the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 24 Jan 2025
| 24 Jan 2025
Towards a high-quality in situ observation network for oxygenated volatile organic compounds (OVOCs) in Europe: transferring metrological traceability to the field
Maitane Iturrate-Garcia
Thérèse Salameh
Paul Schlauri
Annarita Baldan
Martin K. Vollmer
Evdokia Stratigou
Sebastien Dusanter
Jianrong Li
Stefan Persijn
Anja Claude
Rupert Holzinger
Christophe Sutour
Tatiana Macé
Yasin Elshorbany
Andreas Ackermann
Céline Pascale
Stefan Reimann
Volatile organic compounds (VOCs) have a large impact on the oxidising capacity of the troposphere and are major precursors of tropospheric ozone and secondary atmospheric aerosols. Accurate measurements and data comparability of VOCs among monitoring networks are essential to assessing the trends of these secondary air pollutants. Metrological traceability of the measurements to the International System of Units (SI traceability) contributes to both measurement consistency and data comparability. Accurate, stable and SI-traceable reference gas mixtures (RGMs) and working standards are needed to achieve SI traceability through an unbroken chain of calibrations of the analytical instruments used to monitor VOCs. However, for many oxygenated VOCs (OVOCs), such RGMs and working standards are not available at an atmospheric amount of substance fraction levels (< 10 nmol mol−1). Here, we present the protocols developed to transfer SI traceability to the field by producing two types of SI-traceable working standards for selected OVOCs. These working standards, based on RGMs diluted dynamically with dry nitrogen and on certified spiked whole-air samples, were then assessed using a thermal desorber–gas chromatograph–flame ionisation detector (TD–GC–FID) and proton transfer reaction–time of flight–mass spectrometer (PTR–ToF–MS) as analytical methods. For that purpose, we calibrated five analytical instruments using in-house calibration standards and treated the new SI-traceable working standards as samples. Due to analytical limitations, the assessment was only possible for acetaldehyde, acetone, methanol and methyl ethyl ketone (MEK). Relative differences between assigned and measured values were used to assess the working standards based on the dilution of RGMs. The relative differences were within the measurement uncertainty for acetone, MEK, methanol and acetaldehyde at an amount of substance fractions around 10 nmol mol−1. For the working standards based on certified spiked whole-air samples in pressurised cylinders, results showed a good agreement among the laboratories (i.e. differences within the measurement expanded uncertainty (U) ranging between 0.5 and 3.3 nmol mol−1) and with the certified amount of substance fraction for acetaldehyde (15.7 nmol mol−1 ± 3.6 (U) nmol mol−1), acetone (17 nmol mol−1 ± 1.5 (U) nmol mol−1) and MEK (12.3 nmol mol−1 ± 2.3 (U) nmol mol−1). Despite the promising results for the working standards based on the dilution of RGMs and on certified spiked whole-air samples filled into pressurised cylinders, the assessment must be considered with care due to the large measurement uncertainty, particularly for methanol. Active collaboration among the metrological, meteorological and atmospheric chemistry monitoring communities is needed to tackle the challenges of OVOC monitoring, such as the lack of stable and SI-traceable calibration standards (i.e. RGMs and working standards). Besides this collaboration, other research applications, such as modelling and remote sensing, may benefit from the transfer of SI traceability to monitoring stations.
- Article
(1091 KB) - Full-text XML
- BibTeX
- EndNote





Tropospheric ozone plays a key role in the oxidative capacity of the atmosphere (Iglesias-Suarez et al., 2018; Monks et al., 2015; Schultz et al., 2015) through different chemical reactions, such as ozone photodissociation, which is the dominant source of the hydroxyl radical (OH) in the troposphere (e.g. Lelieveld and Dentener, 2000; Zhang et al., 2014). Besides being a strong oxidant with a direct impact on human respiratory health, vegetation growth and crop productivity (Van Dingenen et al., 2009; Schultz et al., 2017; Mills et al., 2018), tropospheric ozone is also a greenhouse gas and a secondary air pollutant (Gaudel et al., 2018; Szopa et al., 2023). In the troposphere, ozone abundance depends on its transport from the stratosphere, formation and destruction through photochemical reactions, and dry deposition (Cooper et al., 2014; Fleming et al., 2018; Jacob, 2000; Stohl et al., 2003; Wild, 2007). Volatile organic compounds (VOCs) – a group of chemical compounds with one or more atoms of carbon and a complex speciation that encompasses thousands of species (Goldstein and Galbally, 2007; Yang et al., 2016) – are one of the major tropospheric ozone precursors (Shao et al., 2009; Xue et al., 2014; Simon et al., 2015). VOC oxidation in the presence of a significant amount of substance fractions of nitrogen oxides (NOx) results in a net production of ozone (Collins et al., 2002; Pugliese et al., 2014).
Oxygenated VOCs (OVOCs) are an important fraction of VOCs, including alcohols, carbonyls (aldehydes and ketones) and carboxylic acid (Legreid et al., 2007; Wu et al., 2020). OVOCs are precursors of tropospheric ozone and secondary organic aerosols and have, thus, an impact on air quality and climate (Boucher et al., 2013; Seinfeld et al., 2016; Shrivastava et al., 2017). OVOCs can be formed by atmospheric photooxidation of hydrocarbons (Atkinson, 2000) and can be emitted directly from vegetation, biomass burning, vehicle exhaust and industrial processes (Placet, 2000; Legreid et al., 2007; Worton et al., 2022). OVOCs with low molecular weights (e.g. methanol; acetone; acetaldehyde; methyl ethyl ketone, MEK) are found at a relatively high amount of substance fractions in the global atmosphere and play an important role in the tropospheric photochemistry. For these OVOCs, the main sinks are oxidation with OH radicals and degradation initiated by photolysis leading to the formation of hydrogen oxide radicals (HOx). For example, oxidation products of methanol are formaldehyde and CO (Bates et al., 2021; Hu et al., 2011), which also impact the oxidation capacity of the troposphere. Acetone, acetaldehyde and MEK are oxidised to peroxy radicals that react with NO2 to form peroxyacetyl nitrate (PAN), which is an important precursor of tropospheric ozone (Millet et al., 2010; Fischer et al., 2012; Khan et al., 2015; Wang et al., 2019) and can lead to the transport of radicals and NO2 over long distances. The production of radicals (e.g. OH, HOx) and PAN further affects the chemistry of the tropospheric ozone (Volkamer et al., 2010; Fischer et al., 2014; Tan et al., 2019; Brewer et al., 2020; Zborowska et al., 2021). Therefore, accurate OVOC monitoring is crucial to assessing tropospheric ozone burdens, trends and variability.
The Tropospheric Ozone Assessment Report, Phase I (TOAR-I), identified uncertainties associated with ozone precursors' emissions, including VOCs, as one of the main contributors to the uncertainty of the modelled spatial and temporal distribution of ozone (Young et al., 2018). Long-term accurate measurements of ozone precursors are required to reduce the uncertainties in their emissions. This need for accurate measurements was also highlighted in TOAR-I as part of the scientific tasks, goals and requirements for tropospheric ozone monitoring (Tarasick et al., 2019). Other programmes and the infrastructure for atmospheric monitoring emphasise the importance of monitoring VOCs, particularly OVOCs, because of their active role and impact on the chemistry of the atmosphere, air quality and climate change. The World Meteorological Organization Global Atmosphere Watch (WMO GAW) programme has listed methanol, ethanol, acetone and formaldehyde as part of reactive gas compounds to be monitored (Schultz et al., 2015). The European Aerosol, Clouds and Trace Gases Research Infrastructure (ACTRIS) (Laj et al., 2024) – through its Centre for Reactive Trace Gases In Situ Measurements (CiGas) – includes OVOCs as one of the four compound clusters to be monitored, together with non-methane hydrocarbons, condensable vapours and NOx (Hoerger et al., 2015; Simon et al., 2023). Metrological traceability of the measurements, ideally to the International System of Units (SI), is essential to guarantee data comparability among the different monitoring networks (Brewer et al., 2018; Güttler and Richter, 2009; Worton et al., 2023).
Metrological traceability is achieved through an unbroken chain of calibrations, each contributing to the uncertainty of measurements (De Bièvre and Taylor, 1997). One way of ensuring SI traceability is to calibrate analytical instruments, which are used to monitor atmospheric compounds, against a primary reference material produced by a national metrology institute (NMI). NMIs prepare these materials following reference procedures, provide complete uncertainty budgets of the assigned values, ensure their stability period and participate in international comparisons with other NMIs to achieve SI traceability (Brewer et al., 2018). However, for certain reactive compounds, such as many OVOCs (e.g. methanol, ethanol), producing a reference material is particularly challenging because of surface, non-linearity and matrix effects, as well as because of stability issues and the low amount of substance fractions (at nmol mol−1 level) required (Grenfell et al., 2010; Leuenberger et al., 2015; Persijn and Baldan, 2023; Rhoderick et al., 2019).
SI-traceable reference gas mixtures (RGMs) have been developed at NMIs for an increasing number of OVOCs in the last decade (e.g. Brown et al., 2013; Worton et al., 2023). Nevertheless, RGMs are only available at an amount of substance fractions higher than atmospheric ones (Rhoderick et al., 2019; Worton et al., 2022). When monitoring atmospheric OVOCs, this higher amount fraction implies that RGMs must be diluted at monitoring stations before calibrating the analytical instruments. Depending on the dilution procedure, SI traceability might be lost because of inadequate dilutions (e.g. using dilution devices such as thermal mass flow controllers, whose calibration is not SI-traceable). Another issue faced by OVOC monitoring stations regarding these RGMs is that the matrix gas of the mixture is not the same as ambient air. Quite often, nitrogen is used as a matrix gas to ensure the inertness of OVOCs like acetaldehyde. The use of dry nitrogen instead of humidified synthetic air may influence the calibration results. The lack of SI traceability and long-term stability of OVOC RGMs produced at low amount fraction levels are other limitations that often have negative effects particularly on long-term OVOC measurements. All these aspects have an impact on data comparability and thus on OVOC trend identification.
Here we present the efforts done between the metrological and atmospheric monitoring communities to transfer SI traceability to the field. For that purpose, protocols to produce two types of SI-traceable working standards – based on the dynamic dilution of RGMs with dry nitrogen and on certified spiked whole-air samples – of selected OVOCs were developed and assessed. OVOCs were selected in close collaboration with stakeholders (e.g. WMO GAW, ACTRIS) based on their relevance for atmospheric and climate research; on their role as tropospheric ozone precursors; and on the lack of accurate, stable and SI-traceable calibration standards. The selected OVOCs were acetaldehyde, acetone, ethanol, methacrolein, methanol, methyl ethyl ketone (MEK) and methyl vinyl ketone (MVK). The amount of substance fractions of the produced working standards were as close as technically feasible to the ambient air amount of substance fractions (<10 nmol mol−1). In this work, we used the quantity amount of substance fraction (a.k.a. amount fraction) – the accepted metrological term (Matschat et al., 2023; Richter, 2007) – instead of the concentration and/or mixing ratio terms. We expressed this quantity in SI units of nmol mol−1, which can be considered equivalent to parts per billion (ppb) under tropospheric conditions (Galbally et al., 2013).
Two types of SI-traceable OVOC working standards were prepared and assessed in this work (Fig. 1): working standards based on the dynamic dilution of SI-traceable reference gas mixtures and working standards based on certified spiked whole-air samples. While for the former a dilution step was needed before assessment, the latter was assessed directly without further dilution. The target amount fraction of each OVOC (acetaldehyde, acetone, ethanol, methacrolein, methanol, MEK and MVK) was 10 nmol mol−1 or lower in order to be as close as possible to the OVOC ambient levels. The assessment of the SI-traceable working standards was performed using several analysers based on two analytical methods (Fig. 1; Appendix A): thermal desorber–gas chromatograph–flame ionisation detector (TD–GC–FID) and proton transfer reaction–time of flight–mass spectrometer (PTR–ToF–MS). The analysers were calibrated with the participants' in-house working standards (Appendix D1). The SI-traceable working standards were treated as samples.
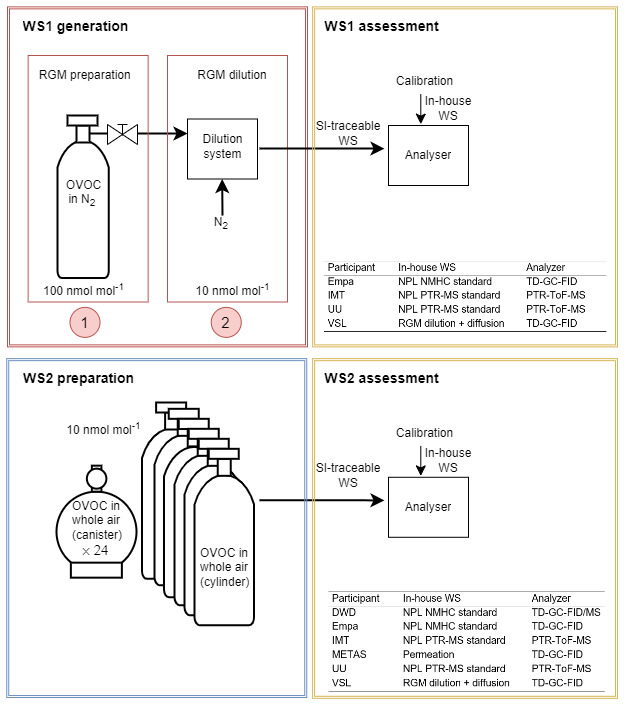
Figure 1Scheme showing the two types of working standards traceable to the International System of Units (SI) prepared in this work, based on the dilution of reference gas mixtures (RGMs) of oxygenated volatile organic compounds (OVOC) in nitrogen (N2) (WS1, working standard; for details, see Sect. 2.1) and on certified spiked whole-air samples (WS2; for details, see Sect. 2.2). Participants in the assessment, analysers (thermal desorber–gas chromatograph–flame ionisation detector, TD–GC–FID; proton transfer reaction–time of flight–mass spectrometer, PTR–ToF–MS) and in-house working standards used to calibrate them are indicated. DWD: Deutscher Wetterdienst, Empa: Swiss Federal Laboratories for Materials Science and Technology, IMT: Institute Mines-Télécom, METAS: Federal Institute of Metrology, NPL: National Physical Laboratory, UU: Utrecht University, VSL: National Metrology Institute. NMHC: non-methane hydrocarbon.
2.1 Generation of SI-traceable working standards based on the dynamic dilution of reference gas mixtures
The first type of SI-traceable working standards developed was based on the dilution of SI-traceable RGMs containing the selected OVOCs at amount fractions of ca. 100 nmol mol−1. To achieve the target amount fraction of 10 nmol mol−1 or lower for the SI-traceable working standards, the dynamic dilution of the produced RGMs was needed (Fig. 1). Dry nitrogen of a high purity (≥99.99990 %) was used as a matrix and dilution gas to prevent any possible reaction (e.g. oxidation) of OVOCs. The potential presence of water and OVOCs in the matrix and the dilution gas was assessed following standard procedures (ISO 19229:2019, 2019).
2.1.1 Gravimetric preparation of RGMs
Four RGMs of OVOCs in dry, high-purity (≥99.99990 %) nitrogen (BIP+, Built-in Purifier, Air Products Inc., PA, USA) were prepared at VSL, the NMI of the Netherlands, in August 2021. For that purpose, the primary gravimetric method was used by means of a high-resolution mass comparator (ISO 6142-1:2015, 2015). In this method, pure liquid compounds are injected in high-pressure gas cylinders. Prior to the injection, the purity of the selected liquid OVOCs was analysed (Appendix B1, Table B1). The steps followed to prepared the gravimetric RGMs are summarised in Fig. 2 and described in the following sections.
- A
Liquid OVOC injection. Known amounts of the pure liquid OVOCs were injected in high-pressure gas cylinders to obtain binary gas mixtures at around 50–100 µmol mol−1 in a first step (Fig. 2a). Besides the injected OVOCs, n-hexane was added as an internal standard to assess RGM stability (Table B1).
- B
Mixture of binaries and further dilution. Then, the binary gas mixtures were combined and further diluted to obtain OVOC RGMs at nominal amount fractions around 100 nmol mol−1 and at a pressure of 12 MPa (Fig. 2b). The RGMs were prepared in 10 L aluminium cylinders (Luxfer Inc., CA, USA) with an Experis® proprietary treatment (Air Products Inc., PA, USA) and a low-dead-volume stainless steel cylinder valve (D304, Rotarex, Luxembourg).
- C
Amount fraction value assignment. The RGM amount fraction value assignment was based on gravimetry, with the exception of methanol and ethanol. For these compounds, the value was assigned by analysis against dynamically prepared OVOC RGMs. Metrological traceability of the gravimetric RGMs was ensured by mass weighing and purity determination, while for methanol and ethanol, it was ensured by mass weighing, volume and purity determination.
- D
Verification.
- D.1
Verification against OVOC gas mixtures. After preparation (between the end of August and mid-September 2021), RGMs were verified against OVOC gas mixtures that contained acetone, ethanol, methacrolein, methanol, MVK and MEK and were generated by a diffusion method (ISO 6145-8:2005, 2005). For acetaldehyde, continuous syringe injection (ISO 6145-4:2004, 2004) and the dynamic dilution of an RGM at a high amount fraction (ISO 6145-7:2018, 2018) were used. The verification process was performed by VSL (Appendix B2.1). For each compound, a response factor was calculated according to Eq. (1), which was used to estimate the compound amount fraction in the gravimetric RGM following Eq. (2). RGM verification was based on the evaluation of the relative difference between the calculated amount fraction and the gravimetric value.
where RFi is the compound i response factor, is the average peak area of compound i in the calibration standard (last five replicates), is the average peak area of compound i in the blanks (last five replicates) and is the amount fraction of compound i in the calibration standard.
where xi is the estimated amount fraction of compound i in the sample, is the average peak area of compound i in the RGM (last five replicates), is the average peak area of compound i in the blanks (last five replicates) and RFi is the response factor of compound i calculated according to Eq. (1).
- D.2
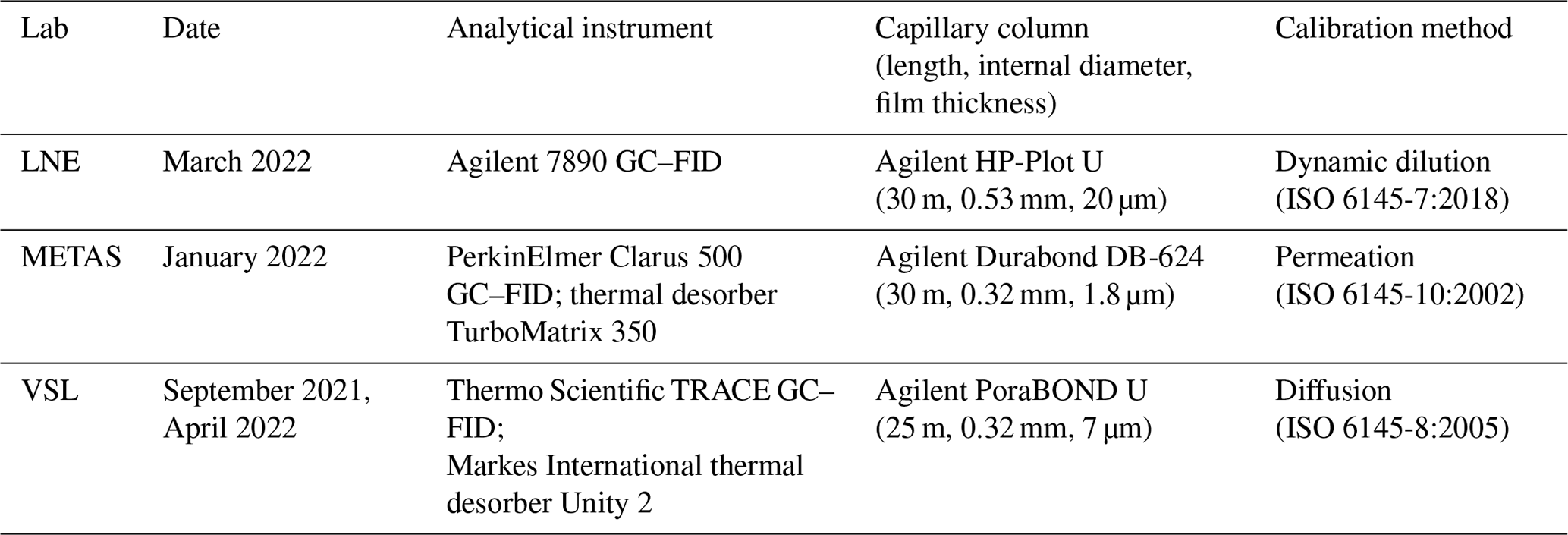
Interlaboratory comparison. A comparison between three laboratories took place to complete the RGM amount fraction verification. During this interlaboratory comparison (Appendix B2.2), one of the verified VSL RGMs (VSL221418) was analysed at VSL and at the NMIs of France (LNE) and Switzerland (METAS) between January and April 2022 using the analytical methods described in Table B3.
- D.1
- E
Long-term stability assessment. In order to assess the long-term stability of the RGMs, repeated analysis with two to three measurement series were performed 5, 7, 13 and 18 months after preparation. Relative differences between averaged measured values for each period and gravimetric values were used as an indicator of temporal stability. The uncertainty of the RGMs, provided together with the assigned value of the amount fraction of each OVOC, was evaluated after the verification and long-term stability assessment. Preparation and verification uncertainty sources were considered to estimate the uncertainty of the RGMs based on the measurement model proposed in ISO 6142-1:2015 (2015). Regarding the preparation sources, uncertainty from weighing, molar masses (Coplen et al., 2020; van der Veen et al., 2021) and the purity of the materials used was propagated using the law of uncertainty propagation (JCGM 100:2008, 2008).
The uncertainty was evaluated using software made in house based on the work described in Alink and Van Der Veen (2000). Uncertainty sources linked to RGM verification included the repeatability of each series of measurements and the spread among the series of measurements. Student's t test was used to determine whether the mean difference between average analytical observed values and gravimetric values was significant. When significant, the uncertainty due to initial loss was included in the uncertainty evaluation (Eq. 3).
where uc is the combined uncertainty of the amount fraction of the compound, u(prep) is the gravimetric preparation standard uncertainty, u(ver) is the analytical verification standard uncertainty and u(loss) is the standard uncertainty due to initial loss.
An additional term was added to the combined uncertainty of the RGMs sent around for SI working standard assessment to account for potential temporal instabilities during the shipment period. The expanded uncertainty was then calculated as the combined uncertainty multiplied by the coverage factor (k=2).
