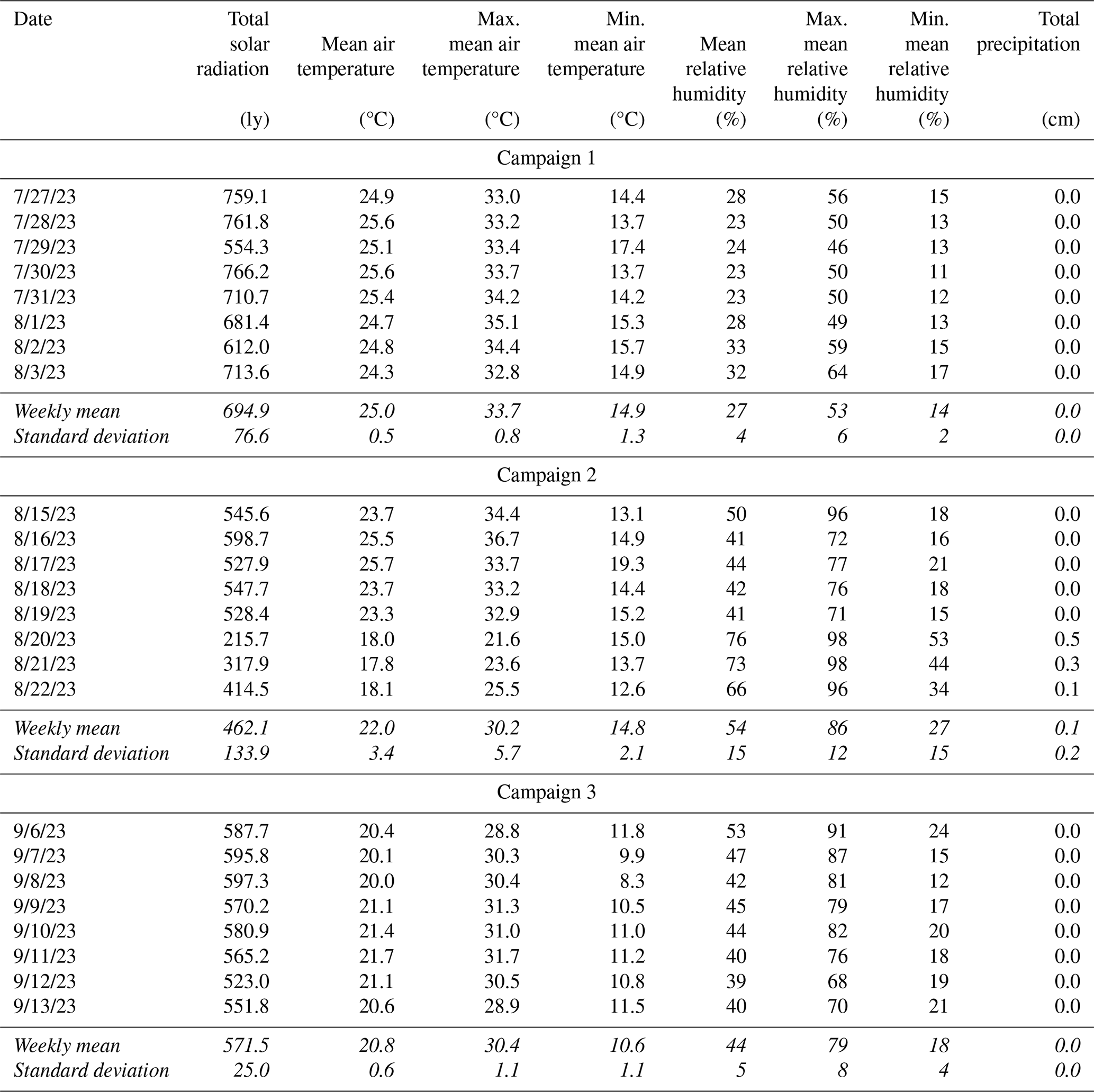

Figure E1(a) Field campaign of candidate materials and CEM in inverted RMAS shields. (b) A close-up of an inverted RMAS shield holding (1) glass traps with candidate materials, (2) PTFE membranes, and (3) breakthrough CEMs.