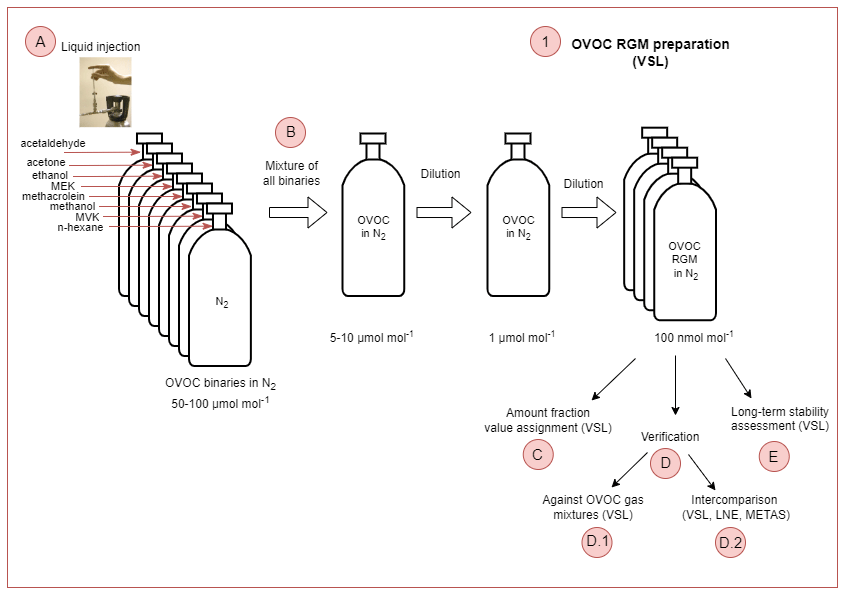
Figure 2Schematic diagram illustrating the steps needed to prepare the reference gas mixtures (RGMs) of the selected oxygenated volatile organic compounds (OVOCs).
2.1.2 RGM dilution
SI-traceable working standards containing OVOCs at atmospheric amount fractions (10 nmol mol−1) were generated by diluting the described RGMs with clean and dry nitrogen using two different dilution systems (Fig. 1). Both dilution systems were warmed up for at least 24 h and flushed with zero gas (i.e. dry high-purity nitrogen) to prevent the presence of water or any other contaminant before the preparation of working standards. The first dilution system was developed by VSL and consisted of one-stage gas dilution with dilution flows ranging from 2–50 L min−1, allowing for dilution ratios up to 1:1000 (Appendix A2). This dilution system was used only during the working standard assessment performed by VSL.
The second dilution system – referred to as VeRDi (Versatile Reactive Gas Diluter) and developed by METAS in collaboration with Swagelok® Switzerland – was a two-stage gas diluter allowing for dilution ratios up to 1:175 000 (Appendix A2). This dilution system was transferred to the institutes assessing the SI-traceable working standards except VSL.
2.2 Preparation of SI-traceable working standards based on certified spiked whole-air samples
The second type of SI-traceable working standards developed consisted of certified whole-air samples that were previously spiked with the selected OVOCs to obtain amount fractions around 10 nmol mol−1. A schematic of the steps given to prepare these SI-traceable working standards is shown in Fig. 3.
- A
Water passivation of the parent cylinders. Two 50 L aluminium cylinders (parent cylinders) were selected and filled with ambient air by the Swiss Federal Laboratories for Materials Science and Technology (Empa). Before filling, both cylinders were evacuated in parallel for 1 h (cylinder pressure <10 hPa) with a membrane pump. Then, to passivate their inner walls with a layer of water to reduce adsorption and surface reactions of the compounds of interest, 0.73 mL of deionised water (Merck Millipore, Germany) was injected individually in each parent cylinder at Empa on 31 March 2021.
- B
OVOC spiking. OVOC spiking was done using a high-pressure cylinder containing an SI-traceable RGM of OVOCs in dry high-pure nitrogen (VSL, the Netherlands) at amount fraction levels between 500 and 1000 nmol mol−1 (Table C1). This SI-traceable RGM was connected to the parent cylinders via a cross connector and a vacuum pump fitted with an on–off valve to isolate the pump from the cylinders. The spiking took place at Empa 3 weeks after the water passivation of the parent cylinders. Both water and OVOC spiking were carried out at room temperature.
- C
Whole-air sampling. One day after the spiking, the two parent cylinders were filled with ambient air at the National Air Pollution Monitoring Network (NABEL) station at Rigi Seebodenalp (ca. 1000 m above sea level; Switzerland) on 22 April 2021. The filling was done using a modified diving compressor (RIX Industries, SA-6). The compressor air inlet was about 2 m above ground and placed upwind of the compressor. Both cylinders were filled in parallel over 3 h to a final pressure of ca. 145 bar. After the sampling and once back in the laboratory, the parent cylinders were stored tilted (ca. 30° inclination) over night with the top facing downward. Then, the two parent cylinders were taken outdoors and stored for another hour at ambient temperature (10 °C) vertically upside down, before the valves were opened to release the liquid water that was potentially formed during the filling. Since the spiking and air filling each took place with the two parent cylinders connected in parallel, it was assumed that OVOC amount fractions in both cylinders were identical (Table C1).
- D
Water passivation of cylinders and canisters. A total of 6 cylinders and 24 canisters (Table C2) were selected for decanting the parent cylinders to produce several identical subsamples (i.e. working standards). Prior to decanting, the working standard cylinders and canisters were spiked with water – following the same procedure described for the parent cylinders – to achieve a 20 % water saturation level.
- E
Filling of cylinders and canisters (decantation). The parent and working standard cylinders, as well as the canisters, were placed in a climate chamber at 40 °C for at least 3 h to ensure thermal equilibration before decanting. The interconnecting tubing was kept as short as possible, and several tanks of the same type were filled simultaneously. After decanting the parent cylinders, the absolute pressure ranges in the working standard cylinders and canisters were 9.9–10.5 and 0.38–0.41 MPa, respectively.
- F
Homogeneity assessment. The homogeneity of the spiked air samples was evaluated before certification (Table C3). For that purpose, seven whole-air samples in different vessel types and the two parent cylinders after decantation were analysed three times using the Empa GC–FID described in Appendix A1. The obtained amount fractions were averaged, and the variations within the same vessel type and among different vessel types were calculated.
- G
Long-term stability assessment. Furthermore, during the certification process, the long-term temporal stability of the whole-air samples in the cylinders was assessed by repeated measurements after 2, 8 and 14 months. Variations due to temporal instability were included in the certified values.
- H
Certification of the spiked whole-air samples. Certification measurements were carried out by VSL and METAS using the two analytical methods described in Table C4 and following the same measurement protocol (Appendix C). Each whole-air sample was analysed at least six times. In total, three series of measurements for whole-air samples in cylinders were performed, but only one measurement series for the samples in canisters was possible due to the limited sample volume. The amount fraction of each compound per whole-air sample was calculated according to Eq. (2). The uncertainty of the assigned amount fraction values included the main uncertainty sources of the sample analysis – such as the spread of the analyser response, background noise, blank issues, potential overlapping of GC peaks and detector drift, among others – and the uncertainty of the analyser calibration (i.e. uncertainty of the RGMs and possible lack of linearity in the measured range: 0–10 nmol mol−1) (Appendix C). The consistency of the assigned amount fraction values for acetone, ethanol, methacrolein, methanol and MVK measured in the same type of vessel was evaluated according to the criterion described by Eq. (4).
where xVSL is the amount fraction value of each OVOC under study assigned by VSL, xMETAS is the amount fraction value of each OVOC under study assigned by METAS, k is the coverage factor (k=2), uVSL is the standard uncertainty of the amount fraction value assigned by VSL according to Eq. (C1) and uMETAS is the standard uncertainty of the amount fraction value assigned by METAS according to Eq. (C1).
Certified reference values for each type of vessel were assigned only when the criterion (Eq. 4) was met for all OVOCs in the same type of vessel. In this case, the certified reference value of each OVOC was the average of VSL- and METAS-assigned values for that compound. The relative uncertainty of the certified reference values was the combined uncertainty of the assigned values provided by VSL and METAS, including the spread of the assigned values due to potential temporal instability (1-year period).
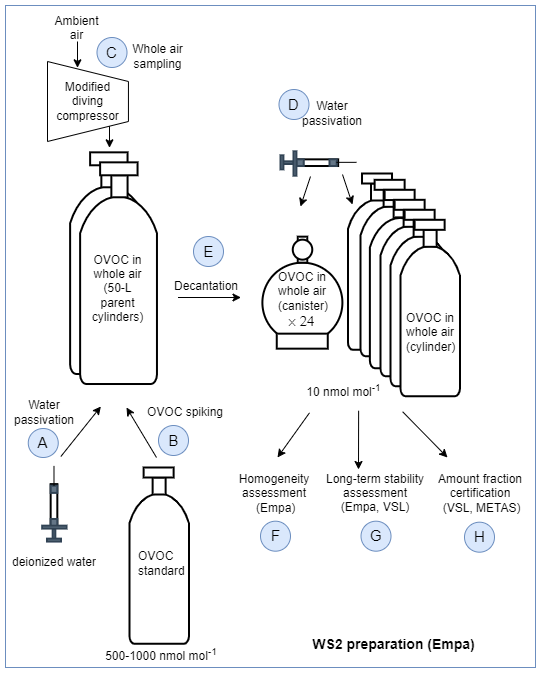
Figure 3Schematic diagram illustrating the steps needed to prepare the SI-traceable working standard based on the certified spiked whole-air sample (WS2).
3.1 Measurement procedure
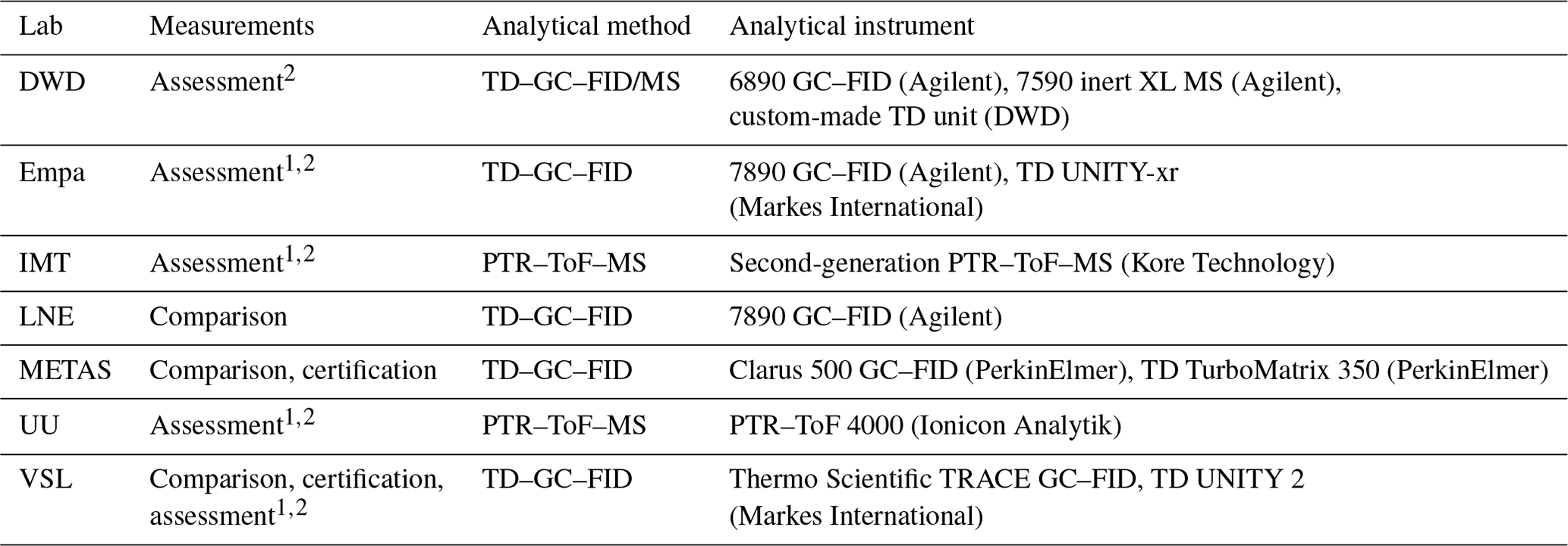
The SI-traceable working standards were assessed by comparing them against in-house working standards (Appendix D1), which were used for routine analyser calibrations by the participants in the assessment (Fig. 1): Deutscher Wetterdienst (DWD), Empa, Institute Mines-Télécom (IMT), METAS, Utrecht University (UU) and VSL (Table 1). For that purpose, the SI-traceable working standards were treated as samples and analysed following the same procedure as for the analyser calibration. The detailed analytical method, calibration standards and measurement procedure to assess both types of SI-traceable working standards are described in Appendix D.
Table 1Information on the assessment of the working standards (WSs) based on the dilution of RGMs with dry nitrogen (WS1) and on the certified spiked whole-air sample (WS2). ECN refers to the effective carbon number. Detailed information on WS2 samples is shown in Table C2. _cyl: cylinder, _can: canister.
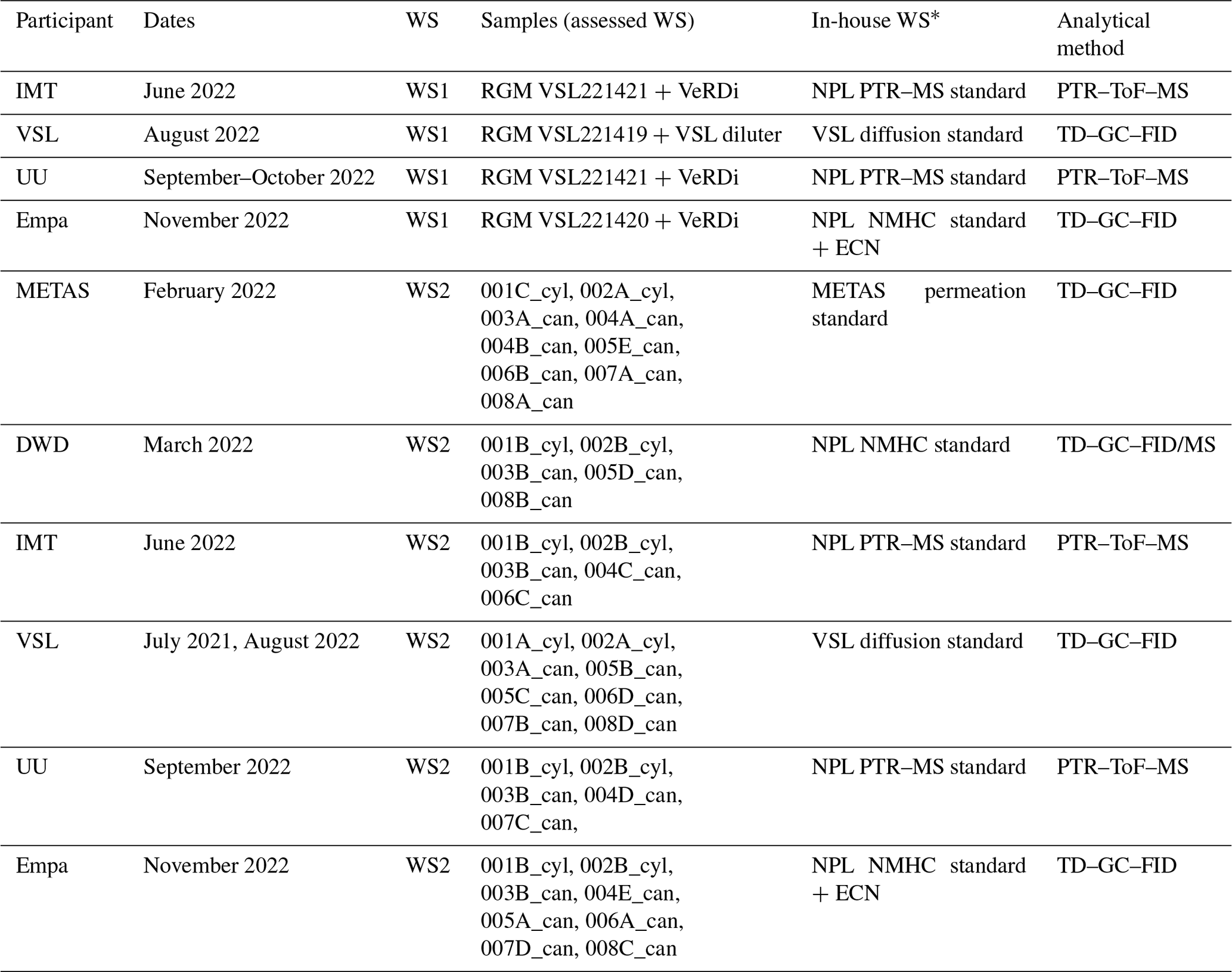
* All the in-house working standards were SI-traceable except for the effective carbon number (ECN).
To assess the SI working standards based on certified spiked whole-air samples, the same air sample cylinders were measured by the participants in the round-robin comparison (Table 1). However, different canisters were sent to the participants because of the low sample volume, which was enough only for one analysis (Table C2).
3.2 Working standard amount fractions and uncertainty
3.2.1 Measured amount fractions and uncertainties
The measured amount fractions of the SI-traceable working standards were calculated using different equations depending on the analytical method and the calibration standard used.
VSL estimated the amount fractions of the SI-traceable working standards based on the dilution of RGMs with dry nitrogen according to Eq. (2), using only the last five measurements for the calculations. Uncertainty of these measured amount fractions was calculated following Eq. (C1).
DWD and Empa followed ACTRIS procedures to estimate the measured OVOC amount fractions and their uncertainties (Reimann et al., 2018). The main uncertainty sources considered by DWD and Empa were the reproducibility of the measurement method (i.e. standard deviation of the multiple measurements of the sample), measurements close to limit of detection and the uncertainty of the in-house working standard (i.e. calibration standard). Sources linked to the uncertainty of the instrument (peak integration uncertainty due to peak overlay, tailing and/or bad peak separation, sampling line artefacts, carryover, and changes in split flow rates) were considered in the standard deviation of the multiple calibration measurements. For OVOCs that were not present in the NPL NMHC standard (Grenfell et al., 2010), Empa used the effective carbon number (ECN; e.g. Sternberg et al., 1962; Apel et al., 1998; Faiola et al., 2012). This assessment procedure led to measurement results that are not metrologically traceable. In addition to the sources of uncertainty described above for DWD and Empa, other uncertainties considered in this approach were the mean relative deviation of the NPL NMHC standard certified uncertainties in the six compounds (ethane, ethene, propane, propene, isobutane and butane) contributing to the carbon response factor (CRF) and the relative standard deviation of the six calculated CRFs.
IMT estimated the amount fractions of the selected OVOCs according to the calibration approach described in de Gouw and Warneke (2007). The combined measurement uncertainty (u(xi)) was calculated as the square root of the sum of quadrats of each relative uncertainty term (Appendix D4). Sources of uncertainty associated with the measured amount fractions included precision of the system and calibration accuracy.
UU followed the method described in Holzinger et al. (2019) to estimate the OVOC amount fractions. The uncertainty of the measured amount fractions was given as the standard deviation of four to six repetitions of the same measurement type.
3.2.2 Assigned amount fractions and uncertainty
For the SI-traceable working standards based on the dilution of RGMs with dry nitrogen, the assigned amount fraction of each sample was estimated according to Eq. (5).
where xth is the assigned amount fraction of the generated SI-traceable working standard (in nmol mol−1), xRGM is the amount fraction of the OVOC under study in the diluted VSL RGM (in nmol mol−1), xres is the amount fraction of the OVOC under study present as a residual in the dilution gas (in nmol mol−1), is the flow rate of the VSL RGM (in mL min−1) and is the flow rate of the dilution gas (in mL min−1).
The uncertainty of the assigned values was calculated following the law of uncertainty propagation (JCGM 100:2008, 2008) according to Eq. (6). Calculations were done using GUM Workbench Professional version 2.4.1.406 (Metrodata GmbH, Germany).
where u(xth) is the uncertainty of the assigned amount fraction of the generated SI-traceable working standard, u(xRGM) is the uncertainty of the VSL RGM used in the comparison (provided in the calibration certificate according to Eq. 3), is the uncertainty of the VSL RGM flow rate, is the uncertainty of the dilution gas flow rate, u(xres) is the uncertainty due to the presence of the compound under study in the dilution and matrix gas as impurity, c1 is the sensitivity coefficient given by the partial derivative of xth with respect to xRGM, c2 is the sensitivity coefficient given by the partial derivative of xth with respect to , c3 is the sensitivity coefficient given by the partial derivative of xth with respect to xres, and c4 is the sensitivity coefficient given by the partial derivative of xth with respect to .
Assigned amount fractions and uncertainty of the working standards based on certified spiked whole-air samples were estimated following the procedure described in Appendix C. The relative expanded uncertainty of the certified reference values was 2 times the combined uncertainty of the assigned values provided by VSL and METAS, including the spread of the assigned values due to potential temporal instability (1-year period) (Eq. C1).
3.2.3 Relative differences between working standards
The assessment of the SI-traceable working standards based on the dilution of RGMs with dry nitrogen was done by calculating the relative difference between the measured and assigned amount fractions described above, while for the SI-traceable working standards based on certified spiked whole-air samples, the relative difference between the measured and the certified amount fractions was calculated.
The expanded uncertainty of each assessment was calculated as 2 times the combined uncertainty (udiff) between the uncertainty of the assigned (Table D1) or certified (Table 3) amount fraction (u(xth)) and the uncertainty of the measured amount fraction u(xi) reported by the participants (Tables D2 and D3) for each compound (Eq. 7).
Results regarding the amount fraction assignment and verification of the RGMs used to generate the SI-traceable working standards based on the dilution of RGMs and of the assessment of these working standards are shown in this section, together with the certification and assessment results of the SI-traceable working standards based on certified spiked whole-air samples.
4.1 Results of the SI-traceable working standards based on the dilution of RGMs
4.1.1 RGM amount fraction assignment, verification and stability evaluation
RGM amount fractions were assigned gravimetrically, taking into consideration the purity of the liquid chemicals injected into the cylinders and results from the mass weighing during the preparation. Results showed purity values >99.9 % for all the liquid compounds, except for methacrolein (98.5 %) and MVK (94.0 %). Water was a common impurity in all the liquid compounds. For methacrolein, MVK and MEK, other organic impurities were found (Table B1). Values of the assigned gravimetric amount fractions ranged between 98 and 105 nmol mol−1 with expanded uncertainties in the preparation ≤5 % (coverage factor k=2) in general (Table 2). However, greater uncertainties were calculated for methanol (5.3 % in VSL221419 and 6.8 % in VSL221420), acetaldehyde (9.6 % in VSL221420 and 9.5 % in VSL221421) and MVK (5.8 % in VSL221421) to take into account initial losses and potential instability of these compounds in the cylinders.
Table 2Gravimetric assigned amount fraction values (xi) for the reference gas mixtures (RGMs) and their expanded uncertainty (U) with a coverage factor of 2 (k=2).
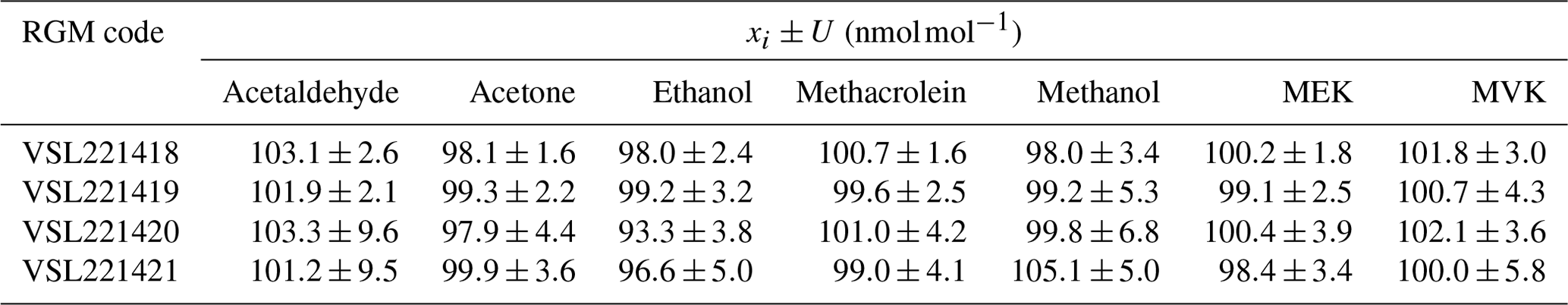
Results from the verification analysis (Table B2), where the prepared RGMs were compared against dynamically generated gas mixtures, showed similar relative differences between analytical and gravimetric values for acetone in the four cylinders (average difference %). These results, similar to the relative differences found for the internal standard (n-hexane), suggest that surface effects (i.e. adsorption losses) were negligible for both compounds. For MEK, the analytical values were also greater than the gravimetric ones and quite similar among different cylinders (average difference < +3.2 %). Analytical values lower than gravimetric ones were found for acetaldehyde, methacrolein and MVK. Average differences were < +2 % and similar among different cylinders for acetaldehyde and methacrolein, which suggests minimal or even negligible adsorption effects with the cylinder wall. The difference was higher for MVK (between −2.5 % and 3.7 %), which might be explained not only by surface effects but also by isomerisation reactions. All the relative differences were within the expanded uncertainty of the verification analysis. The relative differences for ethanol were around −5 %. Compound loss after preparation due to surface effects might explain these differences. Initial losses were also suggested by the great heterogeneity among cylinders for methanol (relative difference between −5.2 % and +3.1 %) as described in Persijn and Baldan (2023).
During the interlaboratory comparison organised as part of the RGM verification process (Appendix B2.2), the three participant laboratories (VSL, METAS and LNE) measured acetone, ethanol and methanol. Results demonstrated very good comparability and a degree of equivalence for acetone (Fig. 4). For methanol, as well as for ethanol, good agreement among laboratories was also found (Fig. B1). However, due to the great expanded uncertainty (37 %) of the ethanol measurement associated with METAS analytical issues, these results were not considered. It can be noted that although different calibration and analytical methods were used, the measurement results of the RGMs were aligned, giving confidence regarding the quality of the work.
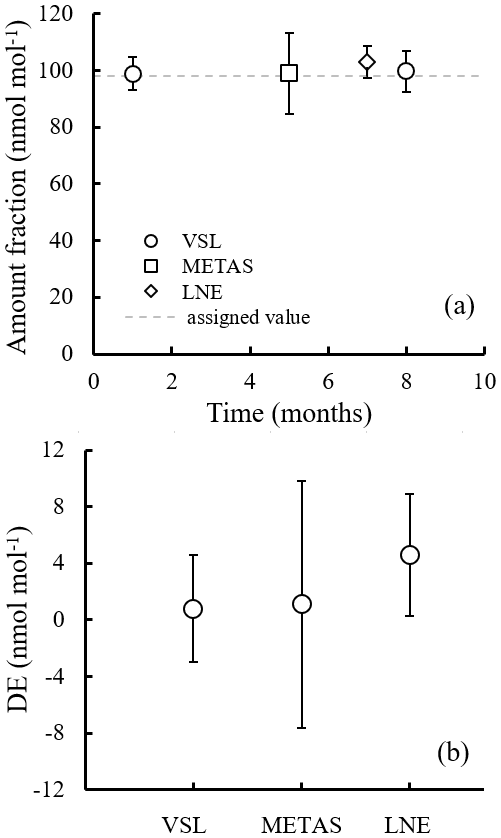
Figure 4Interlaboratory comparison results for (a) acetone and (b) its degree of equivalence (DE; i.e. the deviation of each laboratory from the reference value). For VSL, only the first measurement period was considered (month 1) to estimate the DE. The measured amount fractions reported by the laboratories were the average of five measurements, except for month 1 results, which were the average of three measurements. Error bars show the expanded uncertainty of the measurements (coverage factor k=2). The dashed line indicates the gravimetric amount fraction of the compound.
Long-term stability results (Table B4) suggested very good stability (i.e. relative differences between analytical and gravimetric values < ±5 %) for acetone with relative differences ≤ +2 % even 13–14 months after RGM preparation, although a questionable result (−4.7 %) was obtained at a stability testing period of 18–19 months. Acetone results were similar to those for the internal standard (n-hexane). Good stability was also found for methacrolein. After initial relative differences of ca. −1.5 %, positive values around +0.7 % were found 7–8 months after preparation. The positive values increased up to 3.4 %–3.7 % during the last stability period (18–19 months). MVK and MEK showed, respectively, fluctuating positive (up to +5.7 %) and negative (up to −6.4 %) relative differences most likely due to analytical issues, isomerisation reactions and/or surface effects. Ethanol showed a negative relative difference which remained within the ±5 % threshold, except for one of the measurement results obtained at months 18–19 (−5.1 %). Acetaldehyde and methanol long-term stability had the largest biases. Varying relative differences > ±5 % (positive for acetaldehyde and negative for methanol) were already found after 5–6 months after preparation, which could be explained by analytical issues, matrix effects and initial compound losses due to adsorption effects.
4.1.2 Assessment of SI-traceable working standards based on the dilution of RGMs
The assessment of the SI-traceable working standards based on the dilution of RGMs with dry nitrogen took place over a long period of time (ca. 6 months between the first and last participants). Potential temporal instabilities were considered when comparing results through the certified expanded uncertainty provided with the RGMs (Table 2). The long-term RGM stability of each compound was evaluated and taken into account as an uncertainty term (Table B4). The type of in-house standard, sampling method and analytical instrument used, as well as the amount fraction level of the samples generated, were most likely the parameters explaining the differences found between VSL and the other participants for certain compounds, such as MEK (Fig. 5) and methanol (Fig. 6).
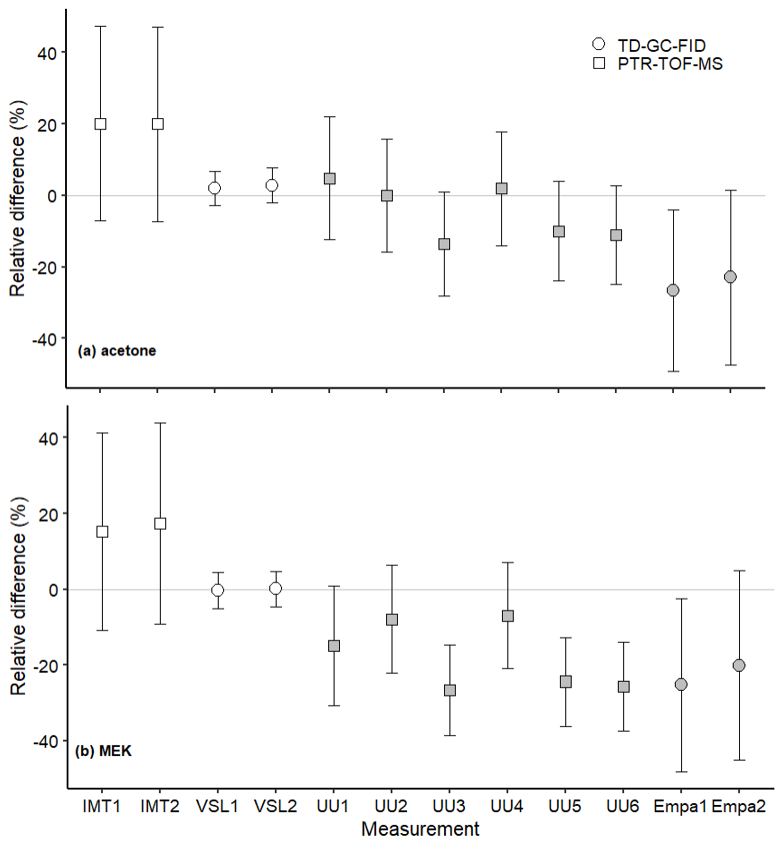
Figure 5Assessment of the SI-traceable working standards based on the dilution of reference gas mixtures with dry nitrogen for (a) acetone and (b) MEK at amount fractions <5 nmol mol−1 (grey symbols) and between 10 and 17 nmol mol−1 (white symbols). Error bars indicate the expanded uncertainty (coverage factor k=2) of the relative difference between in-house and dilution working standards. Measurement labels show the participant and the number of SI-traceable working standards generated by dilution. Measurements were performed in July 2022 (IMT1, IMT2), August 2022 (VSL1, VSL2), September 2022 (UU1–UU6) and November 2022 (Empa1, Empa2).
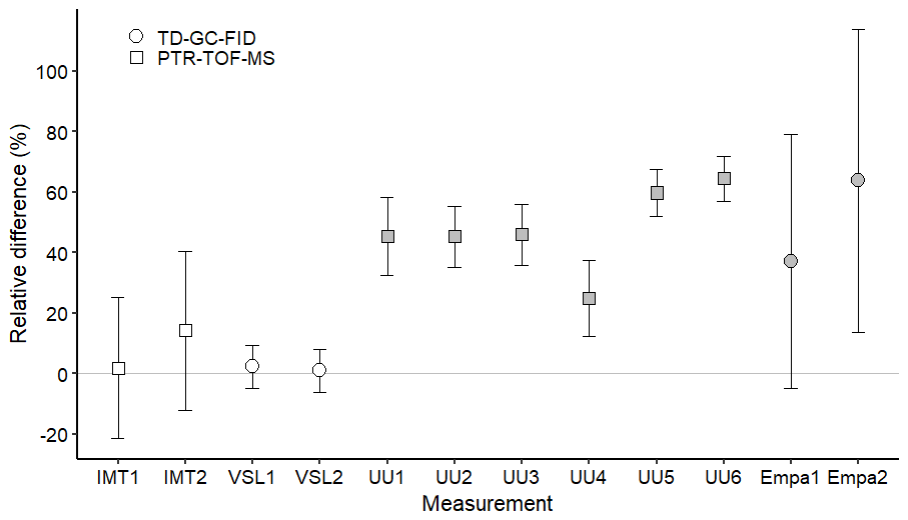
Figure 6Assessment of the SI-traceable working standards based on the dilution of reference gas mixtures with dry nitrogen for methanol at amount fractions <5 nmol mol−1 (grey symbols) and between 10 and 17 nmol mol−1 (white symbols). Error bars indicate the expanded uncertainty (coverage factor k=2) of the relative difference between in-house and dilution working standards. Measurement labels show the participant and the number of SI-traceable working standards generated by dilution. Measurements were performed in July 2022 (IMT1, IMT2), August 2022 (VSL1, VSL2), September 2022 (UU1–UU6) and November 2022 (Empa1, Empa2).
Despite relative differences around ±20 % for IMT and Empa, a good agreement between assigned and analytical values (i.e. relative difference around 0 considering the uncertainty of the difference) was found for acetone, even at amount fractions <5 nmol mol−1 (Fig. 5). This agreement demonstrated the reliability of the dilution systems, RGMs and calibration methods. The great relative differences obtained by Empa for acetone were explained by technical issues with the analytical method (i.e. a leak in the heated valve and flow overshooting when measuring with the Stirling cooling unit). The error was estimated to be around ±30 % and was included in the uncertainty budget. These issues also affected Empa MEK and methanol measurements. Therefore, care should be taken in the interpretation of these results.
Results similar to those of acetone working standards were obtained for MEK at amount fractions around 10 nmol mol−1 (Fig. 5). At levels of lower amount fractions (<5 nmol mol−1), some of the measurements showed analytical fraction values lower than the assigned ones.
Methanol relative differences were relatively small (1 %–14 %) and within the uncertainty range at amount fractions between 10 and 17 nmol mol−1 (Fig. 6). However, at lower amount fractions (<5 nmol mol−1) relative differences were between 25 % and 65 %, suggesting an overestimation of the analytical amount fraction values most likely due to artefacts in the analytical system. Moreover, the temporal instability of methanol within the gas cylinder, with an increase in the amount fraction observed during the first year after preparation for one of the RGMs, might explain part of the overestimation. Methanol instability in gas cylinders was observed in other works (Persijn and Baldan, 2023; Rhoderick et al., 2019). Methanol assessment results suggest, thus, that this OVOC remains a challenging compound to measure.
Measured and assigned acetaldehyde amount fractions showed relatively good agreement; i.e. all the differences were within the uncertainty range (Fig. 7). However, these results must be taken with care because of the large uncertainties. Reactions in the gas cylinders and/or artefacts of the analytical methods might have contributed to analytical amount fractions greater than the theoretical values for acetaldehyde, as well as to uncertainties greater than for the other OVOCs.
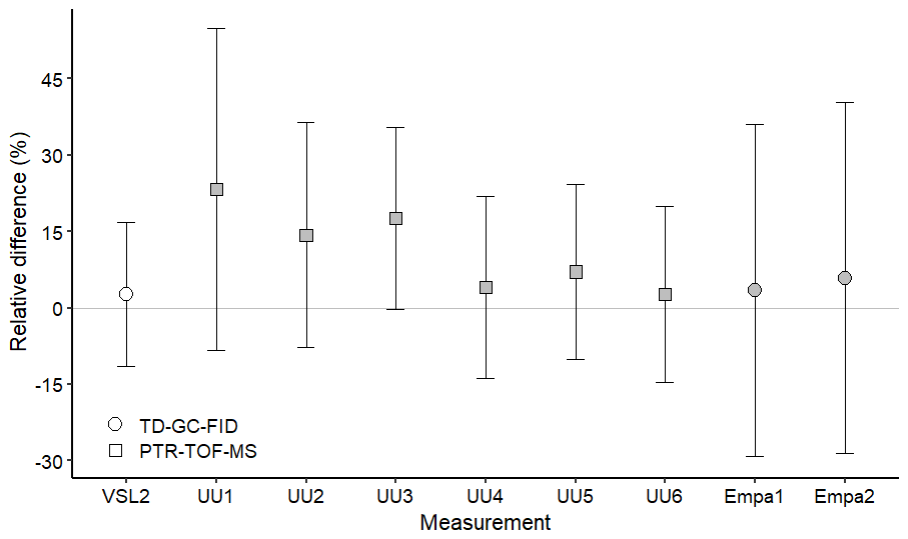
Figure 7Assessment of the SI-traceable working standards based on the dilution of reference gas mixtures with dry nitrogen for acetaldehyde at amount fractions <5 nmol mol−1 (grey symbols) and between 10 and 17 nmol mol−1 (white symbols). Error bars indicate the expanded uncertainty (coverage factor k=2) of the relative difference between in-house and dilution working standards. Measurement labels show the participant and the number of SI-traceable working standards generated by dilution. Measurements were performed in August 2022 (VSL2), September 2022 (UU1–UU6) and November 2022 (Empa1, Empa2).
Assessment results for amount fraction levels around 10 nmol mol−1 suggest that SI-traceable working standards based on the dilution of RGMs can be used as a calibration standard at monitoring stations for key OVOCs, such as acetone, MEK, methanol and acetaldehyde. However, for lower amount fractions (<5 nmol mol−1), suitability of the SI-traceable working standards for MEK, methanol and acetaldehyde is also questionable. The different analytical methods used, the calibration procedure followed and the dilution factors applied to measure and prepare the SI-traceable working standards contributed to that large uncertainty and dispersion of results. Further research where the same methodology is followed – same calibration procedure (e.g. same in-house working standard) and assessment protocol (e.g. setting the same dilution factors to generate SI-traceable working standards at the same amount fractions) – may reduce both uncertainty and dispersion and help to draw conclusions. Moreover, using coated (e.g. SilcoNert 2000) lines – as short as possible – and a low-dead-volume pressure reducer, as well as flushing for a long period of time and performing repeated measurements to guarantee the stability of the analyser and diluter, may reduce the uncertainty of the generated working standards. Even if results are not conclusive, the low RGM uncertainty (<5 %) and long temporal stability (at least up to 18 months after preparation) are promising for providing atmospheric monitoring stations with SI-traceable, accurate OVOC working standards at a very low amount fraction.
4.2 Results of the SI-traceable working standards based on certified spiked whole-air samples
4.2.1 Homogeneity assessment, stability evaluation and amount fraction certification of the spiked whole-air samples
Results of the analysis of variance (ANOVA) performed on the data from the homogeneity test of the subset of vessels filled with the spiked whole-air samples (Table C3) showed good homogeneity (variation <5 %) within the same vessel type for all selected OVOCs. The greatest variation was found for methanol (+3.2 %). For the rest of the OVOCs, the variation was % (e.g. +0.6 % for acetone, +0.9 % for MVK, +1.2 % for MEK, and +1.5 % for acetaldehyde and ethanol). Variation among different vessel types suggested that the vessel material may play a role in the lack of homogeneity particularly for methanol (+22.6 %) and ethanol (+9.7 %). Variation was relatively great also for acetaldehyde (+6.6 %), MEK (+6.6 %) and MVK (+7.0 %). However, good homogeneity was found for acetone (+2.8 %) and toluene (+2.4 %). Although toluene is not an OVOC and, thus, was not spiked into the whole-air sample vessels, the compound was naturally present in the ambient air.
Temporal stability of the air samples was evaluated by Empa considering the ratio between each OVOC and the internal standard (i.e. n-hexane). Ratios of acetone to n-hexane showed good temporal stability (i.e. differences in ratio values among measurements within the uncertainty of the measurement) during the measuring period from August 2021 to September 2022. Except for the uncertainties that were greater, similar results were found for other compounds (methanol, ethanol, acetaldehyde, MVK and MEK). Because the ratio differences observed were within the uncertainty of the measurements and the homogeneity among vessels of the same type was good (variation % except for methanol (+3.2 %)), air samples in the same type of vessel were considered stable.
Certification results obtained for whole-air samples contained in pressurised 10 L aluminium cylinders showed good consistency between the two laboratories performing the certification (i.e. VSL, METAS), with exception of MVK (criterion was not met, Eq. 4; Table C5, Fig. C1). Regarding the other type of pressurised cylinders (3.6 L stainless steel, coated with SilcoNert®), the criterion was not met for MVK. For methanol, the criterion was met only when METAS results were compared against the results obtained for the first measurements performed by VSL (i.e. July 2021). Certified OVOC amount fractions in both cylinders ranged between 7.6 (ethanol) and 17.3 nmol mol−1 (acetone) with expanded uncertainties (k=2) ≤2.6 nmol mol−1 (Table 3). The smallest uncertainties were found for methacrolein and acetone (≤1.5 nmol mol−1). Amount fractions were in line with the estimated spiked values (Table C1) suggesting that, except for acetone, the amount fractions of the selected OVOCs in the sampled air were not significant (close to 0). The higher amount fractions measured for acetone compared to the spiked estimated amount fractions suggested acetone background levels in the sampled whole air of around 6.5 nmol mol−1.
Table 3Certified amount fraction values (x) and their expanded uncertainty (U; coverage factor k=2) estimated for the air samples filled in high-pressure cylinders: 10 L cylinder (MVOC151-001) and 3.6 L (MVOC151-002).
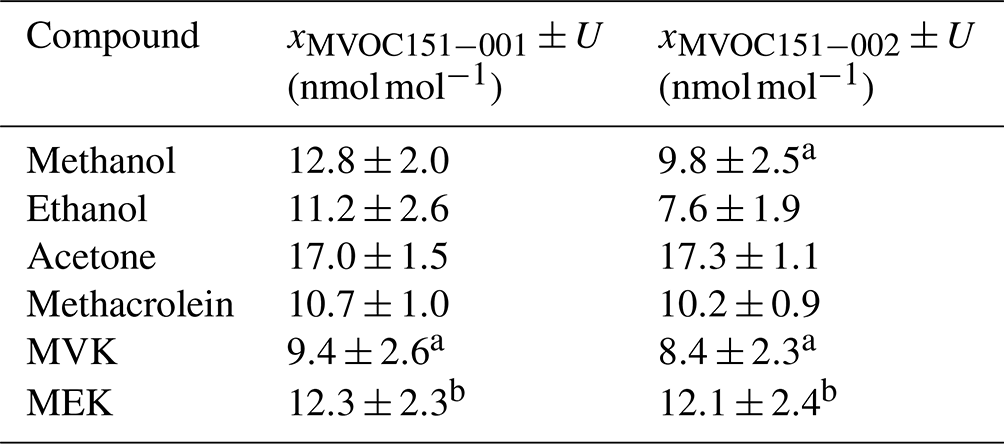
a No compliance with evaluation criterion described in Eq. (4). b Measurement carried out by only one of the laboratories.
Results of the low-pressure canisters were less consistent: the criterion was only met for methacrolein for four canisters (Table C5). For methanol and acetone, the criterion was only met in two canisters. The discrepancy between results for the 15 L canister suggested homogeneity issues for this batch.
4.2.2 Assessment of SI-traceable working standards based on certified spiked whole-air samples
Amount fractions of the OVOCs measured in air samples showed good agreement (Figs. 8–10) among partners. These values were comparable to the certified amount fractions for whole-air samples in cylinders (pressurised at 9.8–10.5 MPa). Only for methanol (Fig. 10) were values more discrepant. Empa results, as for the SI-traceable working standards based on the dilution of RGMs with dry nitrogen, must be interpreted with caution because of the technical issues with the analytical system.
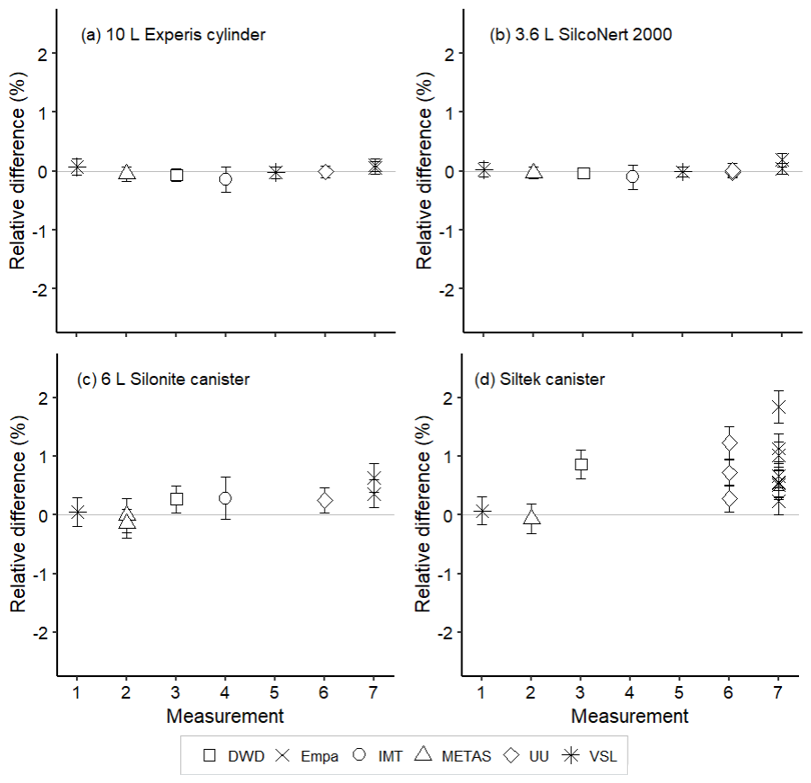
Figure 8Assessment of the SI-traceable working standards based on certified spiked whole-air samples for acetone in (a) 10 L Experis® aluminium cylinders, (b) 3.6 L SilcoNert® 2000 stainless steel cylinders, (c) 6 L Silonite™ stainless steel canisters and (d) 6 L Siltek® stainless steel canisters. Error bars indicate the expanded uncertainty (coverage factor k=2) of the relative difference between the measured and the certified amount fraction values of the working standards. Measurements were performed in July 2021 (1), February 2022 (2), March 2022 (3), June 2022 (4), August 2022 (5), September 2022 (6) and November 2022 (7).