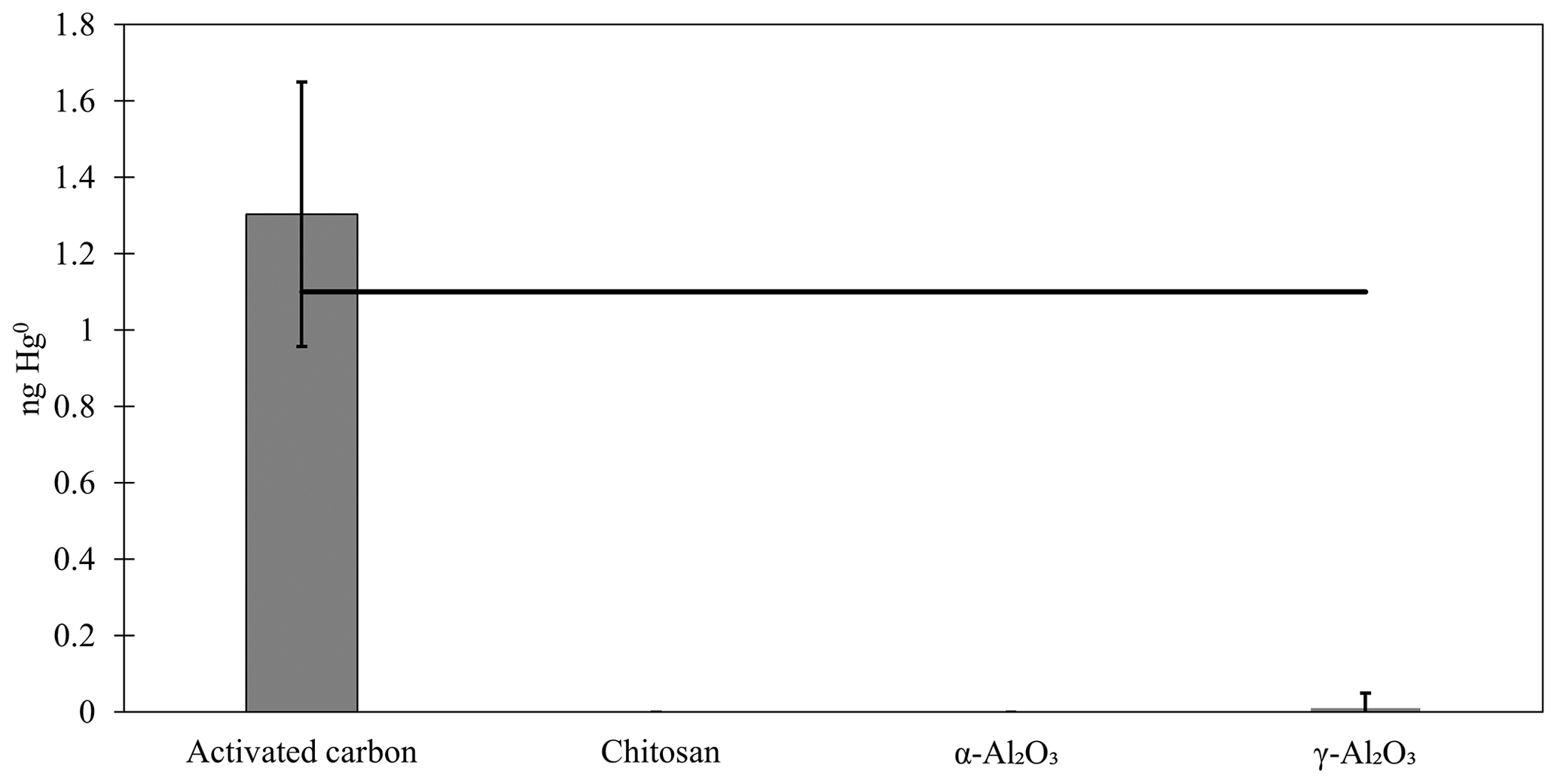
Figure F1Recovery of Hg0 from activated carbon and candidate materials loaded in ambient air by syringe injection. The black line indicates the calculated mass (1.1 ng Hg0) loaded based on the Dumarey equation.
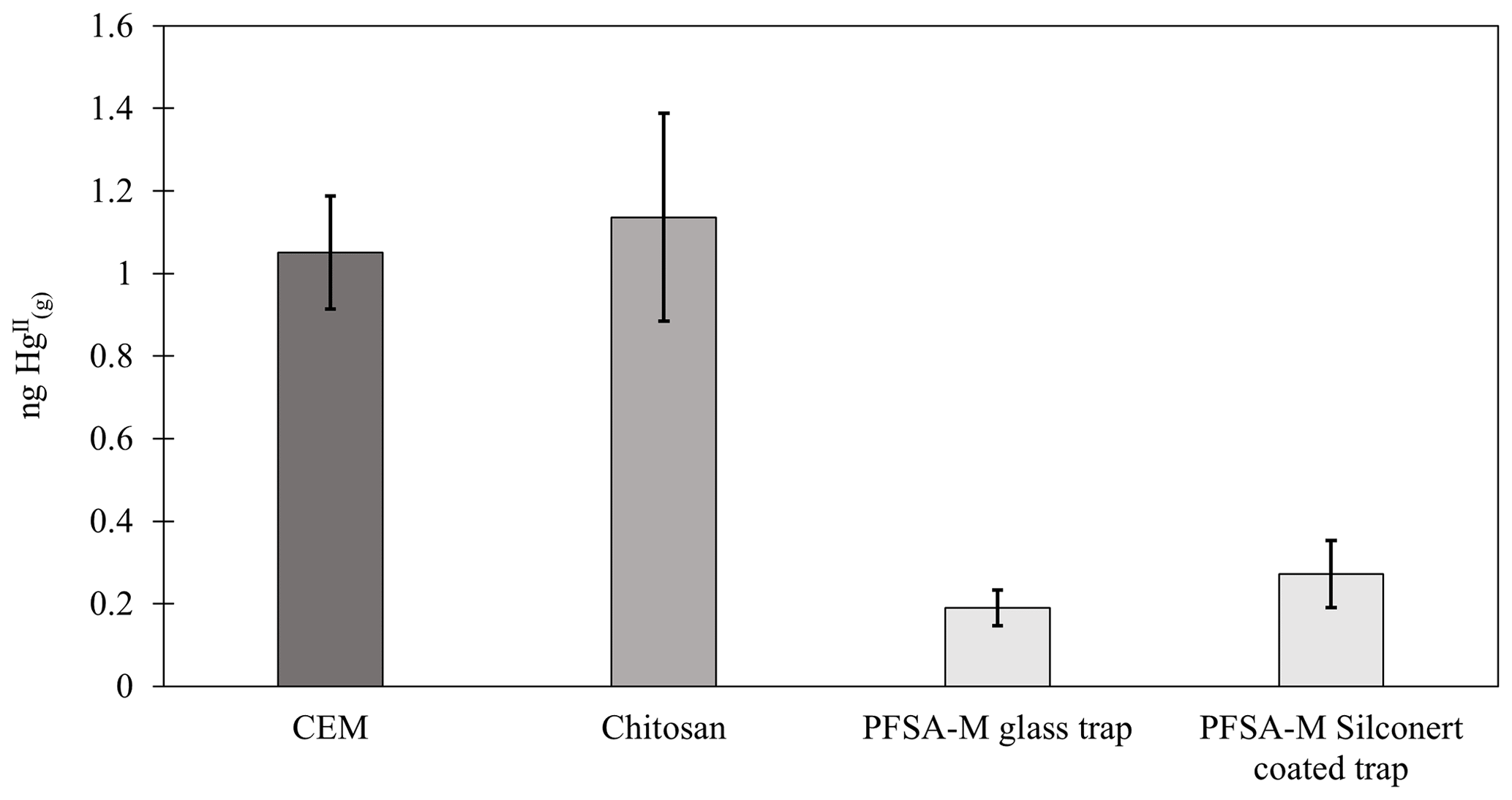
Figure G1 recovery from PFSA-M using deactivated fused-silica-coated and uncoated glass tubes compared to recovery from CEMs and chitosan. The recovery of from PFSA-M was not statistically different between coated and uncoated glass traps (p>0.05, two-sample t test).
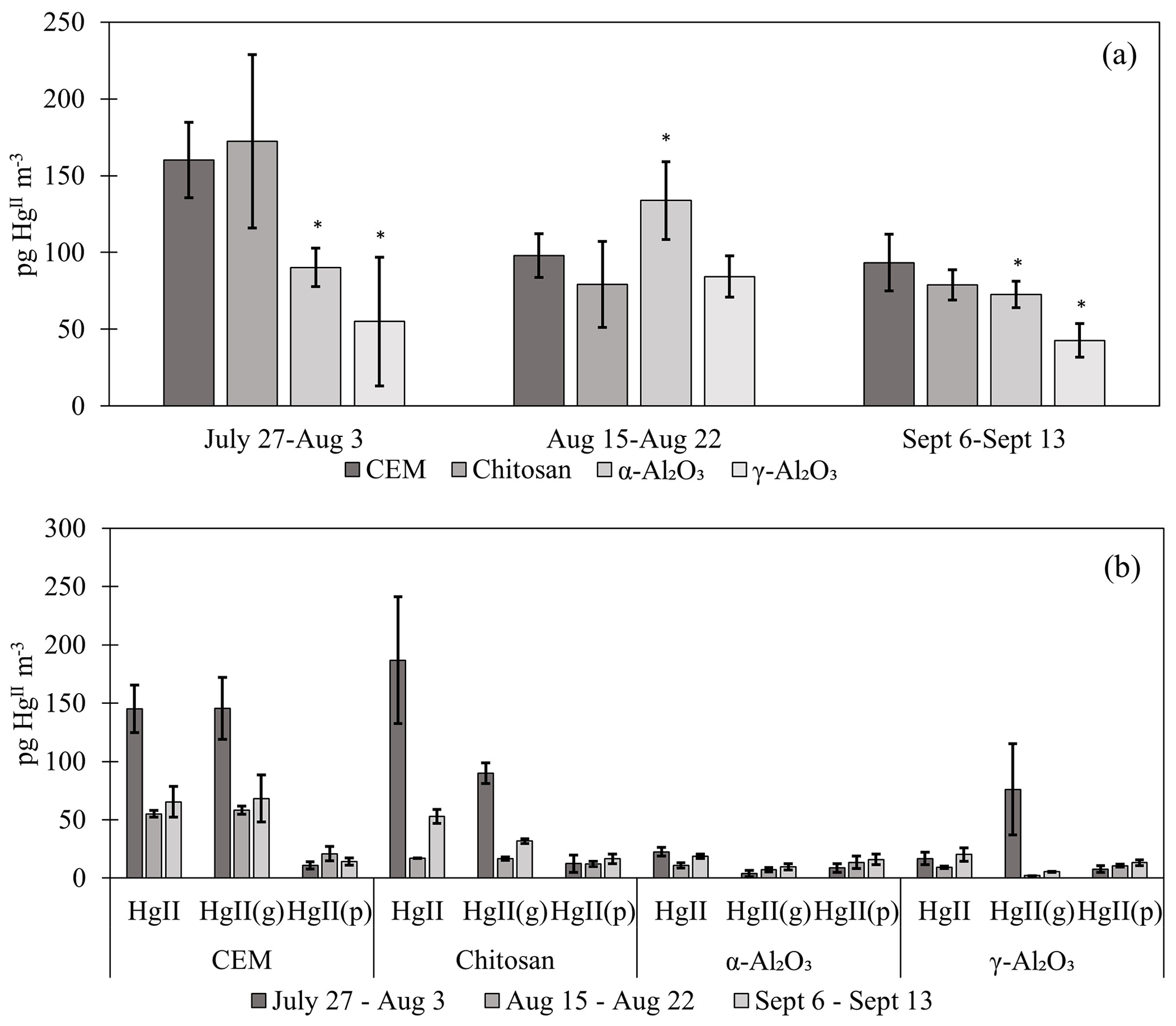
Figure H1(a) Total HgII recovery from the entire trap assembly (PTFE membrane (if present) + candidate material or first-in-line CEM + breakthrough CEM). Data combine traps with and without upstream PTFE membranes for each material type (n=6). Error bars represent 1 standard deviation from the mean. Asterisks (*) indicate a statistically different recovery of HgII on candidate materials compared to CEMs (ANOVA, α≤0.05). (b) Total HgII recovered from candidate materials or first-in-line CEMs without upstream PTFE membranes (HgII; n=3) shown adjacent to downstream candidate materials or CEMs in traps with upstream PTFE (; n=3) and HgII recovered from PTFE membranes (). In theory, .
Table I1Calibrator permeation rates as measured by CEMs.
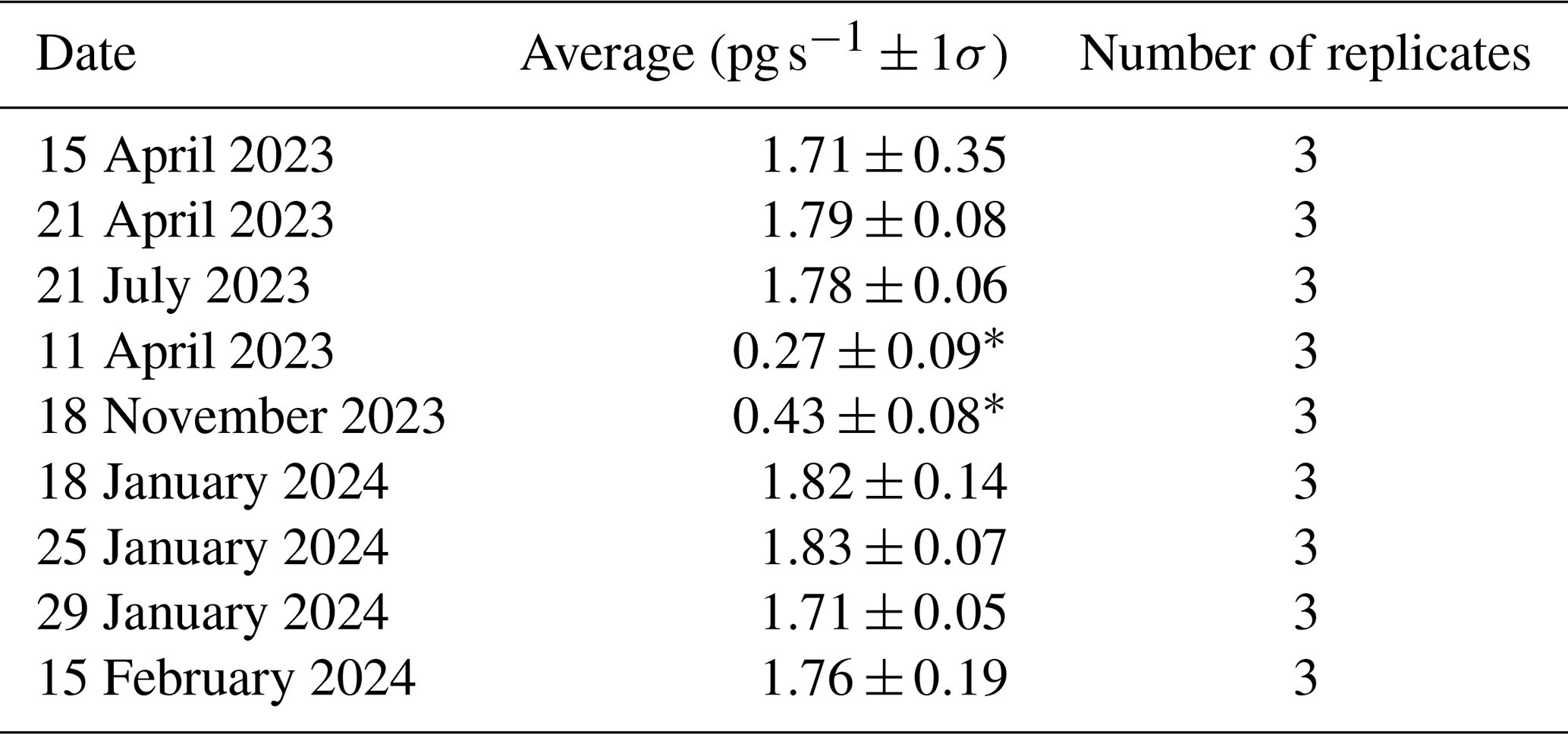
∗ source was not heated during loading.
Table J1Recoveries (pg m−3) of HgII from traps following field deployment. Columns labeled HgII and HgII breakthrough belong to traps deployed without PTFE membranes. Recoveries (pg m−3) of HgII from PTFE are indicated as , recoveries from CEMs or candidate material downstream of PTFE are indicated by , and breakthrough from traps with PTFE membranes is indicated by breakthrough.