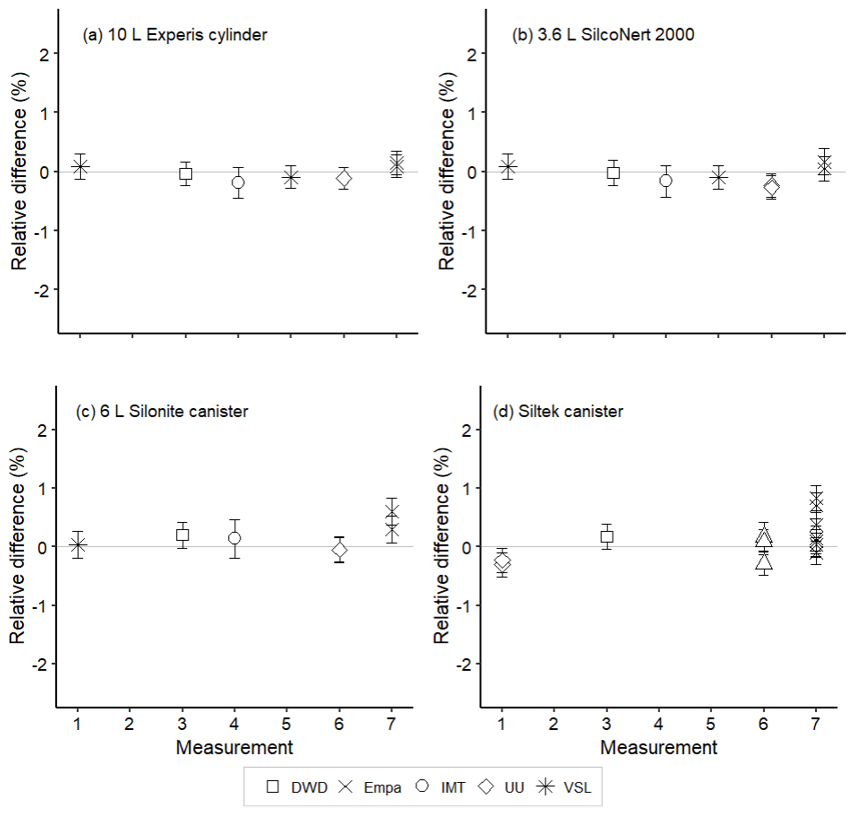
Figure 9Assessment of the SI-traceable working standards based on certified spiked whole-air samples for methyl ethyl ketone (MEK) in (a) 10 L Experis® aluminium cylinders, (b) 3.6 L SilcoNert® 2000 stainless steel cylinders, (c) 6 L Silonite™ stainless steel canisters and (d) 6 L Siltek® stainless steel canisters. Error bars indicate the expanded uncertainty (coverage factor k=2) of the relative difference between the measured and the certified amount fraction values of the working standards. Measurements were performed in July 2021 (1), February 2022 (2), March 2022 (3), June 2022 (4), August 2022 (5), September 2022 (6) and November 2022 (7).
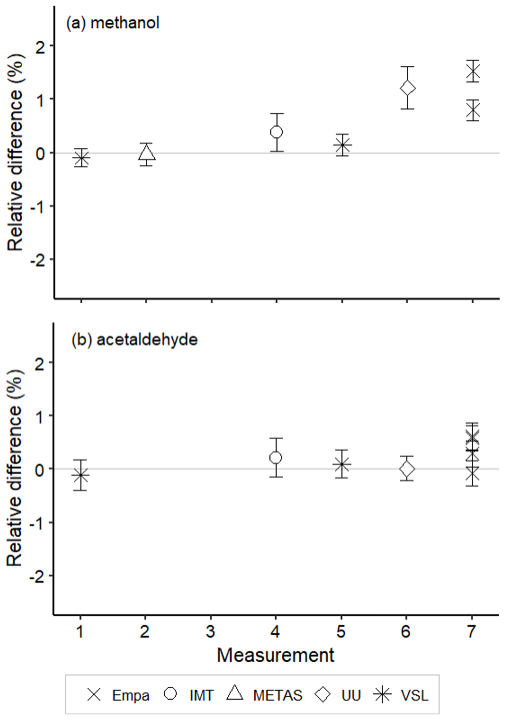
Figure 10Assessment of the SI-traceable working standards based on certified spiked whole-air samples in 10 L Experis® aluminium cylinders for (a) methanol and (b) acetaldehyde. Error bars indicate the expanded uncertainty (coverage factor k=2) of the relative difference between the measured and the certified amount fraction values of the working standards. Measurements were performed in July 2021 (1), February 2022 (2), March 2022 (3), June 2022 (4), August 2022 (5), September 2022 (6) and November 2022 (7).
For whole-air samples in canisters (pressurised at 0.35 MPa), results were quite heterogeneous. Relatively good results were found for acetone (Fig. 8) and MEK (Fig. 9) in the Silonite™ stainless steel canisters. However, for methanol and acetaldehyde, disagreement was found both among most of the participants and with the certified values. Lack of agreement was also observed for air samples in the Siltek® stainless steel canisters. Even if the same cleaning procedure was followed by both type of canisters before filling, the history (i.e. previous fillings) of the Siltek® stainless steel canisters and/or the surface treatment could explain the differences between canister types. History and surface treatment effects on VOC amount fractions have been reported in previous works (e.g. Rhoderick et al., 2019; Persijn and Baldan, 2023). Furthermore, vessel pressure might explain the differences in the agreement of results between cylinders and canisters. Gas pressure effects on the stability of gas mixtures in cylinders have been observed for different compounds, such as CO2 (e.g. Leuenberger et al., 2015; Miller et al., 2015). In these studies, after an initial wall adsorption when the cylinders were filled, desorption took place. This adsorption–desorption process resulted in increasing amount fractions. In Silonite™ canisters, the treatment might have contributed to a lower initial wall adsorption compared to the Siltek® canisters and, therefore, to the lower discrepancies.
Assessment results suggest that certified spiked whole-air samples at a low amount fraction (<20 nmol mol−1) in compressed gas cylinders may be used as SI-traceable working standards for most of the selected OVOCs, except for methanol, at monitoring stations. Using the same matrix gas as the ambient air monitored at atmospheric stations may improve the accuracy of the observations by reducing artefacts and other effects related to the matrix gas.
VOCs are one of the major tropospheric ozone precursors. Despite the importance of performing accurate and comparable VOC measurements to assess tropospheric ozone burdens and trends, several challenges regarding VOC monitoring remain currently open. The lack of stable and SI-traceable gas reference materials for many OVOCs at ambient levels and adapted to constraints of monitoring stations (e.g. limited dilution gas supply) represents some examples of these challenges.
This research has shown that producing SI-traceable RGMs at amount fractions around 100 nmol mol−1, with expanded uncertainties in the preparation of <5 % and temporal stability of at least 14 months, is doable for acetone, methacrolein, MEK, MVK and to some extent ethanol. However, for methanol and acetaldehyde, further research is needed to find suitable cylinder materials and optimal preparation and analytical procedures (e.g. cylinder wall passivation) to minimise surface adsorption and reaction effects, which greatly contributed to the temporal instability of RGMs for both compounds. These stable and accurate RGMs are produced at amount fraction levels greater than ambient levels of the selected OVOCs (i.e. 4–10 nmol mol−1). RGM dilution is thus needed to achieve the amount fraction range required by monitoring stations. To guarantee that SI traceability is maintained, the dilution needs to be done by a dilution system that is traceable. For that purpose, the elements of the dilution system (e.g. thermal mass flow controller) have to be calibrated against traceable flow standards by NMIs and/or accredited calibration laboratories. Moreover, to reduce the uncertainty of the dilution associated with surface effects as much as possible, the components in contact with the RGM should be coated (e.g. SilcoNert® 2000), low-dead-volume pressure reducers should be used, and enough time for reaching stability of the dilution and analytical systems should be recommended. The procedure and recommendations described correspond to the SI-traceable working standards based on RGMs diluted with dry nitrogen described in this work, which can be generated at amount fractions around 10 nmol mol−1 with acceptable relative expanded uncertainties (coverage factor k=2) <10 % (for acetone and MEK, the expanded uncertainty is even lower than 4 %). This first type of SI-traceable working standards seems to be suitable for the calibration of acetone, MEK, methanol and (with larger uncertainties) acetaldehyde at monitoring stations, guaranteeing comparability of the VOC measurements within and among monitoring stations.
Different vessel types were filled with the second type of SI-traceable working standards based on certified whole-air samples: high-pressure (>9.5 MPa) cylinders with different treatments (Experis® and SilcoNert® 2000) and low-pressure (<0.45 MPa) canisters with two different coatings (Silonite™ and Siltek®). Assessment results suggest that certified spiked whole-air samples filled into high-pressure cylinders at amount fractions around 10 nmol mol−1, valid for 12–14 months, might become a valid alternative for calibrating analytical systems measuring acetone, acetaldehyde and MEK at monitoring stations. Even if VOC RGMs in nitrogen are more stable, this second type of SI-traceable working standards will allow for monitoring stations to calibrate their instruments with standards that use a matrix gas similar to the ambient air analysed. Matrix gas effects on the analytical systems are not fully understand yet, but these working standards might provide some insight into the topic. Before going forward with this option, in addition to matrix gas effects on the analytical systems, water passivation and vessel wall effects on the stability of the OVOC amount fractions of these working standards should be explored. Although results of this research suggest that stability might be material dependent, the observed differences might be due to other factors, such as pressure and volume differences among vessels. Specific experiments using new vessels of the same volume and pressure (i.e. vessels that were not previously used) should be designed to find the vessel material performing the best.
Despite these promising findings, conclusions must be driven with caution because of the large values and the broad range obtained for the measurement uncertainties (i.e. 5 %–31 %; coverage factor k=2). Moreover, for both types of working standards, methanol calibration remains challenging.
The RGMs and working standards described in this work are a first step in fulfilling the remaining needs of VOC monitoring. Through an active collaboration among the metrological, meteorological and atmospheric chemistry monitoring communities, harmonisation and comparability among monitoring stations will be promoted (e.g. by estimating uncertainty budgets that are common to the different monitoring programmes). Moreover, this collaboration might provide a better understanding of the impact that pressure, the sampling material, moisture and the matrix have on the preparation of RGMs and working standards. This knowledge may contribute, thus, to improving calibration standards (i.e. RGMs and SI-traceable working standards) and uncertainties in VOC measurements. Furthermore, other research applications, such as modelling and remote sensing, might benefit from the transfer of SI traceability to monitoring stations.
A1 Analytical instruments
A thermal desorber–gas chromatograph–flame ionisation detector (TD–GC–FID) and proton transfer reaction–time of flight–mass spectrometer (PTR–ToF–MS) were the two selected analytical methods in this work. The specific analytical instruments used by the laboratories are summarised in Table A1.
Table A1Information on the analytical instruments used in this work.
1 Assessment SI-traceable working standards based on the dilution of reference gas mixtures (RGMs) with dry nitrogen. 2 Assessment SI-traceable working standards based on certified spiked whole-air samples.
A1.1 DWD (Deutscher Wetterdienst)
DWD deployed a GC–FID/MS system (6890, 7590 inert XL MS, Agilent Technologies Inc., CA, USA), which was coupled to a custom-made sample preconcentration unit that included sampling valves, sampling ports and the preconcentration trap in a box heated to 150 °C. Materials in the sampling path were mainly treated stainless steel or capillaries. Samples were preconcentrated on multibed sorbent tubes (Tenax TA (mesh 60/80), Carbopack X (mesh 40/60) and Carboxen 695 (mesh 20/45) in a in. glass tube, Merck KGaA (Supelco), MO, USA) at 30 °C with a sampling flow of 80 mL min−1. Desorption to a cryo-focus trap (inert capillary cooled to −180 °C) took place at 200 °C with a flow of 10 mL min−1. After heating the cryo-focus to 60 °C, the sample was injected in a splitless fashion onto a BPX5 capillary column (50 m length, 0.32 mm internal diameter, 0.5 µm film thickness, Trajan Scientific and Medical (SGE), Australia). The GC oven was held at 13 °C for 18 min. Then, the oven temperature was increased up to 240 °C at a rate of 6 °C min−1. Hydrogen (H2 5.0 from Air Liquide, France) cleaned using a gas filter (Super Clean gas filter, Restek, PA, USA) was used as a carrier gas at 3.5 mL min−1. Following the separation on the column, the carrier gas flow was split onto the MS and the FID in parallel. For the analysis of the SI-traceable working standards based on spiked whole air, the MS detector was used to achieve sufficient peak separation.
A1.2 Empa (Swiss Federal Laboratories for Materials Science and Technology)
Empa used a GC–FID (7890, Agilent Technologies Inc., CA, USA) coupled to a UNITY-xr (Markes International Ltd., UK) thermal desorber to evaluate the stability and homogeneity of the air samples and to assess the SI-traceable OVOC working standards (Table A1). Samples went through an in-house dehumidifier – consisting of a Stirling cooler (set to −42 °C) and two insulated in-line glass fingers – before sampling, which was done using a UNITY-Air Server (Markes International Ltd., UK) equipped with three ports. From the UNITY-Air Server, samples passed to the thermal desorber, which collected and concentrated the OVOCs under study. The UNITY cold-trap (ozone precursors, cold trap, U-T17O3P-S2; Markes International Ltd., UK) temperature was set to −29 °C before the cold trap was heated up to 250 °C. The two capillary columns were OxyPlot (30 m length, 0.53 mm internal diameter and 10 µm film thickness; Agilent Technologies Inc., CA, USA) and Al2O3 HP-PLOT (50 m length, 0.53 mm internal diameter and 10 µm film thickness; Agilent Technologies Inc., CA, USA). The sample flow was set at 15 mL min−1 for 20 min. The GC oven was held at 40 °C for 3.25 min and then heated up to 200 °C with a temperature ramp of 7 °C min−1. The GC oven was held at 200 °C for 20 min. The carrier gas was helium, which was set at 5 mL min−1 for 20 min and then increased at 25 mL min−1 for 26 min.
A1.3 IMT (Institute Mines-Télécom)
IMT performed the assessment of SI-traceable working standards using a second-generation PTR–ToF–MS (Kore Technology Ltd., UK) (Table A1). Sampling was done through a SilcoNert® 1000 heated line at a flow rate of 200 mL min−1. An in-house system of solenoid valves was coupled to the PTR–ToF–MS to switch automatically between samples and zero air. The measurement time resolution was set to 10 s.
A1.4 LNE (Laboratoire National de Métrologie et d'Essais; NMI of France)
LNE used a GC–FID (7890, Agilent Technologies Inc., CA, USA), equipped with an on-column preconcentration system, during the OVOC RGM comparison (Table A1). The selected capillary column was an HP-Plot U (30 m length, 0.53 mm internal diameter and 20 µm film thickness; Agilent Technologies Inc., CA, USA). The GC oven was held at a constant temperature of 150 °C. The carrier was helium BIP® (Air Products and Chemicals, PA, USA). The sampling was done using a coated (SilcoNert® 2000) sample loop, which injected a sample volume of 60 mL. The preconcentration system was cooled down to −60 °C by a liquid nitrogen cryo-trap system (JAS 66601 CryoTrap, Joint Analytical Systems GmbH, Germany), which was heated up to 150 °C for final injection.
A1.5 METAS (Federal National Metrology Institute; NMI of Switzerland)
METAS used a GC–FID Clarus 500 (PerkinElmer Inc., MA, USA) coupled to a thermal desorber TurboMatrix 350 (PerkinElmer Inc., MA, USA) (Table A1). The capillary column was a Durabond DB-624 (30 m length, 0.32 mm internal diameter and 1.8 µm film thickness; Agilent Technologies Inc., CA, USA). The carrier gas was helium. The system had a Tenax TA sorbent cold trap (PerkinElmer Inc., MA, USA), which was cooled at −30 °C and heated up to 280 °C at a temperature rate of 40 °C s−1. The GC oven was held at 40 °C for 2 min and then heated up to 200 °C at 5 °C min−1. The GC oven was held at 200 °C for 2 min. The sampling was done using conditioned multibed sorbent tubes: Carbograph 2TD (mesh 60/80), Carbograph 1TD (mesh 40/60) and Carbosieve™ SIII (mesh 60/80) (Camsco, TX, USA). Loading of the sorbent tubes was done by means of an in-house loading system at loading volumes between 300 mL (10 min at 30 mL min−1) and 450 mL (15 min at 30 mL min−1).
A1.6 UU (Utrecht University)
UU used a PTR–ToF–MS with a hexapole and ion funnel (PTR–ToF 4000, Ionicon Analytik GmbH, Austria) to assess the SI-traceable working standards (Table A1). A Sulfinert®-coated four-port valve (VICI®, Valco Instruments Co. Inc., TX, USA) kept at 120 °C was used to switch between zero air and a sample inlet. Samples were connected to a PEEK capillary that, depending on the pressure in the cylinders and canisters, produced a flow between 80 and 300 mL min−1.
A1.7 VSL (NMI of the Netherlands)
VSL used a TRACE GC (Thermo Fisher Scientific Inc., PA, USA) coupled to a UNITY 2 (Markes International Ltd., UK) thermal desorber during the OVOC comparison, the certification of air samples and the assessment of SI-traceable working standards (Table A1). A Deans switch in the GC sent the gas sample to two FID detectors and two capillary columns: Stabilwax (30 m length, 0.32 mm internal diameter and 1.0 µm film thickness; Restek Corporation, PA, USA) for MVK and PoraBOND U (25 m length, 0.32 mm internal diameter and 7 µm film thickness; Agilent Technologies Inc., CA, USA) for the other OVOCs. The cold trap filled with a multibed sorbent trap (air toxics, Markes International Ltd., UK) was cooled down to −20 °C and heated up to 300 °C. The sampling flow was set at 20 mL min−1 for 30 min. The GC oven was held at 40 °C for 2 min and then heated up to 230 °C with three temperature ramps of 20 °C min−1 (up to 120 °C), 5 °C min−1 (up to 180 °C) and 10 °C min−1 (up to 230 °C). The GC oven was held at 200 °C for 20 min. The carrier gas was helium.
A2 Dilution systems
Two dilution systems were used to generate the SI-traceable working standards based on the dynamic dilution of RGMs.
The first system, developed by VSL, was a one-stage gas diluter with dilution flows ranging from 2–50 L min−1, allowing for dilution ratios up to 1:1000. Flows of the RGM (0.1 L min−1) and of the dilution gas (nitrogen, AP BIP Plus grade 6.0) were accurately controlled using three mass flow controllers (MFCs) (EL-FLOW® Select series, Bronkhorst, the Netherlands), operating at up to 10 and 25 L min−1. The dilution system was mostly built in inert glass. Other materials in contact with the OVOC gas mixtures were polytetrafluoroethylene (PTFE), 316 stainless steel (SS) (small surfaces) or coated 316 SS (SilcoNert® 2000, SilcoTek, PA, USA). A coated (SilcoNert® 2000) pressure reducer was connected to the RGMs and flushed thoroughly before attaching it to the dilution system. For the purpose of this assessment, the MFCs were set and calibrated using two mercury piston prover volumeters (Bronkhorst, the Netherlands), which were in turn calibrated at the VSL Flow Department, at working ranges of 0–0.5 and 0–10 L min−1. Temperature and pressure were measured by equipment calibrated at the VSL Temperature Department and Pressure Department to convert flow to conditions of standard temperature and pressure (STP) (293.15 K, 101.3 kPa).
The second system (VeRDi, Versatile Reactive Gas Diluter), developed by METAS in collaboration with Swagelok® Switzerland, was a two-stage gas dilution system allowing for dilution ratios up to 1:175 000. The main components of this dilution system were four MFCs (two MFCs at up to 0.1 L min−1 (red-y, Vögtlin Instruments, GmbH, Switzerland) and two MFCs at up to 5 L min−1 (Sensirion AG, Switzerland)), two pressure controllers (Bronkhorst High-Tech B.V., the Netherlands), a valve terminal (MPA-L, Festo Beteiligungen GmbH & Co. KG, Germany) and a vacuum pump. Elements in contact with RGM flow were coated (SilcoNert® 2000), including all the stainless steel tubing of in. internal diameter used to build VeRDi. The tubes were welded, instead of joined through fittings, in order to reduce dead volumes and potential leaks. MFCs and pressure regulators were calibrated using clean and dry nitrogen against METAS primary standards to ensure traceability of the dilution. The VeRDi software controlling was developed in LabVIEW (National Instruments, Austin, TX).
B1 Purity analysis
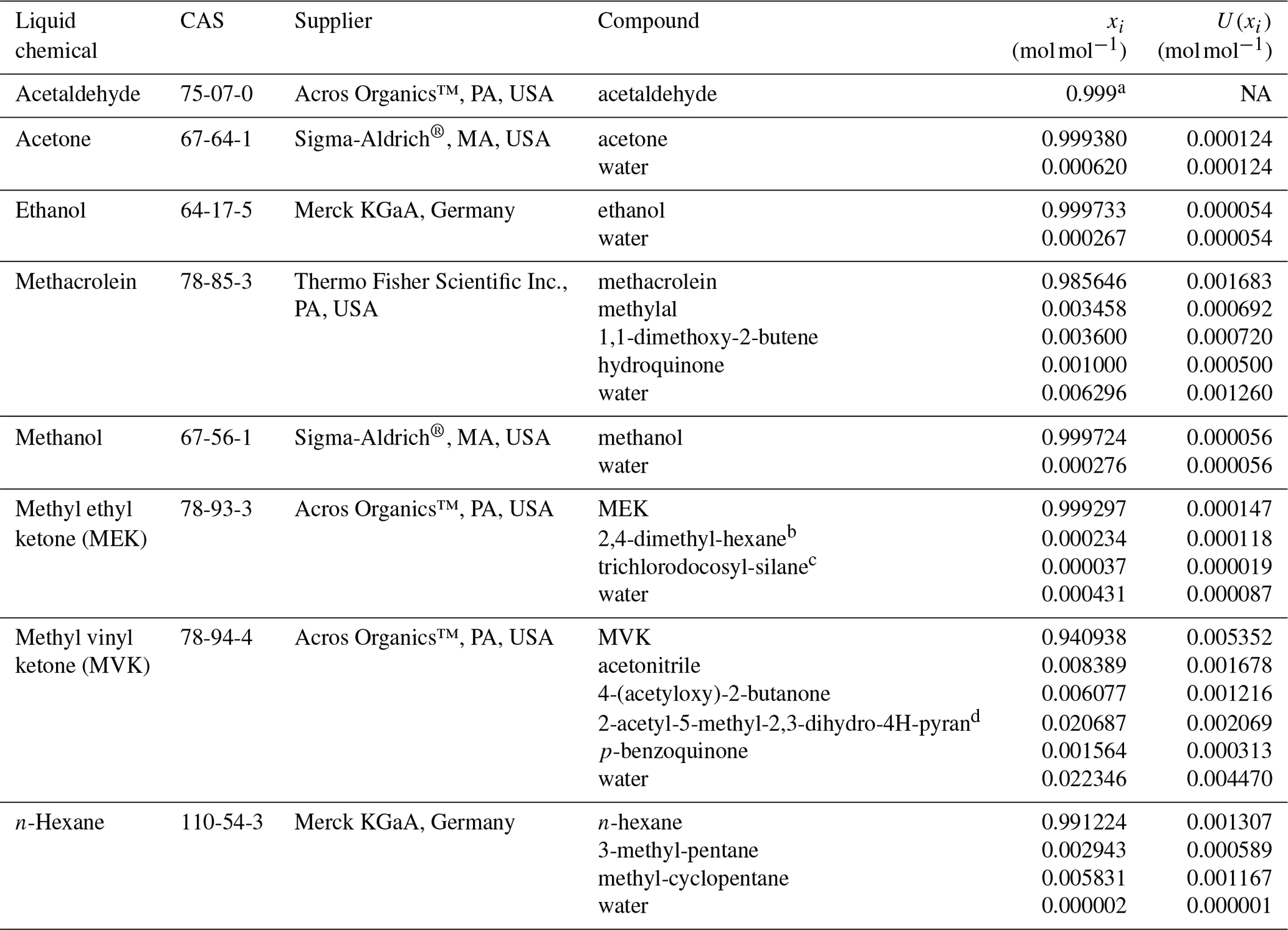
Prior to their injection in the pressurised cylinders, the pure liquid oxygenated volatile organic compounds (OVOCs), selected to prepare the gravimetric RGMs, were analysed to determine their purity according to ISO 19229:2019 (2019). For that purpose, VSL (the NMI of the Netherlands) used a gas chromatograph (GC) system (6890, Agilent Technologies Inc., CA, USA) with a mass spectrometer (MS) and a flame ionisation detector (FID) equipped with a GS-GasPro capillary column (60 m length, 0.32 mm internal diameter and 0.25 µm film thickness; Agilent Technologies Inc., CA, USA). For acetaldehyde, it was not possible to perform the purity analysis because of the physical properties of the liquid chemical, which made its handling difficult. The water content in the liquid OVOCs was determined by Karl Fischer titration (coulometric Karl Fischer titrator, Metrohm). Results of the purity analysis are included in Table B1.