All data are included in the article and Appendix.
LL suggested materials, designed and executed the experiments, performed data analysis and interpretation, and prepared the manuscript. SMDC suggested materials, supervised the experiments and data analysis, and edited the manuscript. SNL built and consulted on the use of the HgBr2 calibrator, consulted on other experiments, and edited the manuscript. MSG conceived the project and acquired funding, suggested materials, supervised experiments, and edited the manuscript.
The contact author has declared that none of the authors has any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
The authors would like to thank Igor Slowing for suggesting poly(1,4-phenylene) sulfide in the early stages of the project and Jan Gačnik for suggesting we invert RMAS shields to deploy powder materials. Thanks also go to undergraduate research assistants Mitch Aiken, Nicole Choma, Chris Ford, Ryan Murphy, and Morgan Yeager for help with maintaining the trace-clean glassware and equipment used in this work. Thanks to the two anonymous reviewers who provided helpful feedback.
This research has been supported by the NSF Division of Atmospheric and Geospace Sciences (grant nos. 2043042, 2044537, and 1951513).
This paper was edited by Rebecca Washenfelder and reviewed by two anonymous referees.
Ali, Z., Ahmad, R., Khan, A., and Adalata, B.: Complexation of Hg(II) ions with a functionalized adsorbent: A thermodynamic and kinetic approach, Prog. Nucl. Energ., 105, 146–152, https://doi.org/10.1016/j.pnucene.2018.01.004, 2018.
Allen, N., Gačnik, J., Dunham-Cheatham, S. M., and Gustin, M. S.: Interaction of reactive mercury with surfaces and implications for atmospheric mercury speciation measurements, Atmos. Environ., 318, 120240, https://doi.org/10.1016/j.atmosenv.2023.120240, 2024.
Ariya, P. A., Amyot, M., Dastoor, A., Deeds, D., Feinberg, A., Kos, G., Poulain, A., Ryjkov, A., Semeniuk, K., Subir, M., and Toyota, K.: Mercury physicochemical and biogeochemical transformation in the atmosphere and at atmospheric interfaces: A review and future directions, Chem. Rev., 115, 3760–3802, https://doi.org/10.1021/cr500667e, 2015.
Baronskiy, M. G., Tsybulya, S. V., Kostyukov, A. I., Zhuzhgov, A. V., and Snytnikov, V. N.: Structural properties investigation of different alumina polymorphs (η-, γ-, χ-, θ-, α-Al2O3) using Cr3+ as a luminescent probe, J. Lumin., 242, 118554, https://doi.org/10.1016/j.jlumin.2021.118554, 2022.
Deeds, D. A., Ghoshdastidar, A., Raofie, F., Guérette, E. A., Tessier, A., and Ariya, P. A.: Development of a particle-trap preconcentration-soft ionization mass spectrometric technique for the quantification of mercury halides in air, Anal. Chem., 87, 5109–5116, https://doi.org/10.1021/ac504545w, 2015.
Driscoll, C. T., Mason, R. P., Chan, H. M., Jacob, D. J., and Pirrone, N.: Mercury as a global pollutant: sources, pathways, and effects, Environ. Sci. Technol., 47, 4967–4983, https://doi.org/10.1021/es305071v, 2013.
Dumarey, R., Brown, R. J. C., Corns, W. T., Brown, A. S., and Stockwell, P. B.: Elemental mercury vapour in air: the origins and validation of the “Dumarey equation” describing the mass concentration at saturation, Accredit. Qual. Assur., 15, 409–414, https://doi.org/10.1007/s00769-010-0645-1, 2010.
Dunham-Cheatham, S. M., Lyman, S., and Gustin, M. S.: Evaluation of sorption surface materials for reactive mercury compounds, Atmos. Environ., 242, 117836, https://doi.org/10.1016/j.atmosenv.2020.117836, 2020.
Dunham-Cheatham, S. M., Lyman, S., and Gustin, M. S.: Comparison and calibration of methods for ambient reactive mercury quantification, Sci. Total Environ., 856, 159219, https://doi.org/10.1016/j.scitotenv.2022.159219, 2023.
Elgiar, T. R., Lyman, S. N., Andron, T. D., Gratz, L., Hallar, A. G., Horvat, M., Vijayakumaran Nair, S., O'Neil, T., Volkamer, R., and Živković, I.: Traceable Calibration of Atmospheric Oxidized Mercury Measurements, Environ. Sci. Technol., 58, 10706–10716, https://doi.org/10.1021/acs.est.4c02209, 2024.
Gačnik, J., Živković, I., Ribeiro, S. G., Kotnik, J., Berisha, S., Nair, S. V., Jurov, A., Cvelbar, U., and Horvat, M.: Calibration approach for gaseous oxidized mercury based on nonthermal plasma oxidation of elemental mercury, Anal. Chem., 94, 8234–8240, https://doi.org/10.1021/acs.analchem.2c00260, 2022.
Gačnik, J., Lyman, S. N., Dunham-Cheatham, S. M., and Gustin, M. S.: Limitations and insights regarding atmospheric mercury sampling using gold, Anal. Chim. Acta, 1319, 342956, https://doi.org/10.1016/j.aca.2024.342956, 2024.
Gustin, M. S., Dunham-Cheatham, S. M., and Zhang, L.: Comparison of 4 methods for measurement of reactive, gaseous oxidized, and particulate bound mercury, Environ. Sci. Technol., 53, 14489–14495, https://doi.org/10.1021/acs.est.9b04648, 2019.
Gustin, M. S., Dunham-Cheatham, S. M., Zhang, L., Lyman, S., Choma, N., and Castro, M.: Use of membranes and detailed HYSPLIT analyses to understand atmospheric particulate, gaseous oxidized, and reactive mercury chemistry, Environ. Sci. Technol., 55, 893–901, https://doi.org/10.1021/acs.est.0c07876, 2021.
Gustin, M. S., Dunham-Cheatham, S. M., Allen, N., Choma, N., Johnson, W., Lopez, S., Russell, A., Mei, E., Magand, O., Dommergue, A., and Elgiar, T.: Observations of the chemistry and concentrations of reactive Hg at locations with different ambient air chemistry, Sci. Total Environ., 904, 166184, https://doi.org/10.1016/j.scitotenv.2023.166184, 2023.
Holmes, C. D., Jacob, D. J., Corbitt, E. S., Mao, J., Yang, X., Talbot, R., and Slemr, F.: Global atmospheric model for mercury including oxidation by bromine atoms, Atmos. Chem. Phys., 10, 12037–12057, https://doi.org/10.5194/acp-10-12037-2010, 2010.
Horowitz, H. M., Jacob, D. J., Zhang, Y., Dibble, T. S., Slemr, F., Amos, H. M., Schmidt, J. A., Corbitt, E. S., Marais, E. A., and Sunderland, E. M.: A new mechanism for atmospheric mercury redox chemistry: implications for the global mercury budget, Atmos. Chem. Phys., 17, 6353–6371, https://doi.org/10.5194/acp-17-6353-2017, 2017.
Huang, J. and Gustin, M. S.: Uncertainties of gaseous oxidized mercury measurements using KCl-coated denuders, cation-exchange membranes, and nylon membranes: humidity influences, Environ. Sci. Technol., 49, 6102–6108, https://doi.org/10.1021/acs.est.5b00098, 2015.
Huang, J., Miller, M. B., Weiss-Penzias, P., and Gustin, M. S.: Comparison of gaseous oxidized Hg measured by KCl-coated denuders, and nylon and cation exchange membranes, Environ. Sci. Technol., 47, 7307–7316, https://doi.org/10.1021/es4012349, 2013.