Table B1Purity of the liquid OVOCs used to prepare the gravimetric reference gas mixtures including the amount fraction of compounds and impurities (xi) and its expanded uncertainty (U(xi); coverage factor k=2). CAS refers to the chemical abstract service registry number. The purity analysis of acetaldehyde was not possible because of handling difficulties associated with the physical properties of the liquid chemical.
a Purity value provided by the manufacturer. b According to the MS database, the first hit with the highest probability is 2,4-dimethyl-hexane, but the probability is only around 10 %. c According to the MS database, the first hit with the highest probability is trichlorodocosyl-silane, but the probability is only around 15 %. d The impurity might also be MVK dimer.
B2 RGM verification
B2.1 Verification measurement results
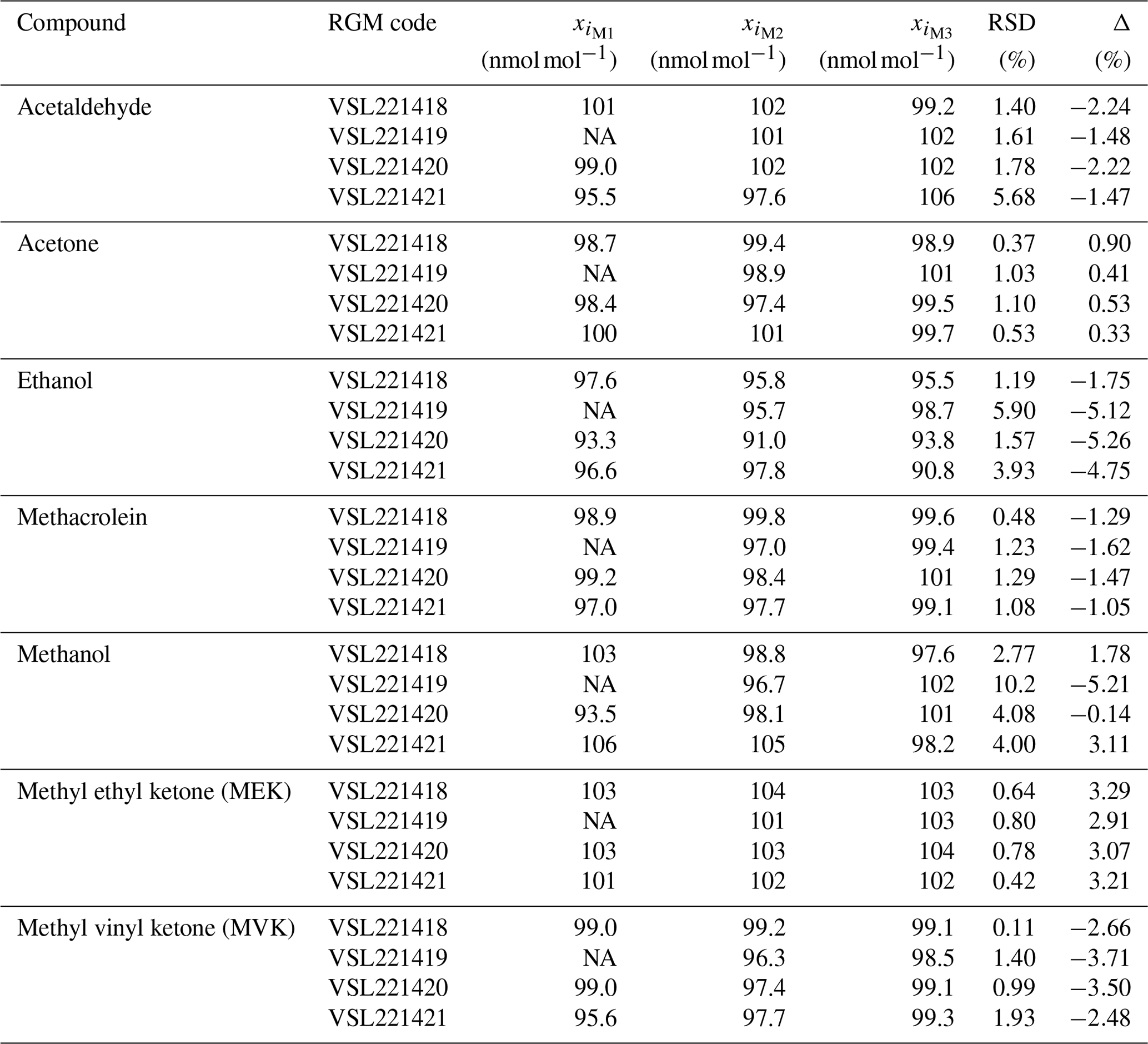
The verification process was repeated three times using the VSL thermal desorber (TD)–GC–FID described in Appendix A1. The RGMs were connected to an autosampler built by VSL, therefore sharing the same pressure reducer. Lines and the pressure reducer were coated (SilcoNert® 2000). To guarantee the same sampling conditions (20 mL min−1 sampling flow for 15 min, total volume 300 mL at 293 K and 101.3 kPa) for gravimetric and dynamically prepared RGMs, the mass flow controller (MFC) of the thermal desorber was operated in a light vacuum mode by means of a pump. Each gas mixture was analysed 20 times. Results of the verification measurements performed 1 month after preparation of the RGMs, estimated according to Eq. (2), are shown in Table B2. Three verification measurements were carried out for each RGM.
Table B2Verification results obtained 1 month after preparation of the reference gas mixtures (RGMs) gravimetrically prepared RGMs at the NMI of the Netherlands (VSL). Three verification measurements (M1, M2 and M3) of the amount fraction of each compound (xi) were performed per RGM. The relative standard deviation (RSD) and the relative difference between analytical and gravimetric values (Δ) are also shown. NA indicates data that are not available due to an analytical issue during a measurement.
B2.2 RGM interlaboratory comparison
The national metrology institutes (NMIs) of France (LNE), Switzerland (METAS) and the Netherlands (VSL) took part in an interlaboratory comparison to verify the produced RGMs. Three different thermal desorber–gas chromatograph–flame ionisation detector (TD–GC–FID) systems and calibration methods (Table B3) were used to analyse the amount fraction of acetone, ethanol and methanol in the RGM sent around (VSL221418).
Table B3Information on the interlaboratory comparison measurements of one of the OVOC reference gas mixtures (RGMs; VSL221418) prepared by VSL to generate SI-traceable working standards based on its dilution using dry nitrogen.
The same coated (SilcoNert® 2000) pressure reducer (RX 2400, Rotarex, Luxembourg) and line ( in. coated line of 1 m length) were used, for at least one series measurements, by LNE and METAS. VSL used an autosampler (VSL spin) equipped with a multi-position valve (VICI AG International, Switzerland), a coated (SilcoNert® 2000) pressure reducer (Tescom, TX, USA) and coated lines (SilcoNert® 2000, in. diameter, ca. 1 m length). Five series of measurements were performed by LNE and by METAS. VSL performed three series of measurements before shipping the comparison standard to the other laboratories (September 2021) and five series of measurements after the shipment (April 2022). At least five replicates per series were analysed. Individual measurement sequences consisted in the analysis of blank samples (at the beginning and end of each measurement), calibration standard samples (at two amount fraction levels) and comparison standard samples (which were analysed between the calibration standards to minimise drift effects and prevent biases). LNE sampling was done through a coated (SilcoNert® 2000) sample loop of 20 mL volume; the total sample volume was 60 mL. VSL sampling was done by means of the autosampler (Unity 2, Markes International, Ltd., UK) coupled to the TD–GC–FID at a sampling flow rate of 20 mL min−1 for 15 min (300 mL sample volume). Multibed sorbent tubes (Carbograph 2 (mesh 60/80), Carbograph 1 (mesh 40/60), Carbosieve™ SIII (mesh 60/80); Camsco, TX, USA) were used for sampling by METAS; the loading volume ranged between 150 and 450 mL.
Amount fraction values of the comparison standard were assigned applying Eqs. (1) and (2). The degree of equivalence of each laboratory for acetone was estimated as the difference between analytical measurement values obtained by each laboratory and the gravimetric reference value provided by VSL, following standard procedures used in key comparisons.
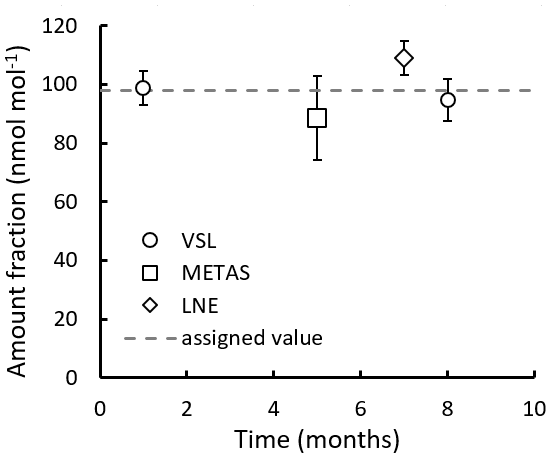
Figure B1Interlaboratory comparison results for methanol. Reported values were the average of five measurements, except for month 1 results, which were the average of three measurements. Error bars show the expanded uncertainty of the measurements (coverage factor k=2). The dashed line indicates the gravimetric amount fraction of the compound.
B3 RGM stability evaluation
Results of the long-term stability evaluation for two of the prepared RGMs (VSL221418 and VSL221419) are shown in Table B4. The evaluation was carried out immediately after preparation (0–1 month). Other stability periods considered were 5–6, 7–8, 13–14 and 18–19 months after preparation of the RGMs.
Table B4Temporal stability of two of the gravimetric RGMs. Results are expressed as the relative difference (Δ) of the average analytical value with respect to the gravimetric value. Deviations larger than ±5 % are in bold. The stability period is indicated as the number of months after RGM preparation. NA indicates data that are not available due to an analytical issue during a measurement.
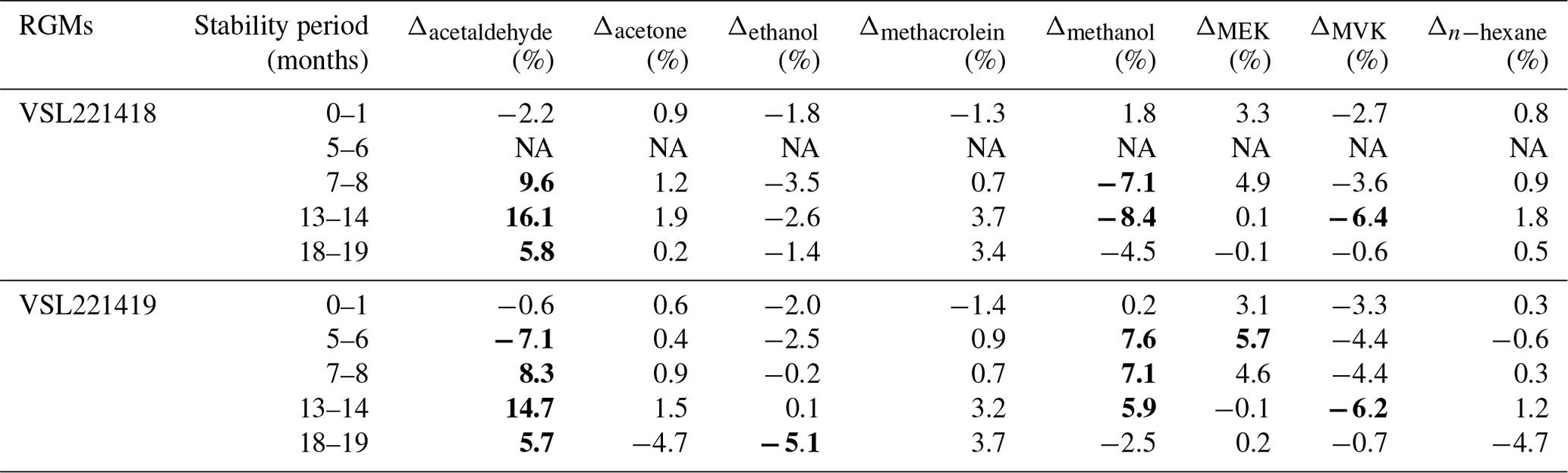
To spike the two parent cylinders with the selected oxygenated volatile organic compounds (OVOCs), a certified reference gas mixture (RGM) filled into a high-pressure 5 L aluminium cylinder (D249650, VSL, the Netherlands) was used. The RGM contained acetaldehyde, acetone, ethanol, methacrolein, methanol, methyl ethyl ketone (MEK), methyl vinyl ketone (MVK), benzene, n-hexane and propane in dry nitrogen at amount fractions between 500 and 1004 nmol mol−1 (Table C1). The cylinder content was transferred to the parent cylinders through a cross connector joined to the outlet of the RGM cylinder (that was heated to avoid condensation), to the parent cylinders and to the vacuum pump used to evacuate the RGM cylinder. Because dilution factors of around 0.011 were expected after whole-air filling of the parent cylinders, the RGM cylinder was fully evacuated into the parent cylinders to reach OVOC spiked values between 5 and 10 nmol mol−1 (Table C1).
Table C1Amount fraction (xcyl) and certified expanded uncertainty (U(xcyl)) of the OVOCs contained in the gas cylinder used for spiking the air samples. Estimated spiked amount fraction (xspiked) and uncertainty (U(xspiked)) of the parent cylinders are also included. The coverage factor of the uncertainty is 2 (k=2).
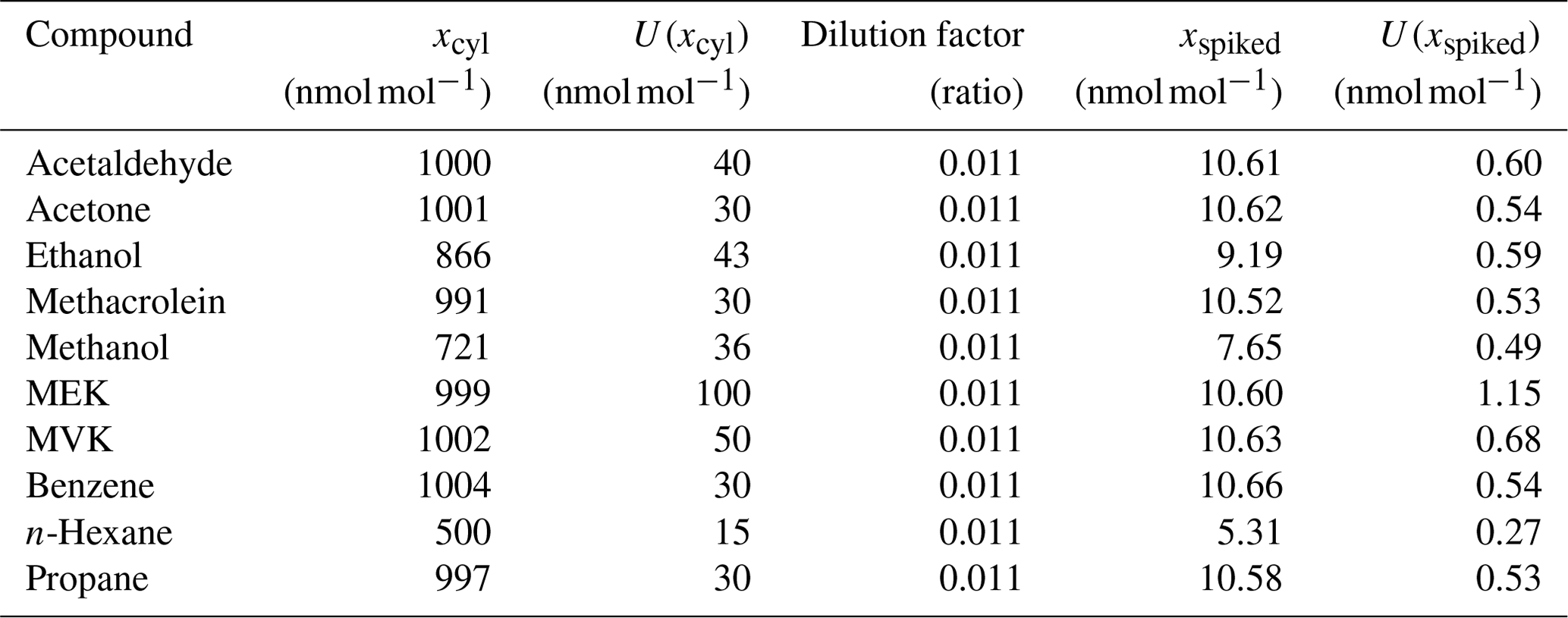
Table C2Air sample cylinders (_cyl) and canisters (_can) used to perform one of the described actions: certification (C), assessment (A) and stability (S).
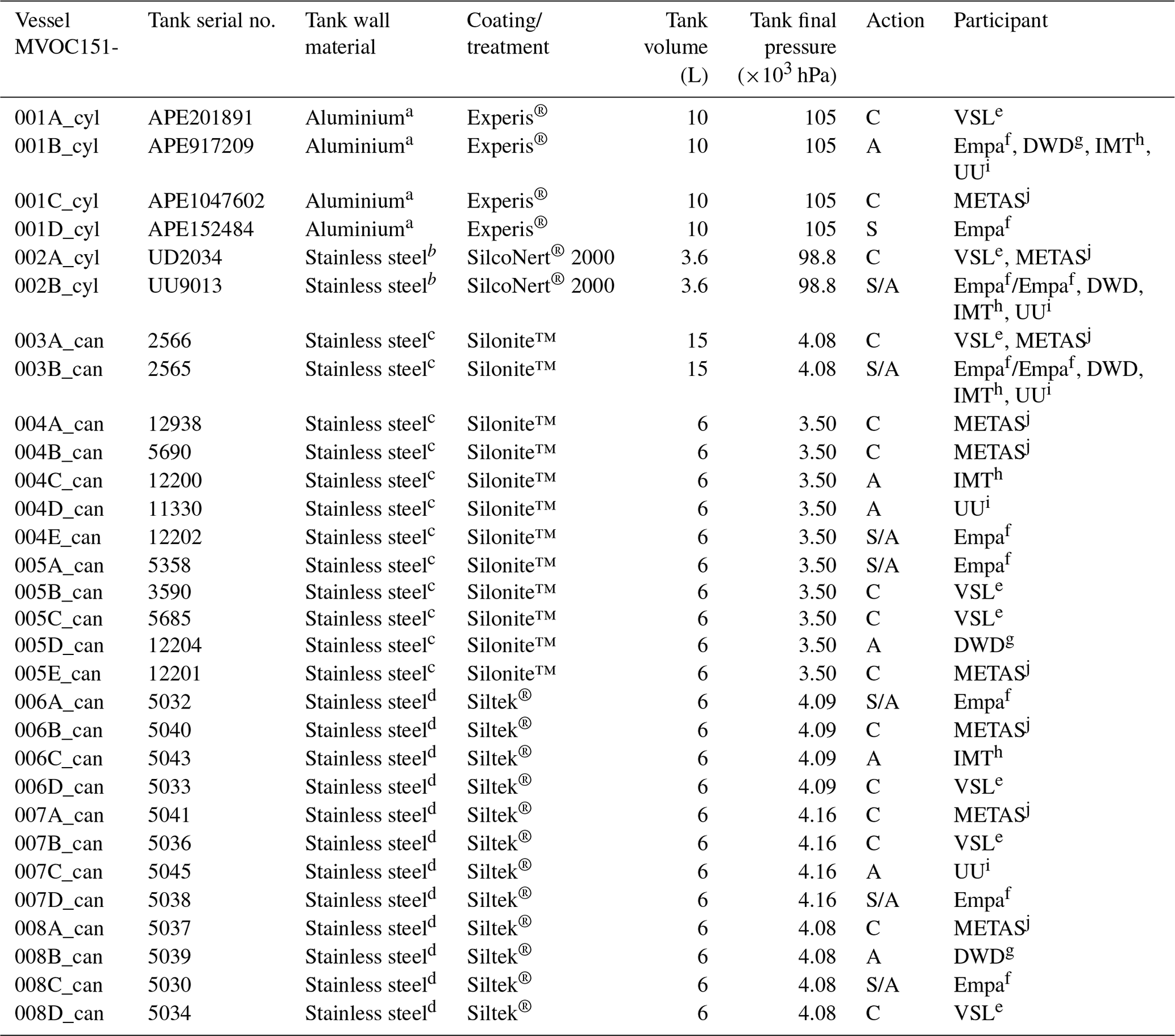
a Air Products. b Swagelok. c Entech Instruments. d Restek. e NMI of the Netherlands. f Swiss Federal Laboratories for Materials Science and Technology. g Deutscher Wetterdienst. h Institute Mines-Télécom. i Utrecht University. j NMI of Switzerland.
To produce the SI-traceable working standards of certified spiked whole-air samples, 6 cylinders and 24 canisters (Table C2) were filled with the spiked whole air contained in the two parent cylinders. For that purpose, the parent cylinders were decanted into the selected cylinders and canisters to produce several identical subsamples (i.e. working standards). Four cylinders were 10 L aluminium cylinders with Experis® treatment for non-methane hydrocarbon (NMHC) VOC (Air Products Inc., PA, USA), and two were 3.6 L coated (SilcoNert® 2000) stainless steel cylinders (Swagelok Co., OH, USA). Of the selected canisters, 12 were coated with Silonite™ (ten 6 L stainless steel canisters and two 15 L stainless steel canisters; Entech Instruments, CA, USA), and 12 were coated with Silcosteel® (6 L stainless steel; Restek Corporation, PA, USA).