Jones, C. P., Lyman, S. N., Jaffe, D. A., Allen, T., and O'Neil, T. L.: Detection and quantification of gas-phase oxidized mercury compounds by GC/MS, Atmos. Meas. Tech., 9, 2195–2205, https://doi.org/10.5194/amt-9-2195-2016, 2016.
Kaulfus, A. S., Nair, U., Holmes, C. D., and Landing, W. M.: Mercury wet scavenging and deposition differences by precipitation type, Environ. Sci. Technol., 51, 2628–2634, https://doi.org/10.1021/acs.est.6b04187, 2017.
Khalizov, A. F., Guzman, F. J., Cooper, M., Mao, N., Antley, J., and Bozzelli, J.: Direct detection of gas-phase mercuric chloride by ion drift-chemical ionization mass spectrometry, Atmos. Environ., 238, 117687, https://doi.org/10.1016/j.atmosenv.2020.117687, 2020.
Landis, M. S., Stevens, R. K., Schaedlich, F., and Prestbo, E. M.: Development and characterization of an annular denuder methodology for the measurement of divalent inorganic reactive gaseous mercury in ambient air, Environ. Sci. Technol., 36, 3000–3009, https://doi.org/10.1021/es015887t, 2002.
Laurier, F. J., Mason, R. P., Whalin, L., and Kato, S.: Reactive gaseous mercury formation in the North Pacific Ocean's marine boundary layer: A potential role of halogen chemistry, J. Geophys. Res.-Atmos., 108, 4529, https://doi.org/10.1029/2003JD003625, 2003.
Luippold, A., Gustin, M. S., Dunham-Cheatham, S. M., Castro, M., Luke, W., Lyman, S., and Zhang, L.: Use of multiple lines of evidence to understand reactive mercury concentrations and chemistry in Hawai'i, Nevada, Maryland, and Utah, USA, Environ. Sci. Technol., 54, 7922–7931, https://doi.org/10.1021/acs.est.0c02283, 2020a.
Luippold, A., Gustin, M. S., Dunham-Cheatham, S. M., and Zhang, L.: Improvement of quantification and identification of atmospheric reactive mercury, Atmos. Environ., 224, 117307, https://doi.org/10.1016/j.atmosenv.2020.117307, 2020b.
Lyman, S., Jones, C., O'Neil, T., Allen, T., Miller, M., Gustin, M. S., Pierce, A. M., Luke, W., Ren, X., and Kelley, P.: Automated calibration of atmospheric oxidized mercury measurements, Environ. Sci. Technol., 50, 12921–12927, https://doi.org/10.1021/acs.est.6b04211, 2016.
Lyman, S. N. and Jaffe, D. A.: Formation and fate of oxidized mercury in the upper troposphere and lower stratosphere, Nat. Geosci., 5, 114–117, https://doi.org/10.1038/ngeo1353, 2012.
Lyman, S. N., Cheng, I., Gratz, L. E., Weiss-Penzias, P., and Zhang, L.: An updated review of atmospheric mercury, Sci. Total Environ., 707, 135575, https://doi.org/10.1016/j.scitotenv.2019.135575, 2020a.
Lyman, S. N., Gratz, L. E., Dunham-Cheatham, S. M., Gustin, M. S., and Luippold, A.: Improvements to the accuracy of atmospheric oxidized mercury measurements, Environ. Sci. Technol., 54, 13379–13388, https://doi.org/10.1021/acs.est.0c02747, 2020b.
Manos, M. J. and Kanatzidis, M. G.: Metal sulfide ion exchangers: superior sorbents for the capture of toxic and nuclear waste-related metal ions, Chem. Sci., 7, 4804–4824, https://doi.org/10.1039/C6SC01039C, 2016.
Mao, N. and Khalizov, A.: Exchange reactions alter molecular speciation of gaseous oxidized mercury, ACS Earth Space Chemistry, 5, 1842–1853, https://doi.org/10.1021/acsearthspacechem.1c00178, 2021.
McClure, C. D., Jaffe, D. A., and Edgerton, E. S.: Evaluation of the KCl denuder method for gaseous oxidized mercury using HgBr2 at an in-service AMNet Site, Environ. Sci. Technol., 48, 11437–11444, https://doi.org/10.1021/es502545k, 2014.
Miller, M. B., Dunham-Cheatham, S. M., Gustin, M. S., and Edwards, G. C.: Evaluation of cation exchange membrane performance under exposure to high Hg0 and HgBr2 concentrations, Atmos. Meas. Tech., 12, 1207–1217, https://doi.org/10.5194/amt-12-1207-2019, 2019.
R Core Team: R: A language and environment for statistical computing, https://www.R-project.org/ (last access: 25 October 2024), 2023.