Table C3Results of the homogeneity test performed on a subset of vessels filled with the SI-traceable working standards based on certified spiked whole-air samples. The amount fraction (xa) and standard deviation (SD) correspond to three replicates analysed by Empa for each vessel, including the parent cylinders (E-202A, E-202B). Number in italics and with ∗ refers to an outlier due to analytical issues that was removed for the data analysis.
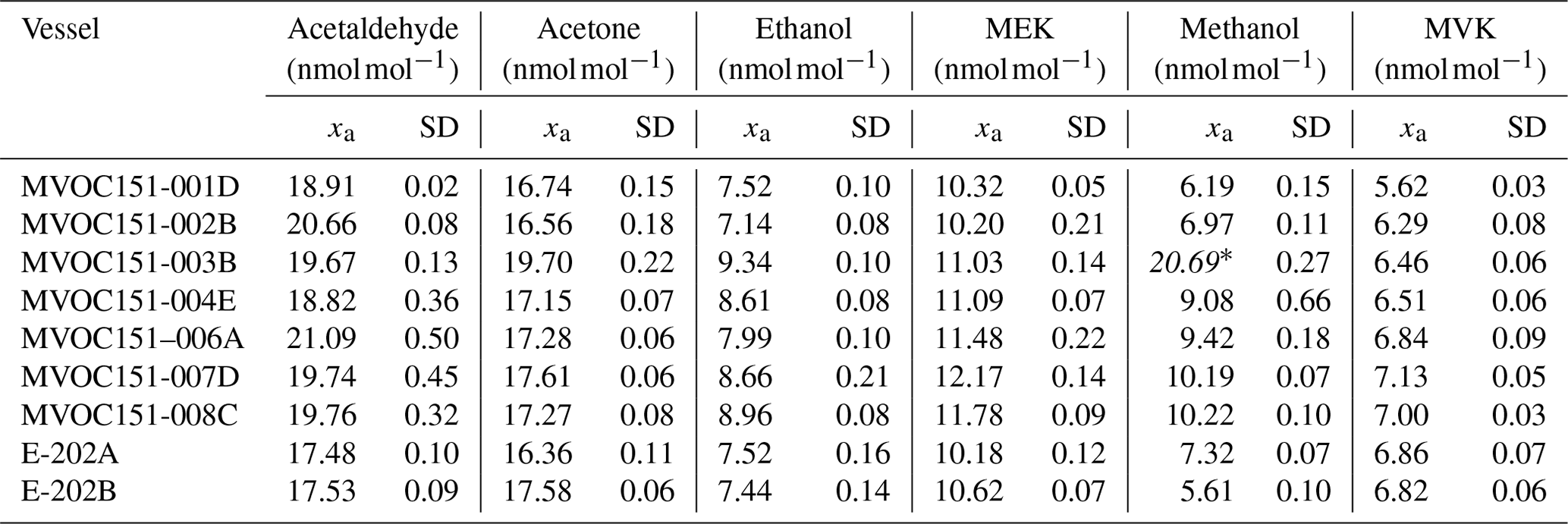
Table C4Analytical methods used for the certification of air samples.
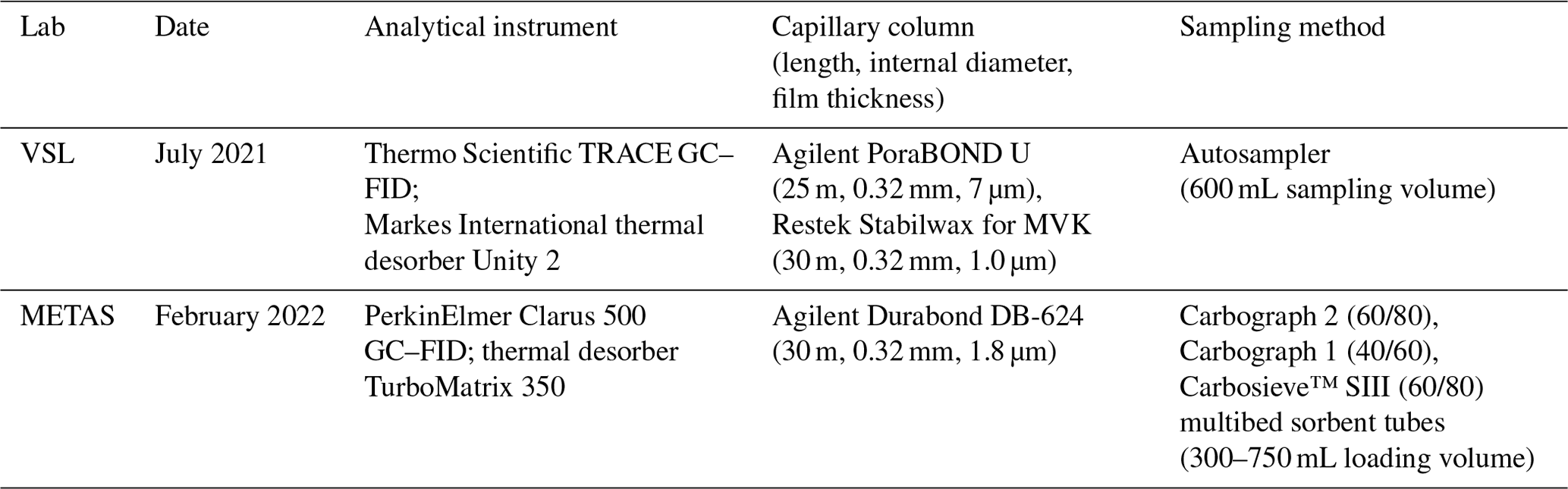
Certification of the air samples was done using two thermal desorber–gas chromatograph–flame ionisation detector (TD–GC–FID) systems (Table C4) and following the same measurement sequence: blanks, air sample, calibration standard at the level with a low amount fraction (1–24 nmol mol−1, depending on the compound), air sample and calibration standard at the level with a high amount fraction (10–45 nmol mol−1, depending on the compound). VSL calibration standards consisted of two multi-compound gas mixtures at 2 and 10 nmol mol−1 for acetone, methanol, ethanol, acetaldehyde, methacrolein, methyl vinyl ketone (MVK) and methyl ethyl ketone (MEK) in nitrogen. The calibration standards were prepared by diluting two gravimetric RGMs containing these OVOCs in nitrogen, as well as n-hexane and propane, at 100 and 1000 nmol mol−1. An additional calibration standard containing acetone, ethanol, methanol and n-hexane in clean and dry air at ca. 10 nmol mol−1 was obtained by diffusion. METAS generated calibration standards containing acetaldehyde, acetone, ethanol, methacrolein, methanol and MVK in nitrogen at around 10 nmol mol−1 by the permeation method (ISO 6145-10:2002, ISO, 2002) using a magnetic suspension balance (Waters, DE, USA) and a portable generator (Pascale et al., 2017).
The uncertainty of the assigned amount fraction of each compound and air sample was the result of multiplying the combined uncertainty of each air sample by the coverage factor (k=2). The combined uncertainty was estimated as the combination of the uncertainty of the calibration standards, the mean standard deviation of the measurements results and the pooled standard deviation of the measurements (Eq. C1).
where u(xsample) is the uncertainty of the assigned amount fraction of the compound in the air sample, xsample is the assigned amount fraction of the compound in the air sample, u(xcal) is the uncertainty of the amount fraction of the compound in the calibration standard, xcal is the amount fraction of the compound in the calibration standard, is the mean standard deviation of the response factor of the compound calibration standard, is the average response factor of the compound calibration standard (average of three measurements), is the mean standard deviation of the GC–FID compound responses (average of three measurements), is the average GC–FID compound response (average of three measurements) and u(pooledsd) is the pooled standard deviation of the measurement results.
Table C5Analytical amount fraction (xa) and its expanded uncertainty (U; coverage factor k=2) obtained by the two national metrology institutes (NMIs) certifying the air samples: VSL and METAS.
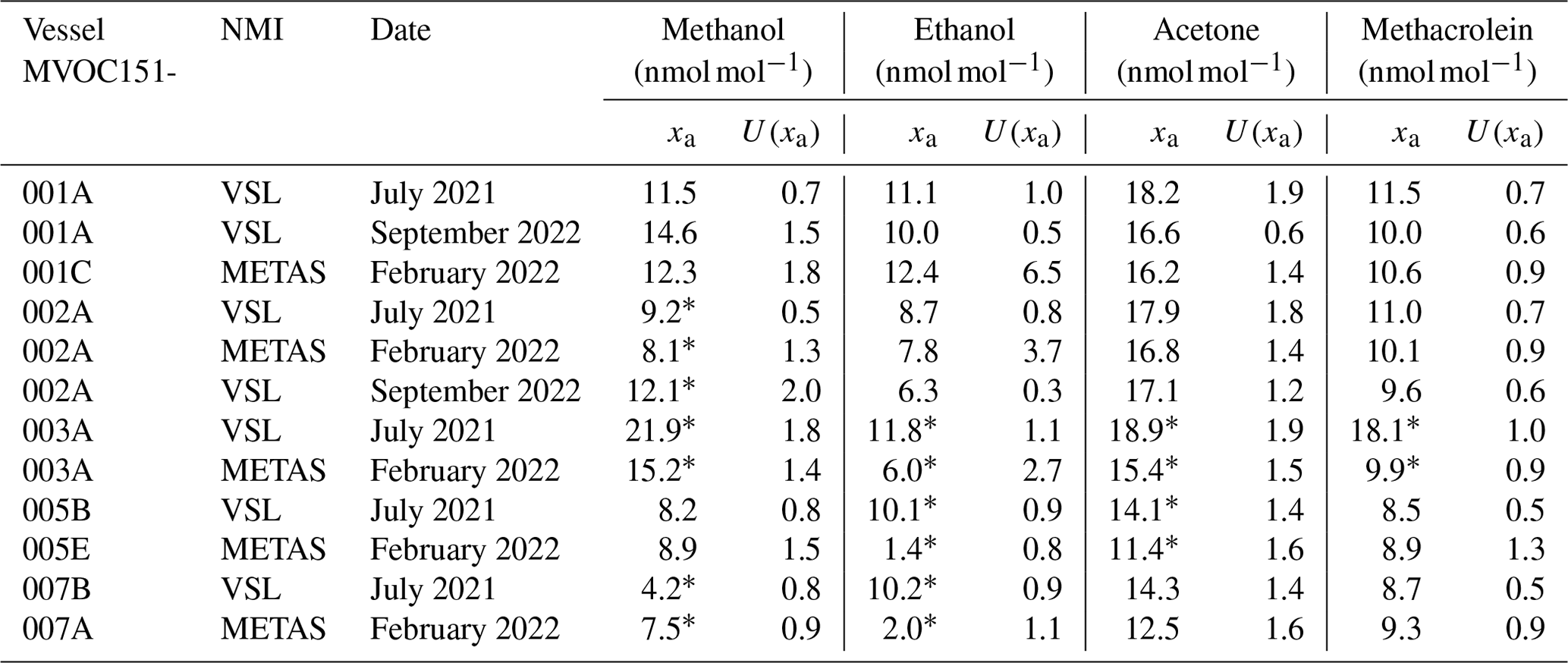
∗ Values for which the criterion described in Eq. (4) was not fulfilled.
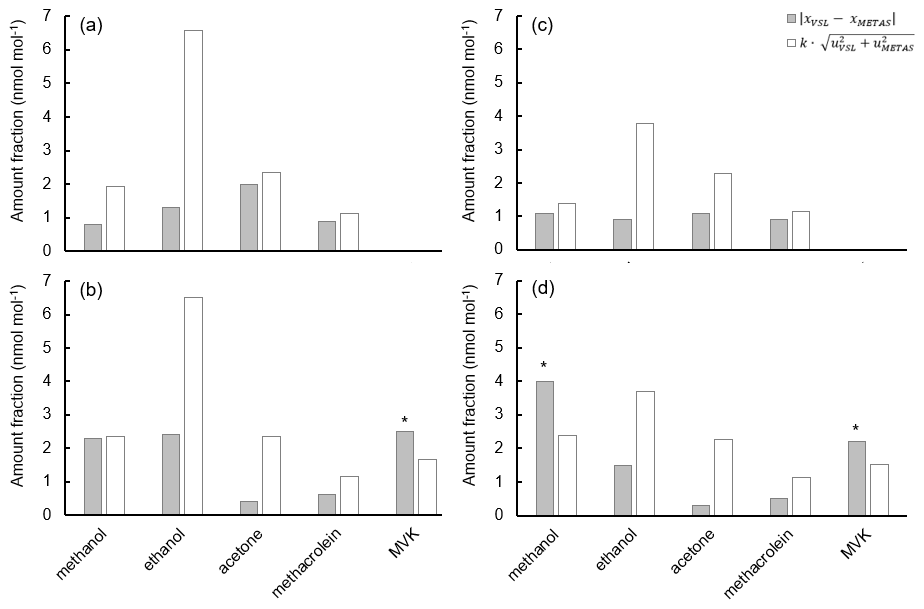
Figure C1Representation of the consistency of the amount fraction values assigned to the 10 L Experis cylinders according the criterion described in Eq. (4). METAS measurements (vessel 001C and 002A) performed in February 2022 were compared to the VSL measurements carried out in July 2021 on vessel (a) 001A and (c) 002A and in September 2022 on vessel (b) 001A and (d) 002A. The asterisks show those measurements for which the absolute difference in the measured amount fraction (xVSL, xMETAS) was greater than twice (coverage factor k=2) the square root of the sum of squares of the measurement standard uncertainties (uVSL,uMETAS).
D1 In-house working standards
The analytical instruments selected to assess the SI-traceable working standards (Appendix A, Table A1) were calibrated with in-house working standards generated using different methods.
The thermal desorber–gas chromatograph–flame ionisation detector/mass spectrometer (TD–GC–FID/MS) system used by DWD (Deutscher Wetterdienst) to assess the working standards based on certified spiked whole-air samples was calibrated using one of the reference gas mixtures (RGMs) prepared by the NMI of the Netherlands (VSL) for this work (oxygenated volatile organic compounds (OVOCs) in nitrogen at 100 nmol mol−1) without further dilution. In addition, the DWD TD–GC–FID/MS system was calibrated using a primary reference material containing 30 non-methane hydrocarbons (NMHCs) considered ozone precursors at amount fraction levels of 2 nmol mol−1 (NPL NMHC standard; Grenfell et al., 2010). The same type of standard (NPL NMHC) was used to calibrate the Empa TD–GC–FID.
The ion transmission curves of the proton transfer reaction–time of flight–mass spectrometer (PTR–ToF–MS) were determined using a SI-traceable certified reference material produced by the National Physical Laboratory (NPL), the NMI of the United Kingdom (Worton et al., 2023), as in-house working standards (NPL PTR–MS standard). The in-house working standards (NPL D961410 used by Institute Mines-Télécom (IMT) and D961397 used by Utrecht University (UU)) contained 20 compounds at amount fractions around 1 µmol mol−1 covering a mass spectrum from 33 to 671. Prior to instrument calibration, the in-house working standards were diluted with zero air (i.e. dry nitrogen) down to amount fractions <10 nmol mol−1.
The VSL TD–GC–FID was calibrated using RGMs based on the diffusion method (dynamic preparation method ISO 6145-8) as in-house working standards for acetone, ethanol, methacrolein, methanol, methyl ethyl ketone (MEK) and methyl vinyl ketone (MVK). For acetaldehyde, an in-house working standard was obtained by the dynamic dilution of a 1 µmol mol−1 multi-component RGM containing acetaldehyde, acetone, ethanol, methacrolein, methanol, MEK, MVK and propane in nitrogen. Three to six in-house working standards were prepared in the range of 4–20 nmol mol−1.
D2 Measurement procedure for assessing the working standards based on dynamic dilution of RGMs
Samples were prepared by the dynamic dilution of RGMs. VSL, IMT and UU generated two samples. VSL set the same dilution factor for both samples (10 times dilution to obtain OVOC amount fractions close to 10 nmol mol−1), while the Swiss Federal Laboratories for Materials Science and Technology (Empa) and IMT used different dilution factors (Table D1). UU prepared six samples using different dilution factors (Table D1).
Table D1Flow rates (in mL min−1) and relative expanded uncertainty (coverage factor k=2) of the dilution systems used to dilute VSL SI-traceable RGM during the assessment of SI-traceable working standards by each laboratory. Gas flow rates correspond to the flow rate of the VSL SI-traceable RGM (), first-step dilution flow rate (), split flow rate () and second-step dilution flow rate (). The assigned amount fractions of the selected compounds are shown together with their expanded uncertainties (k=2), both expressed in nmol mol−1.
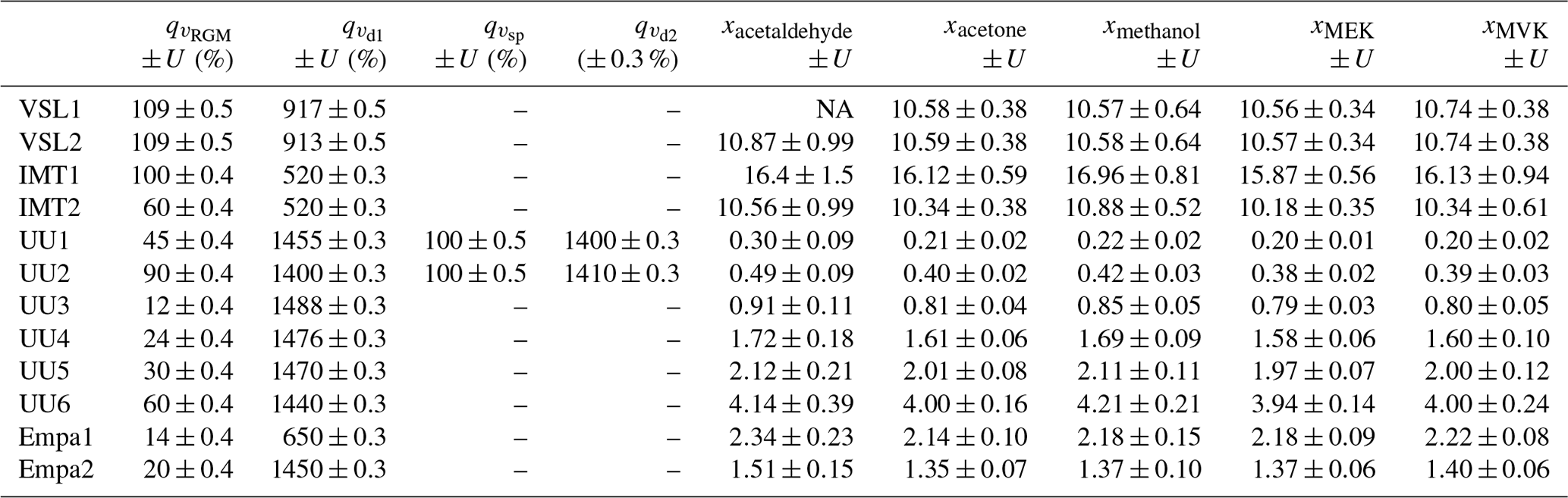
Before performing the measurement sequence, VSL sampled and analysed 15 times the pure nitrogen used for dilution to clean the analytical system and to assess the system blank. Each in-house standard (three to six in total) and sample (i.e. SI-traceable working standard) was sampled at a flow rate of 20 mL min−1 for 30 min.
Empa ran 5 to 10 GC runs with a sample of a similar humidity level and composition for the matrix gas to condition the GC–FID. After the conditioning, six consecutive runs without injecting any sample were measured to estimate the system blank. Then, six in-house working standard runs were followed by six runs for each sample (i.e. SI-traceable working standard). In-house working standard and blank runs (12 runs in total) were repeated after the last sample measurement. The sampling volume was set at 300 mL (20 min at 15 mL min−1).
IMT measurement sequence started with 30 min of zero-air sampling to quantify background signals and to verify signal stability. The zero air was obtained using a catalytic converter containing platinum wool (high-sensitivity catalyst for a total organic carbon (TOC) analyser, Shimadzu Corporation, Japan), which was heated up to 350 °C. Blank measurements were performed before and after each new sample test and calibration. After the first blank measurements, the calibration took place by analysing in-house working standards for 60–90 min. The in-house working standards (5 mL min−1) were diluted with a zero-air flow rate of 1 L min−1. Flows were regulated by MFCs in a gas calibration unit (GCU; Ionicon Analytik GmbH, Austria). Then, each sample was analysed for 90 min. The same sampling line coated with SilcoNert® 1000 and sampling flow rate of 100 mL min−1 were used for blanks, calibration standards and samples.
UU measured each sample two to four times for at least 30 s. Before and after each sample measurement, UU analysed blanks (i.e. zero air produced by a heated platinum catalyst) and the in-house working standard (NPL PTR–MS standard). Blanks, in-house working standards and samples were injected through a sample loop (250 µL volume) according the procedure described in Holzinger et al. (2019). The in-house working standards (loop flow of 10 mL min−1) were diluted with a zero-air flow rate of 240 mL min−1. Sample flows, depending on the pressure in cylinders and canisters, were produced between 80 and 300 mL min−1.
D3 Measurement procedure for assessing working standards based on certified whole-air samples
The same air sample cylinders were assessed by the participants (round-robin comparison). However, different canisters were sent to the participants because of the low sample volume, which was enough only for one analysis (Table C2). Participants followed a measurement sequence similar to the measurement procedure described for the SI-traceable working standards based on RGM dilution. After some blank measurements (six times for GC–FID and 30 min for PTR–ToF–MS), in-house working standards were measured at a minimum of two amount fraction levels (six times per level for GC–FID and for PTR–ToF–MS, IMT measured for 90 min and UU measured for 1 min). Samples were measured between calibration levels (six times each sample for GC–FID and PTR–ToF–MS measurements of 90 min per sample at IMT and 1 min at UU). Blank measurements were performed again after the second amount fraction level of the calibration.
D4 IMT-measured amount fractions
IMT estimated the amount fractions of the selected OVOCs according to the calibration approach described in de Gouw and Warneke (2007) and following Eq. (D1). In practice, a sensitivity factor of H3O+ normalised to 106 cps (counts per second; Sn(RH+)) is derived for each targeted compound during calibration experiments. This sensitivity factor comprises the following parameters: kPTR, Δt, T(RH+) and T(H3O+). The approach used in de Gouw and Warneke (2007) to account for humidity-dependent sensitivities was applied in this work.
where xi is the amount fraction of the compound R (i.e. OVOC under study), kPTR is the proton transfer reaction rate coefficient of , Δt is the reaction time in the drift tube, I(RH+) is the observed signal (counts per second, cps) for the protonated ion RH+, I(H3O+) is the observed signal (cps) for the reagent ion H3O+, T(RH+) is the transmission efficiency for RH+, T(H3O+) is the transmission efficiency for H3O+ and SN(RH+) is the sensitivity factor of H3O+ normalised to 106 cps.