Shah, V., Jacob, D. J., Thackray, C. P., Wang, X., Sunderland, E. M., Dibble, T. S., Saiz-Lopez, A., Černušák, I., Kellö, V., Castro, P. J., Wu, R., and Wang, C.: Improved mechanistic model of the atmospheric redox chemistry of mercury, Environ. Sci. Technol., 55, 14445–14456, https://doi.org/10.1021/acs.est.1c03160, 2021.
Song, Z., Huang, S., Zhang, P., Yuan, T., and Zhang, Y.: Isotope Data Constrains Redox Chemistry of Atmospheric Mercury, Environ. Sci. Technol., 58, 13307–13317, https://doi.org/10.1021/acs.est.4c02600, 2024.
Steffen, A., Douglas, T., Amyot, M., Ariya, P., Aspmo, K., Berg, T., Bottenheim, J., Brooks, S., Cobbett, F., Dastoor, A., Dommergue, A., Ebinghaus, R., Ferrari, C., Gardfeldt, K., Goodsite, M. E., Lean, D., Poulain, A. J., Scherz, C., Skov, H., Sommar, J., and Temme, C.: A synthesis of atmospheric mercury depletion event chemistry in the atmosphere and snow, Atmos. Chem. Phys., 8, 1445–1482, https://doi.org/10.5194/acp-8-1445-2008, 2008.
Szymańska, E. and Winnicka, K.: Stability of chitosan – a challenge for pharmaceutical and biomedical applications, Mar. Drugs, 13, 1819–1846, https://doi.org/10.3390/md13041819, 2015.
United States Environmental Protection Agency: Method 3052, Revision 0: Microwave assisted acid digestion of siliceous and organically based matrices, EPA publication SW-846, 1996.
United States Environmental Protection Agency: Appendix to Method 1631, Total mercury in tissue, sludge, sediment, and soil by acid digestion and BrCl oxidation, 2001.
United States Environmental Protection Agency: Method 1631, Revision E: Mercury in water by oxidation, purge and trap, and cold vapor atomic fluorescence spectrometry, 2002.
United States Environmental Protection Agency: Method 7473, Revision 0: Mercury in Solids and Solutions by thermal decomposition, amalgamation, and atomic absorption spectrophotometry, EPA publication SW-846, 2007.
Vieira, R. S. and Beppu, M. M.: Interaction of natural and crosslinked chitosan membranes with Hg(II) ions, Colloid. Surface. A, 279, 196–207, https://doi.org/10.1016/j.colsurfa.2006.01.026, 2006.
Western Regional Climate Center: https://raws.dri.edu/, last access: 2 February 2024.
Yu, J. G., Yue, B. Y., Wu, X. W., Liu, Q., Jiao, F. P., Jiang, X. Y., and Chen, X. Q.: Removal of mercury by adsorption: a review, Environ. Sci. Pollut. R., 23, 5056–5076, https://doi.org/10.1007/s11356-015-5880-x, 2016.
Zheng, Y., Duan, Y., Tang, H., Li, C., Li, J., Zhu, C., and Liu, S.: Experimental research on selective adsorption of gaseous mercury (II) over SiO2, TiO2 and γ-Al2O3, Fuel, 237, 202–208, https://doi.org/10.1016/j.fuel.2018.09.153, 2019.
Zimmermann, T., Von Der Au, M., Reese, A., Klein, O., Hildebrandt, L., and Pröfrock, D.: Substituting HF by HBF4 – an optimized digestion method for multi-elemental sediment analysis via ICP-MS/MS, Anal. Methods-UK, 12, 3778–3787, https://doi.org/10.1039/D0AY01049A, 2020.
- Abstract
- Introduction
- Methods
- Results and discussion
- Conclusions
- Appendix A: Preliminary assessment of gaseous elemental mercury sorption in an argon atmosphere to poly(1,4-phenylene sulfide), chitosan, perfluorosulfonic acid, and α-Al2O3
- Appendix B: Preliminary assessment of sorption to PFSA-M and PPS in an argon atmosphere
- Appendix C: Appendix to EPA Method 1631
- Appendix D
- Appendix E
- Appendix F
- Appendix G
- Appendix H
- Appendix I
- Appendix J
- Code and data availability
- Author contributions
- Competing interests
- Disclaimer
- Acknowledgements
- Financial support
- Review statement
- References
- Abstract
- Introduction
- Methods
- Results and discussion
- Conclusions
- Appendix A: Preliminary assessment of gaseous elemental mercury sorption in an argon atmosphere to poly(1,4-phenylene sulfide), chitosan, perfluorosulfonic acid, and α-Al2O3
- Appendix B: Preliminary assessment of sorption to PFSA-M and PPS in an argon atmosphere
- Appendix C: Appendix to EPA Method 1631
- Appendix D
- Appendix E
- Appendix F
- Appendix G
- Appendix H
- Appendix I
- Appendix J
- Code and data availability
- Author contributions
- Competing interests
- Disclaimer
- Acknowledgements
- Financial support
- Review statement
- References