Sources of uncertainty associated with the measured amount fractions included the precision of the system and the calibration accuracy. The uncertainty linked to the precision of the system (uprec) was calculated according Eq. (D2). The uncertainty associated with the calibration accuracy () was estimated applying Eq. (D3).
where uprec is the measurement precision expressed as the amount fraction, Im(RH+) is the RH+ signal (cps) observed when a sample was measured, Iz(RH+) is the RH+ signal (cps) observed when zeroing the instrument, I(H3O+) is the observed signal (cps) for the reagent ion H3O+ and SN(RH+) is the sensitivity factor of H3O+ normalised to 106 cps.
where is the relative combined uncertainty of the calibration accuracy, xcal is the OVOC amount fraction generated after the dilution of the calibration standard, u(xcyl) is the standard uncertainty of the OVOC amount fraction in the calibration standard (calibration certificate), xcyl is the OVOC amount fraction in the calibration standard (calibration certificate), is the flow rate of the calibration standard, is the flow rate of the dilution gas, is the standard uncertainty of the calibration standard flow rate and is the standard uncertainty of the dilution gas flow rate.
D5 Uncertainty of the measurements performed to assess the SI-traceable working standards
Tables D2 and D3 show the amount fraction results of the measurements performed by the participants for the assessment of the SI-traceable working standards for each of the selected OVOCs.
Table D2Amount fraction (xi) of the selected OVOCs measured by the participants of the assessment of the SI-traceable working standards based on the dilution of RGMs. Expanded uncertainty (U, coverage factor k=2) of the measurements are indicated together with the amount fractions, both expressed in nmol mol−1.
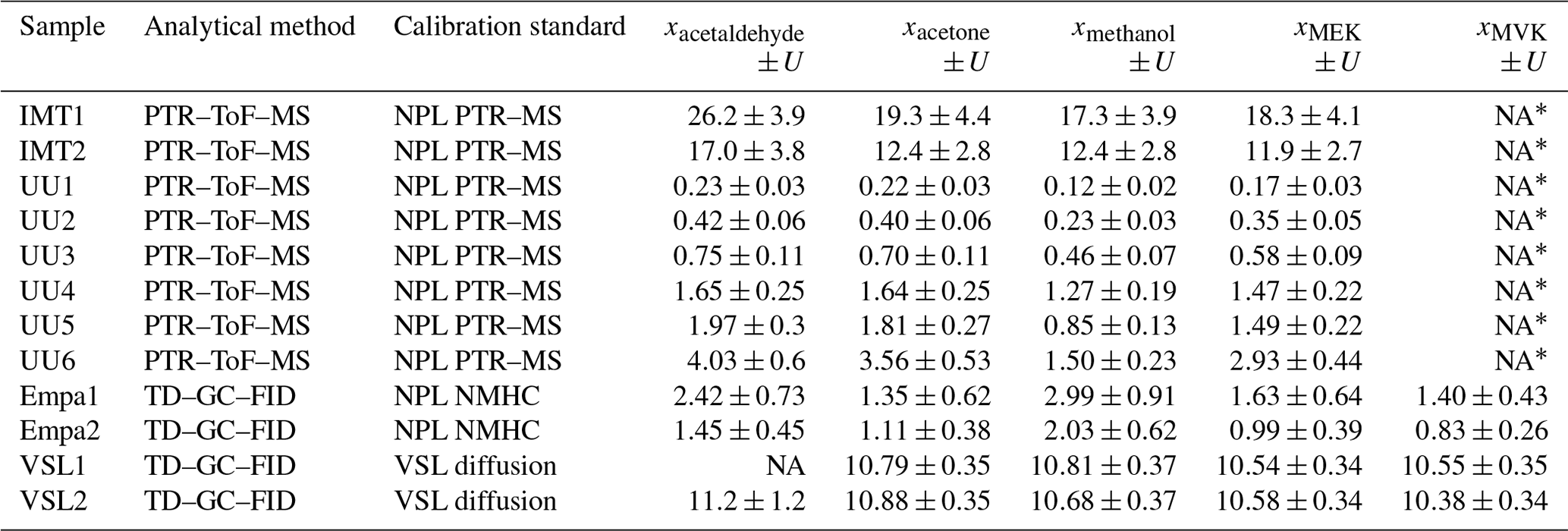
∗ MVK data not available because PTR–ToF–MS can only provide the sum of MVK and methacrolein.
Table D3Amount fraction (xi) of the selected OVOCs measured by the participants of the assessment of the SI-traceable working-standards-based certified spiked whole-air samples. Expanded uncertainty (U, coverage factor k=2) of the measurements is indicated together with the amount fractions, both expressed in nmol mol−1. The analytical methods correspond to TD–GC–FID (AM1) and PTR–ToF–MS (AM2), and the calibration standards correspond to the NPL NMHC standard (Std1), METAS permeation standard (Std2), VSL diffusion standard (Std3) and NPL PTR–MS standard (Std4).
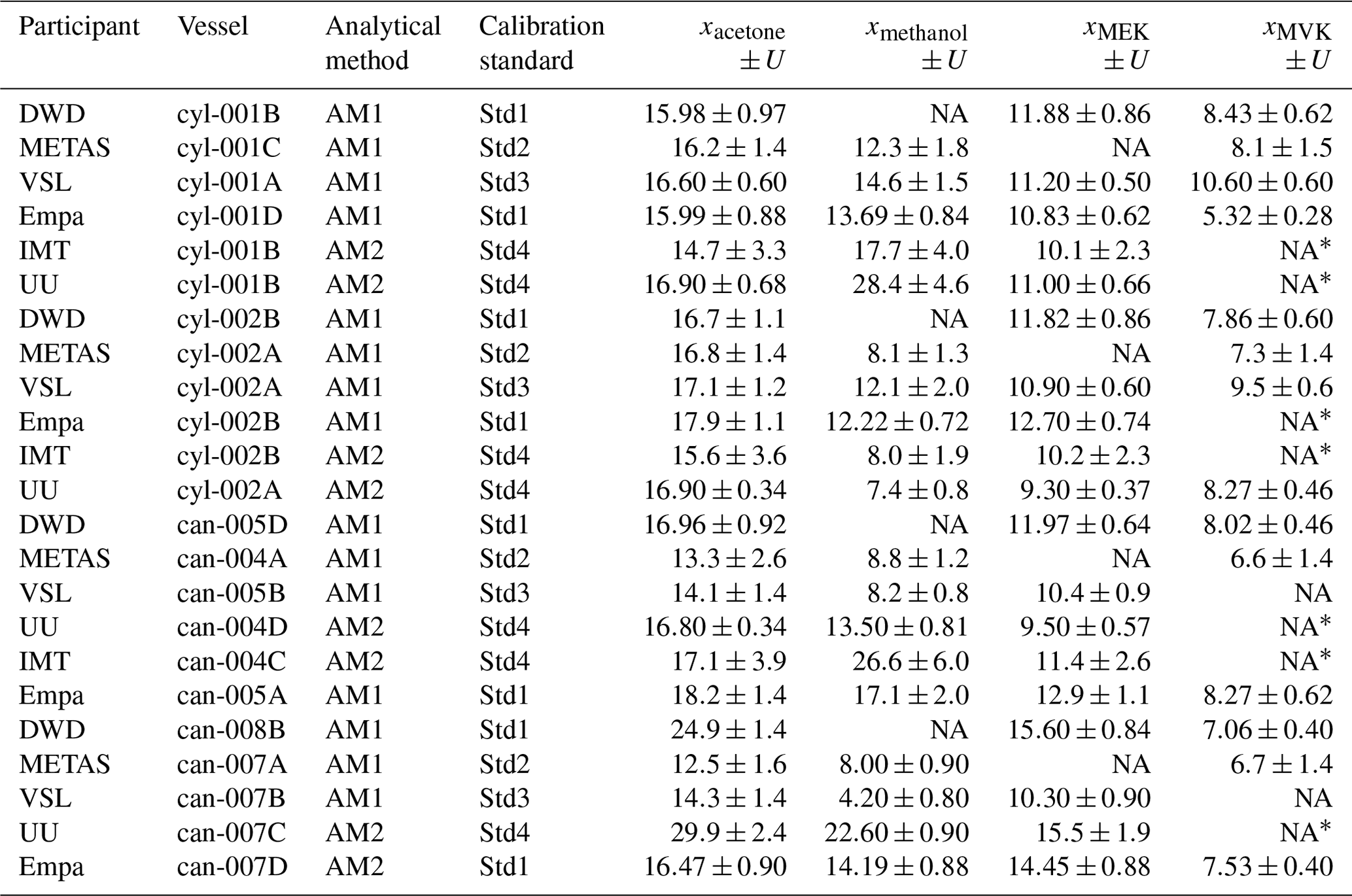
∗ MVK data not available because PTR–ToF–MS can only provide the sum of MVK and methacrolein.
Data used in this work are available from the MetClimVOC community on Zenodo at https://doi.org/10.5281/zenodo.14178374 (Iturrate-Garcia et al., 2024).
SR, PS, CP, MIG, AB, TS, AC and RH designed the study. AB, JL and SP prepared the reference gas mixtures (RGMs) used for generated SI-traceable working standards based on the dilution of RGMs. PS and MKV prepared the SI-traceable working standards based on air samples, which were certified by AB and MIG. AA designed and coordinated the development of the VeRDi dilution system, with supervision of CP. For the assessment measurements, ES, SD, TS and RH performed the PTR–ToF–MS measurements and PS, AB and AC performed the TD–GC–FID measurements. AB, JL, CS and MIG carried out the OVOC RGM comparison. MIG wrote the paper with assistance from all authors, who provided information on instrument description and data. All authors reviewed and approved the latest version of the paper.
The contact author has declared that none of the author has any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
This article is part of the special issue “Tropospheric Ozone Assessment Report Phase II (TOAR-II) Community Special Issue (ACP/AMT/BG/GMD inter-journal SI)”. It is not associated with a conference.
We acknowledge the technical staff at ACTRIS CiGas units (IMT Nord Europe, Empa, DWD) and the participating laboratories for their support. Special thanks go to the Tropospheric Ozone Precursors focus Working Group for their valuable comments that improved the paper.
This research was performed within the framework of the project 19ENV06 MetClimVOC, which has received funding from the EMPIR programme cofinanced by the Participating States and from the European Union's Horizon 2020 research and innovation programme. In addition to the support provided by the European Association of National Metrology Institutes (grant no. 19ENV06, MetClimVOC) to this research, Utrecht University received support from the Ruisdael Observatory, a scientific infrastructure cofinanced by the Dutch Research Council (NWO; grant no. 184.034.015).
This paper was edited by Mingjin Tang and reviewed by three anonymous referees.
Alink, A. and Van Der Veen, A. M. H.: Uncertainty calculations for the preparation of primary gas mixtures Part 1: Gravimetry, Metrologia, 37, 641–650, 2000.
Apel, E. C., Calvert, J. G., Greenberg, J. P., Riemer, D., Zika, R., Kleindienst, T. E., Lonneman, W. A., Fung, K., and Fujita, E.: Generation and validation of oxygenated volatile organic carbon standards for the 1995 Southern Oxidants Study Nashville Intensive, J. Geophys. Res.-Atmos., 103, 22281–22294, https://doi.org/10.1029/98JD01383, 1998.
Atkinson, R.: Atmospheric chemistry of VOCs and NOx, Atmos. Environ., 34, 2063–2101, https://doi.org/10.1016/S1352-2310(99)00460-4, 2000.
Bates, K. H., Jacob, D. J., Wang, S., Hornbrook, R. S., Apel, E. C., Kim, M. J., Millet, D. B., Wells, K. C., Chen, X., Brewer, J. F., Ray, E. A., Commane, R., Diskin, G. S., and Wofsy, S. C.: The Global Budget of Atmospheric Methanol: New Constraints on Secondary, Oceanic, and Terrestrial Sources, J. Geophys. Res.-Atmos., 126, e2020JD033439, https://doi.org/10.1029/2020JD033439, 2021.
Boucher, O., Randall, D., Artaxo, P., Bretherton, C., Feingold, G., Forster, P., Kerminen, V.-M., Kondo, Y., Liao, H., Lohmann, U., Rasch, P., Satheesh, S. K., Sherwood, S., Stevens, B., and Zhang, X. Y.: Clouds and aerosols, in: Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, edited by: Stocker, T. F., Qin, D., Plattner, G.-K., Tignor, M., Allen, S. K., Doschung, J., Nauels, A., Xia, Y., Bex, V., and Midgley, P. M., Cambridge University Press, 571–657, https://doi.org/10.1017/CBO9781107415324.016, 2013.
Brewer, J. F., Fischer, E. V., Commane, R., Wofsy, S. C., Daube, B. C., Apel, E. C., Hills, A. J., Hornbrook, R. S., Barletta, B., Meinardi, S., Blake, D. R., Ray, E. A., and Ravishankara, A. R.: Evidence for an Oceanic Source of Methyl Ethyl Ketone to the Atmosphere, Geophys. Res. Lett., 47, e2019GL086045, https://doi.org/10.1029/2019GL086045, 2020.
Brewer, P. J., Brown, R. J. C., Tarasova, O. A., Hall, B., Rhoderick, G. C., and Wielgosz, R. I.: SI traceability and scales for underpinning atmospheric monitoring of greenhouse gases, Metrologia, 55, S174–S181, https://doi.org/10.1088/1681-7575/aad830, 2018.
Brown, A. S., Milton, M. J. T., Brookes, C., Vargha, G. M., Downey, M. L., Uehara, S., Augusto, C. R., Fioravante, A. de L., Sobrinho, D. G., Dias, F., Woo, J. C., Kim, B. M., Kim, J. S., Mace, T., Fükö, J. T., Qiao, H., Guenther, F., Rhoderick, J., Gameson, L., Botha, A., Tshilongo, J., Ntsasa, N. G., Val'ková, M., Durisova, Z., Kustikov, Y., Konopelko, L., Fatina, O., and Wessel, R.: Final report on CCQM-K93: Preparative comparison of ethanol in nitrogen, Metrologia, 50, 08025–08025, https://doi.org/10.1088/0026-1394/50/1A/08025, 2013.
Collins, W. J., Derwent, R. G., Johnson, C. E., and Stevenson, D. S.: The oxidation of organic compounds in the troposphere and their global warming potentials, Climatic Change, 52, 453–479, https://doi.org/10.1023/A:1014221225434, 2002.
Cooper, O. R., Parrish, D. D., Ziemke, J., Balashov, N. V., Cupeiro, M., Galbally, I. E., Gilge, S., Horowitz, L., Jensen, N. R., Lamarque, J. F., Naik, V., Oltmans, S. J., Schwab, J., Shindell, D. T., Thompson, A. M., Thouret, V., Wang, Y., and Zbinden, R. M.: Global distribution and trends of tropospheric ozone: An observation-based review, Elementa: Science of the Anthropocene, 2, 000029, https://doi.org/10.12952/journal.elementa.000029, 2014.
Coplen, T. B., Holden, N. E., Ding, T., Meijer, H. A. J., Vogl, J., and Zhu, X. The table of standard atomic weights – An exercise in consensus, Rapid Commun. Mass Sp., 36, e8864, https://doi.org/10.1002/rcm.8864, 2020.
De Bièvre, P. and Taylor, P. D. P.: Traceability to the SI of amount-of-substance measurements: From ignoring to realizing, a chemist's view, Metrologia, 34, 67–75, https://doi.org/10.1088/0026-1394/34/1/10, 1997.
de Gouw, J. and Warneke, C.: Measurements of volatile organic compounds in the earth's atmosphere using proton-transfer-reaction mass spectrometry, Mass Spectrom. Rev., 26, 223–257, https://doi.org/10.1002/mas.20119, 2007.
Faiola, C. L., Erickson, M. H., Fricaud, V. L., Jobson, B. T., and VanReken, T. M.: Quantification of biogenic volatile organic compounds with a flame ionization detector using the effective carbon number concept, Atmos. Meas. Tech., 5, 1911–1923, https://doi.org/10.5194/amt-5-1911-2012, 2012.
Fischer, E. V., Jacob, D. J., Millet, D. B., Yantosca, R. M., and Mao, J.: The role of the ocean in the global atmospheric budget of acetone, Geophys. Res. Lett., 39, L01807, https://doi.org/10.1029/2011GL050086, 2012.
Fischer, E. V., Jacob, D. J., Yantosca, R. M., Sulprizio, M. P., Millet, D. B., Mao, J., Paulot, F., Singh, H. B., Roiger, A., Ries, L., Talbot, R. W., Dzepina, K., and Pandey Deolal, S.: Atmospheric peroxyacetyl nitrate (PAN): a global budget and source attribution, Atmos. Chem. Phys., 14, 2679–2698, https://doi.org/10.5194/acp-14-2679-2014, 2014.
Fleming, Z. L., Doherty, R. M., Von Schneidemesser, E., Malley, C. S., Cooper, O. R., Pinto, J. P., Colette, A., Xu, X., Simpson, D., Schultz, M. G., Lefohn, A. S., Hamad, S., Moolla, R., Solberg, S., and Feng, Z.: Tropospheric Ozone Assessment Report: Present-day ozone distribution and trends relevant to human health, Elementa: Science of the Anthropocene, 6, 12, https://doi.org/10.1525/elementa.273, 2018.
Galbally, I. E., Schultz, M. G., Buchmann, B., Gilge, S., Guenther, F., Koide, H., Oltmans, S., Patrick, L., Scheel, H.-E., Smit, H., Steinbacher, M., Steinbrecht, W., Tarasova, O., Viallon, J., Volz-Thomas, A., Weber, M., Wielgosz, R., and Zellweger, C.: Guidelines for Continuous Measurement of Ozone in the Troposphere, GAW Report No. 209, Publication WMO-No. 1110, WMO, Geneva, 76 pp., ISBN: 978-92-63-11110-4, https://library.wmo.int/idurl/4/49557 (last access: 8 November 2024), 2013.
Gaudel, A., Cooper, O. R., Ancellet, G., Barret, B., Boynard, A., Burrows, J. P., Clerbaux, C., Coheur, P. F., Cuesta, J., Cuevas, E., Doniki, S., Dufour, G., Ebojie, F., Foret, G., Garcia, O., Granados-Muñoz, M. J., Hannigan, J. W., Hase, F., Hassler, B., Huang, G., Hurtmans, D., Jaffe, D., Jones, N., Kalabokas, P., Kerridge, B., Kulawik, S., Latter, B., Leblanc, T., Le Flochmoën, E., Lin, W., Liu, J., Liu, X., Mahieu, E., McClure-Begley, A., Neu, J. L., Osman, M., Palm, M., Petetin, H., Petropavlovskikh, I., Querel, R., Rahpoe, N., Rozanov, A., Schultz, M. G., Schwab, J., Siddans, R., Smale, D., Steinbacher, M., Tanimoto, H., Tarasick, D. W., Thouret, V., Thompson, A. M., Trickl, T., Weatherhead, E., Wespes, C., Worden, H. M., Vigouroux, C., Xu, X., Zeng, G., and Ziemke, J.: Tropospheric Ozone Assessment Report: Present-day distribution and trends of tropospheric ozone relevant to climate and global atmospheric chemistry model evaluation, Elementa: Science of the Anthropocene, 6, 39, https://doi.org/10.1525/elementa.291, 2018.
Goldstein, A. H. and Galbally, I. E.: Known and Unexplored Organic Constituents in the Earth's Atmosphere, Environ. Sci. Technol., 41, 1502–1800, https://doi.org/10.1021/es072476p, 2007.
Grenfell, R. J. P., Milton, M. J. T., Harling, A. M., Vargha, G. M., Brookes, C., Quincey, P. G., and Woods, P. T.: Standard mixtures of ambient volatile organic compounds in synthetic and whole air with stable reference values, J. Geophys. Res.-Atmos., 115, D14302, https://doi.org/10.1029/2009JD012933, 2010.
Güttler, B. and Richter, W.: Traceability of chemical measurement results, Chimia, 63, 619–623, https://doi.org/10.2533/chimia.2009.619, 2009.
Hoerger, C. C., Claude, A., Plass-Duelmer, C., Reimann, S., Eckart, E., Steinbrecher, R., Aalto, J., Arduini, J., Bonnaire, N., Cape, J. N., Colomb, A., Connolly, R., Diskova, J., Dumitrean, P., Ehlers, C., Gros, V., Hakola, H., Hill, M., Hopkins, J. R., Jäger, J., Junek, R., Kajos, M. K., Klemp, D., Leuchner, M., Lewis, A. C., Locoge, N., Maione, M., Martin, D., Michl, K., Nemitz, E., O'Doherty, S., Pérez Ballesta, P., Ruuskanen, T. M., Sauvage, S., Schmidbauer, N., Spain, T. G., Straube, E., Vana, M., Vollmer, M. K., Wegener, R., and Wenger, A.: ACTRIS non-methane hydrocarbon intercomparison experiment in Europe to support WMO GAW and EMEP observation networks, Atmos. Meas. Tech., 8, 2715–2736, https://doi.org/10.5194/amt-8-2715-2015, 2015.
Holzinger, R., Acton, W. J. F., Bloss, W. J., Breitenlechner, M., Crilley, L. R., Dusanter, S., Gonin, M., Gros, V., Keutsch, F. N., Kiendler-Scharr, A., Kramer, L. J., Krechmer, J. E., Languille, B., Locoge, N., Lopez-Hilfiker, F., Materić, D., Moreno, S., Nemitz, E., Quéléver, L. L. J., Sarda Esteve, R., Sauvage, S., Schallhart, S., Sommariva, R., Tillmann, R., Wedel, S., Worton, D. R., Xu, K., and Zaytsev, A.: Validity and limitations of simple reaction kinetics to calculate concentrations of organic compounds from ion counts in PTR-MS, Atmos. Meas. Tech., 12, 6193–6208, https://doi.org/10.5194/amt-12-6193-2019, 2019.
Hu, L., Millet, D. B., Mohr, M. J., Wells, K. C., Griffis, T. J., and Helmig, D.: Sources and seasonality of atmospheric methanol based on tall tower measurements in the US Upper Midwest, Atmos. Chem. Phys., 11, 11145–11156, https://doi.org/10.5194/acp-11-11145-2011, 2011.
Iglesias-Suarez, F., Kinnison, D. E., Rap, A., Maycock, A. C., Wild, O., and Young, P. J.: Key drivers of ozone change and its radiative forcing over the 21st century, Atmos. Chem. Phys., 18, 6121–6139, https://doi.org/10.5194/acp-18-6121-2018, 2018.
ISO 19229:2019: Gas analysis – Purity analysis and the treatment of purity data, 2nd edn., International Organization for Standardization (ISO), Geneva, Switzerland, 18 pp., https://iso.org/standard/72010.html (last access: 20 October 2023), 2019.
ISO 6142-1:2015 Gas analysis – Preparation of calibration gas mixtures – Part 1: Gravimetric method for Class I mixtures, 1st edn., International Organization for Standardization (ISO), Geneva, Switzerland, 39 pp., https://iso.org/standard/59631.html (last access: 20 October 2023), 2015.
ISO 6145-10:2002 Gas analysis – Preparation of calibration gas mixtures using dynamic volumetric methods – Part 10: Permeation method, 1st edn., International Organization for Standardization (ISO), Geneva, Switzerland, 16 pp., https://iso.org/standard/25916.html (last access: 20 October 2023), 2002.
ISO 6145-4:2004 Gas analysis – Preparation of calibration gas mixtures using dynamic volumetric methods – Part 4: Continuous syringe injection method, 2nd edn., International Organization for Standardization (ISO), Geneva, Switzerland, 15 pp., https://iso.org/standard/36478.html (last access: 20 October 2023), 2004.
ISO 6145-7:2018 Gas analysis – Preparation of calibration gas mixtures using dynamic methods – Part 7: Thermal mass-flow controllers, 3rd edn., International Organization for Standardization (ISO), 14 pp., https://iso.org/standard/73212.html (last access: 20 October 2023), 2018.
ISO 6145-8:2005 Gas analysis – Preparation of calibration gas mixtures using dynamic volumetric methods – Part 8: Diffusion method, 1st edn., International Organization for Standardization (ISO), 19 pp., https://iso.org/standard/36480.html (last access: 20 October 2023), 2005.
Iturrate-Garcia, M., Salameh, T., Schlauri, P., Baldan, A., Vollmer, M. K., Stratigou, E., Dusanter, S., Li, J., Persijn, S., Claude, A., Holzinger, R., Sutour, C., and Reimann, S.: Data from: Towards a high quality in-situ observation network for oxygenated volatile organic compounds (OVOCs) in Europe: transferring traceability to the International System of Units (SI) to the field, Version 1.0.0, Zenodo [data set], https://doi.org/10.5281/zenodo.14178374, 2024.
Jacob, D. J.: Heterogeneous chemistry and tropospheric ozone, Atmos. Environ., 34, 2131–2159, https://doi.org/10.1016/S1352-2310(99)00462-8, 2000.
JCGM 100:2008: BIPM, IEC, IFCC, ILAC, ISO, IUPAC, IUPAP and OIML. Evaluation of measurement data – Guide to the expression of uncertainty in measurement (GUM). Joint Committee for Guides in Metrology (JCGM), 134 pp., https://www.bipm.org/documents/20126/2071204/JCGM_100_2008_E.pdf (last access: 20 October 2023), 2008.
Khan, M. A. H., Cooke, M. C., Utembe, S. R., Archibald, A. T., Maxwell, P., Morris, W. C., Xiao, P., Derwent, R. G., Jenkin, M. E., Percival, C. J., Walsh, R. C., Young, T. D. S., Simmonds, P. G., Nickless, G., O'Doherty, S., and Shallcross, D. E.: A study of global atmospheric budget and distribution of acetone using global atmospheric model STOCHEM-CRI, Atmos. Environ., 112, 269–277, https://doi.org/10.1016/j.atmosenv.2015.04.056, 2015.
Laj, P., Lund Myhre, C., Riffault, V., Amiridis, V., Fuchs, H., Eleftheriadis, K., Petäjä, T., Salameh, T., Kivekäs, N., Juurola, E., Saponaro, G., Philippin, S., Cornacchia, C., Alados Arboledas, L., Baars, H., Claude, A., De Mazière, M., Dils, B., Dufresne, M., Evangeliou, N., Favez, O., Fiebig, M., Haeffelin, M., Herrmann, H., Höhler, K., Illmann, N., Kreuter, A., Ludewig, E., Marinou, E., Möhler, O., Mona, L., Murberg, L. E., Nicolae, D., Novelli, A., O'Connor, E., Ohneiser, K., Petracca Altieri, R. M., Picquet-Varrault, B., van Pinxteren, D., Pospichal, B., Putaud, J. P., Reimann, S., Siomos, N., Stachlewska, I., Tillmann, R., Avoudouri, K. A., Wandinger, U., Wiedenshohler, A., Apituley, A., Comerón, A., Gysel-Beer, M., Mihalopoulos, N., Nikolova, N., Pietruczuk, A., Sauvage, S., Sciare, J., Skov, H., Svendby, T., Swietlicki, E., Tonev, D., Vaughan, G., Zdimal, V., Baltensperger, U., Doussin, J.-F., Kulmala, M., Pappalardo, G., Sorvari S. S., and Vana, M.: Aerosol, Clouds and Trace Gases Research Infrastructure – ACTRIS, the European research infrastructure supporting atmospheric science, B. Am. Meterol Soc., 105, E1098–E1136, https://doi.org/10.1175/BAMS-D-23-0064.1, 2024.
Legreid, G., Lööv, J. B., Staehelin, J., Hueglin, C., Hill, M., Buchmann, B., Prevot, A. S. H., and Reimann, S.: Oxygenated volatile organic compounds (OVOCs) at an urban background site in Zürich (Europe): Seasonal variation and source allocation, Atmos. Environ., 41, 8409–8423, https://doi.org/10.1016/j.atmosenv.2007.07.026, 2007.
Lelieveld, J. and Dentener, F. J.: What controls tropospheric ozone?, J. Geophys. Res.-Atmos., 105, 3531–3551, https://doi.org/10.1029/1999JD901011, 2000.
Leuenberger, M. C., Schibig, M. F., and Nyfeler, P.: Gas adsorption and desorption effects on cylinders and their importance for long-term gas records, Atmos. Meas. Tech., 8, 5289–5299, https://doi.org/10.5194/amt-8-5289-2015, 2015.
Matschat, R., Richter, S., Vogl, J., and Kipphardt, H.: On the way to SI traceable primary transfer standards for amount of substance measurements in inorganic chemical analysis, Anal. Bioanal. Chem., 415, 3057–3071, https://doi.org/10.1007/s00216-023-04660-4, 2023.
Miller, W. R., Rhoderick, G. C., and Guenther, F. R.: Investigating adsorption/desorption of carbon dioxide in aluminum compressed gas cylinders, Anal. Chem., 87, 1957–1962, https://doi.org/10.1021/ac504351b, 2015.
Millet, D. B., Guenther, A., Siegel, D. A., Nelson, N. B., Singh, H. B., de Gouw, J. A., Warneke, C., Williams, J., Eerdekens, G., Sinha, V., Karl, T., Flocke, F., Apel, E., Riemer, D. D., Palmer, P. I., and Barkley, M.: Global atmospheric budget of acetaldehyde: 3-D model analysis and constraints from in-situ and satellite observations, Atmos. Chem. Phys., 10, 3405–3425, https://doi.org/10.5194/acp-10-3405-2010, 2010.
Mills, G., Pleijel, H., Malley, C. S., Sinha, B., Cooper, O. R., Schultz, M. G., Neufeld, H. S., Simpson, D., Sharps, K., Feng, Z., Gerosa, G., Harmens, H., Kobayashi, K., Saxena, P., Paoletti, E., Sinha, V., and Xu, X.: Tropospheric ozone assessment report: Present-day tropospheric ozone distribution and trends relevant to vegetation, Elementa: Science of the Anthropocene, 6, 74, https://doi.org/10.1525/elementa.302, 2018.
Monks, P. S., Archibald, A. T., Colette, A., Cooper, O., Coyle, M., Derwent, R., Fowler, D., Granier, C., Law, K. S., Mills, G. E., Stevenson, D. S., Tarasova, O., Thouret, V., von Schneidemesser, E., Sommariva, R., Wild, O., and Williams, M. L.: Tropospheric ozone and its precursors from the urban to the global scale from air quality to short-lived climate forcer, Atmos. Chem. Phys., 15, 8889–8973, https://doi.org/10.5194/acp-15-8889-2015, 2015.
Pascale, C., Guillevic, M., Ackermann, A., Leuenberger, D., and Niederhauser, B.: Two generators to produce SI-traceable reference gas mixtures for reactive compounds at atmospheric levels, Meas. Sci. Technol., 28, 124002, https://doi.org/10.1088/1361-6501/aa870c, 2017.
Persijn, S. T. and Baldan, A.: A new look at the sorption kinetics in reference gas standards, Meas. Sci. Technol., 34, 115018, https://doi.org/10.1088/1361-6501/ace9ee, 2023.
Placet, M.: Emissions of ozone precursors from stationary sources: a critical review, Atmos. Environ., 34, 2183–2204, https://doi.org/10.1016/S1352-2310(99)00464-1, 2000.
Pugliese, S. C., Murphy, J. G., Geddes, J. A., and Wang, J. M.: The impacts of precursor reduction and meteorology on ground-level ozone in the Greater Toronto Area, Atmos. Chem. Phys., 14, 8197–8207, https://doi.org/10.5194/acp-14-8197-2014, 2014.
Reimann, S., Wegener, R., Claude, A., and Sauvage, S.: Deliverable 3.17, Updated measurement guideline for NOx and VOCs, ACTRIS report, 103 pp., https://actris.eu/sites/default/files/Documents/ACTRIS-2/Deliverables/WP3_D3.17_M42.pdf (last access: 20 October 2023), 2018.
Rhoderick, G. C., Cecelski, C. E., Miller, W. R., Worton, D. R., Moreno, S., Brewer, P. J., Viallon, J., Idrees, F., Moussay, P., Kim, Y. D., Kim, D., Lee, S., Baldan, A., and Li, J.: Stability of gaseous volatile organic compounds contained in gas cylinders with different internal wall treatments, Elementa: Science of the Anthropocene, 7, 28, https://doi.org/10.1525/elementa.366, 2019.
Richter, W.: Recommendations on quantities, symbols and measurement units for publications in ACQUAL, Accredit. Qual. Assur., 12, 497–498, https://doi.org/10.1007/s00769-007-0273-6, 2007.
Seinfeld, J. H., Bretherton, C., Carslaw, K. S., Coe, H., DeMott, P. J., Dunlea, E. J., Feingold, G., Ghan, S., Guenther, A. B., Kahn, R., Kraucunas, I., Kreidenweis, S. M., Molina, M. J., Nenes, A., Penner, J. E., Prather, K. A., Ramanathan, V., Ramaswamy, V., Rasch, P. J., Ravishankara, A. R., Rosenfeld, D., Stephens, G., and Wood, R.: Improving our fundamental understanding of the role of aerosol-cloud interactions in the climate system, P. Natl. Acad. Sci. USA, 113, 5781–5790, https://doi.org/10.1073/pnas.1514043113, 2016.
Schultz, M. G., Akimoto, H., Bottenheim, J., Buchmann, B., Galbally, I. E., Gilge, S., Helmig, D., Koide, H., Lewis, A. C., Novelli, P. C., Plass-Dölmer, C., Ryerson, T. B., Steinbacher, M., Steinbrecher, R., Tarasova, O., Tørseth, K., Thouret, V., and Zellweger, C.: The Global Atmosphere Watch reactive gases measurement network, Elementa: Science of the Anthropocene, 3, 000067, https://doi.org/10.12952/journal.elementa.000067, 2015.
Schultz, M. G., Schröder, S., Lyapina, O., Cooper, O. R., Galbally, I., Petropavlovskikh, I., Von Schneidemesser, E., Tanimoto, H., Elshorbany, Y., Naja, M., Seguel, R. J., Dauert, U., Eckhardt, P., Feigenspan, S., Fiebig, M., Hjellbrekke, A. G., Hong, Y. D., Kjeld, P. C., Koide, H., Lear, G., Tarasick, D., Ueno, M., Wallasch, M., Baumgardner, D., Chuang, M. T., Gillett, R., Lee, M., Molloy, S., Moolla, R., Wang, T., Sharps, K., Adame, J. A., Ancellet, G., Apadula, F., Artaxo, P., Barlasina, M. E., Bogucka, M., Bonasoni, P., Chang, L., Colomb, A., Cuevas-Agulló, E., Cupeiro, M., Degorska, A., Ding, A., Fröhlich, M., Frolova, M., Gadhavi, H., Gheusi, F., Gilge, S., Gonzalez, M. Y., Gros, V., Hamad, S. H., Helmig, D., Henriques, D., Hermansen, O., Holla, R., Hueber, J., Im, U., Jaffe, D. A., Komala, N., Kubistin, D., Lam, K. S., Laurila, T., Lee, H., Levy, I., Mazzoleni, C., Mazzoleni, L. R., McClure-Begley, A., Mohamad, M., Murovec, M., Navarro-Comas, M., Nicodim, F., Parrish, D., Read, K. A., Reid, N., Ries, L., Saxena, P., Schwab, J. J., Scorgie, Y., Senik, I., Simmonds, P., Sinha, V., Skorokhod, A. I., Spain, G., Spangl, W., Spoor, R., Springston, S. R., Steer, K., Steinbacher, M., Suharguniyawan, E., Torre, P., Trickl, T., Weili, L., Weller, R., Xiaobin, X., Xue, L., and Zhiqiang, M.: Tropospheric Ozone Assessment Report: Database and metrics data of global surface ozone observations, Elementa: Science of the Anthropocene, 5, 58, https://doi.org/10.1525/elementa.244, 2017.
Shao, M., Lu, S., Liu, Y., Xie, X., Chang, C., Huang, S., and Chen, Z.: Volatile organic compounds measured in summer in Beijing and their role in ground-level ozone formation, J. Geophys. Res.-Atmos., 114, D00G06, https://doi.org/10.1029/2008JD010863, 2009.
Shrivastava, M., Cappa, C. D., Fan, J., Goldstein, A. H., Guenther, A. B., Jimenez, J. L, Kuang, C., Laskin, A., Martin, S. T., Ng, N. L., Petaja, T., Pierce, J. R., Rasch, P. J., Roldin, P., Seinfeld, J. H., Shilling, J, Smith, J. N., Thornton, J. A., Volkamer, R., Wang, J., Worsnop, D. R., Zaveri, R. A., Zelenyuk, A., and Zhang, Q.: Recent advances in understanding secondary organic aerosol: Implications for global climate forcing, Rev. Geophys., 55, 509–559, https://doi.org/10.1002/2016RG000540, 2017.
Simon, H., Reff, A., Wells, B., Xing, J., and Frank, N.: Ozone trends across the United States over a period of decreasing NOx and VOC emissions, Environ. Sci. Technol., 49, 186–195, https://doi.org/10.1021/es504514z, 2015.
Simon, L., Gros, V., Petit, J.-E., Truong, F., Sarda-Estève, R., Kalalian, C., Baudic, A., Marchand, C., and Favez, O.: Two years of volatile organic compound online in situ measurements at the Site Instrumental de Recherche par Télédétection Atmosphérique (Paris region, France) using proton-transfer-reaction mass spectrometry, Earth Syst. Sci. Data, 15, 1947–1968, https://doi.org/10.5194/essd-15-1947-2023, 2023.
Sternberg, J. C., Gallaway, W. S., and Jones, D. T. L.: The mechanism of response of flame ionization detectors, in: Gas Chromatography: Third International Symposium Held Under the Auspices of the Analysis Instrumentation Division of the Instrument Society of America, edited by: Brenner, N., Callen, J. E., and Weiss, M. D., Academic Press, New York and London, 231–267, 1962.
Stohl, A., Bonasoni, P., Cristofanelli, P., Collins, W., Feichter, J., Frank, A., Forster, C., Gerasopoulos, E., Gäggeler, H., James, P., Kentarchos, T., Kromp-Kolb, H., Krüger, B., Land, C., Meloen, J., Papayannis, A., Priller, A., Seibert, P., Sprenger, M., Roelofs, G. J., Scheel, H. E., Schnabel, C., Siegmund, P., Tobler, L., Trickl, T., Wernli, H., Wirth, V., Zanis, P., and Zerefos, C.: Stratosphere-troposphere exchange: A review, and what we have learned from STACCATO, J. Geophys. Res.-Atmos., 108, 8516, https://doi.org/10.1029/2002jd002490, 2003.
Szopa, S., Naik, V., Ahikary, B., Artaxo, P., Berntsen, T., Collins, W., Fuzzi, S., Gallardo, L., Kiendler-Scharr, A., Kimont, Z., Liao, H., Unger, N., and Zanis, P.: Short-lived Climate Forcers, in: Climate Change 2021 – The Physical Science Basis. Contribution of Working Group I to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge University Press, 817–922, https://doi.org/10.1017/9781009157896.008, 2023.
Tan, Z., Lu, K., Hofzumahaus, A., Fuchs, H., Bohn, B., Holland, F., Liu, Y., Rohrer, F., Shao, M., Sun, K., Wu, Y., Zeng, L., Zhang, Y., Zou, Q., Kiendler-Scharr, A., Wahner, A., and Zhang, Y.: Experimental budgets of OH, HO2, and RO2 radicals and implications for ozone formation in the Pearl River Delta in China 2014, Atmos. Chem. Phys., 19, 7129–7150, https://doi.org/10.5194/acp-19-7129-2019, 2019.
Tarasick, D., Galbally, I. E., Cooper, O. R., Schultz, M. G., Ancellet, G., Leblanc, T., Wallington, T. J., Ziemke, J., Liu, X., Steinbacher, M., Staehelin, J., Vigouroux, C., Hannigan, J. W., García, O., Foret, G., Zanis, P., Weatherhead, E., Petropavlovskikh, I., Worden, H., Osman, M., Liu, J., Chang, K. L., Gaudel, A., Lin, M., Granados-Muñoz, M., Thompson, A. M., Oltmans, S. J., Cuesta, J., Dufour, G., Thouret, V., Hassler, B., Trickl, T., and Neu, J. L.: Tropospheric ozone assessment report: Tropospheric ozone from 1877 to 2016, observed levels, trends and uncertainties, Elementa: Science of the Anthropocene, 7, 39, https://doi.org/10.1525/elementa.376, 2019.
van der Veen, A.M.H, Meija, J., Possolo, A., and Hibbert, D.B.: Interpretation and use of standard atomic weights (IUPAC Technical Report), Pure Appl. Chem., 93, 629–646, https://doi.org/10.1515/pac-2017-1002, 2021.
Van Dingenen, R., Dentener, F. J., Raes, F., Krol, M. C., Emberson, L., and Cofala, J.: The global impact of ozone on agricultural crop yields under current and future air quality legislation, Atmos. Environ., 43, 604–618, https://doi.org/10.1016/j.atmosenv.2008.10.033, 2009.
Volkamer, R., Sheehy, P., Molina, L. T., and Molina, M. J.: Oxidative capacity of the Mexico City atmosphere – Part 1: A radical source perspective, Atmos. Chem. Phys., 10, 6969–6991, https://doi.org/10.5194/acp-10-6969-2010, 2010.
Wang, S., Hornbrook, R. S., Hills, A., Emmons, L. K., Tilmes, S., Lamarque, J. F., Jimenez, J. L., Campuzano-Jost, P., Nault, B. A., Crounse, J. D., Wennberg, P. O., Kim, M., Allen, H., Ryerson, T. B., Thompson, C. R., Peischl, J., Moore, F., Nance, D., Hall, B., Elkins, J., Tanner, D., Huey, L. G., Hall, S. R., Ullmann, K., Orlando, J. J., Tyndall, G. S., Flocke, F. M., Ray, E., Hanisco, T. F., Wolfe, G. M., St. Clair, J., Commane, R., Daube, B., Barletta, B., Blake, D. R., Weinzierl, B., Dollner, M., Conley, A., Vitt, F., Wofsy, S. C., Riemer, D. D., and Apel, E. C.: Atmospheric Acetaldehyde: Importance of Air-Sea Exchange and a Missing Source in the Remote Troposphere, Geophys. Res. Lett., 46, 5601–5613, https://doi.org/10.1029/2019GL082034, 2019.
Wild, O.: Modelling the global tropospheric ozone budget: exploring the variability in current models, Atmos. Chem. Phys., 7, 2643–2660, https://doi.org/10.5194/acp-7-2643-2007, 2007.
Worton, D. R., Moreno, S., Brewer, P. J., Li, J., Baldan, A., and van der Veen, A. M. H.: Bilateral comparison of primary reference materials (PRMs) containing methanol, ethanol and acetone in nitrogen, Accredit. Qual. Assur., 27, 265–274, https://doi.org/10.1007/s00769-022-01513-y, 2022.
Worton, D. R., Moreno, S., O'Daly, K., and Holzinger, R.: Development of an International System of Units (SI)-traceable transmission curve reference material to improve the quantitation and comparability of proton-transfer-reaction mass-spectrometry measurements, Atmos. Meas. Tech., 16, 1061–1072, https://doi.org/10.5194/amt-16-1061-2023, 2023.
Wu, C., Wang, C., Wang, S., Wang, W., Yuan, B., Qi, J., Wang, B., Wang, H., Wang, C., Song, W., Wang, X., Hu, W., Lou, S., Ye, C., Peng, Y., Wang, Z., Huangfu, Y., Xie, Y., Zhu, M., Zheng, J., Wang, X., Jiang, B., Zhang, Z., and Shao, M.: Measurement report: Important contributions of oxygenated compounds to emissions and chemistry of volatile organic compounds in urban air, Atmos. Chem. Phys., 20, 14769–14785, https://doi.org/10.5194/acp-20-14769-2020, 2020.
Xue, L. K., Wang, T., Gao, J., Ding, A. J., Zhou, X. H., Blake, D. R., Wang, X. F., Saunders, S. M., Fan, S. J., Zuo, H. C., Zhang, Q. Z., and Wang, W. X.: Ground-level ozone in four Chinese cities: precursors, regional transport and heterogeneous processes, Atmos. Chem. Phys., 14, 13175–13188, https://doi.org/10.5194/acp-14-13175-2014, 2014.
Yang, Y., Shao, M., Wang, X., Nölscher, A. C., Kessel, S., Guenther, A., and Williams, J.: Towards a quantitative understanding of total OH reactivity: A review, Atmos. Environ., 134, 147–161, https://doi.org/10.1016/j.atmosenv.2016.03.010, 2016.
Young, P. J., Naik, V., Fiore, A. M., Gaudel, A., Guo, J., Lin, M. Y., Neu, J. L., Parrish, D. D., Rieder, H. E., Schnell, J. L., Tilmes, S., Wild, O., Zhang, L., Ziemke, J., Brandt, J., Delcloo, A., Doherty, R. M., Geels, C., Hegglin, M. I., Hu, L., Im, U., Kumar, R., Luhar, A., Murray, L., Plummer, D., Rodriguez, J., Saiz-Lopez, A., Schultz, M. G., Woodhouse, M. T., and Zeng, G.: Tropospheric Ozone Assessment Report: Assessment of global-scale model performance for global and regional ozone distributions, variability, and trends, Elementa: Science of the Anthropocene, 6, 49, https://doi.org/10.1525/elementa.265, 2018.
Zborowska, A. G., MacInnis, C. Y., Ye, C. Z., and Osthoff, H. D.: On the photolysis branching ratio of methyl ethyl ketone, Atmos. Environ., 254, 118383, https://doi.org/10.1016/j.atmosenv.2021.118383, 2021.
Zhang, H., Wu, S., Huang, Y., and Wang, Y.: Effects of stratospheric ozone recovery on photochemistry and ozone air quality in the troposphere, Atmos. Chem. Phys., 14, 4079–4086, https://doi.org/10.5194/acp-14-4079-2014, 2014.
- Abstract
- Introduction
- Working standards traceable to the International System of Units (SI)
- Assessment of the SI-traceable working standards
- Results
- Conclusions
- Appendix A: Analytical instruments and dilution systems
- Appendix B: Purity analysis, stability evaluation and verification of the reference gas mixtures (RGMs)
- Appendix C: Whole-air sample spiking and certification
- Appendix D: Assessment of the working standards traceable to the International System of Units (SI)
- Data availability
- Author contributions
- Competing interests
- Disclaimer
- Special issue statement
- Acknowledgements
- Financial support
- Review statement
- References
- Abstract
- Introduction
- Working standards traceable to the International System of Units (SI)
- Assessment of the SI-traceable working standards
- Results
- Conclusions
- Appendix A: Analytical instruments and dilution systems
- Appendix B: Purity analysis, stability evaluation and verification of the reference gas mixtures (RGMs)
- Appendix C: Whole-air sample spiking and certification
- Appendix D: Assessment of the working standards traceable to the International System of Units (SI)
- Data availability
- Author contributions
- Competing interests
- Disclaimer
- Special issue statement
- Acknowledgements
- Financial support
- Review statement
- References