the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 07 Jun 2018
| 07 Jun 2018
The sensitivity of benzene cluster cation chemical ionization mass spectrometry to select biogenic terpenes
Avi Lavi
Michael P. Vermeuel
Gordon A. Novak
Timothy H. Bertram
Benzene cluster cations are a sensitive and selective reagent ion for chemical ionization of select biogenic volatile organic compounds. We have previously reported the sensitivity of a field deployable chemical ionization time-of-flight mass spectrometer (CI-ToFMS), using benzene cluster cation ion chemistry, for detection of dimethyl sulfide, isoprene and α-pinene. Here, we present laboratory measurements of the sensitivity of the same instrument to a series of terpenes, including isoprene, α-pinene, β-pinene, D-limonene, ocimene, β-myrcene, farnesene, α-humulene, β-caryophyllene, and isolongifolene at atmospherically relevant mixing ratios (< 100 pptv). In addition, we determine the dependence of CI-ToFMS sensitivity on the reagent ion neutral delivery concentration and water vapor concentration. We show that isoprene is primarily detected as an adduct (C5H8 ⋅ ) with a sensitivity ranging between 4 and 10 ncps ppt−1, which depends strongly on the reagent ion precursor concentration, de-clustering voltages, and specific humidity (SH). Monoterpenes are detected primarily as the molecular ion () with an average sensitivity, across the five measured compounds, of 14 ± 3 ncps ppt−1 for SH between 7 and 14 g kg−1, typical of the boreal forest during summer. Sesquiterpenes are detected primarily as the molecular ion () with an average sensitivity, across the four measured compounds, of 9.6 ± 2.3 ncps ppt−1, that is also independent of specific humidity. Comparable sensitivities across broad classes of terpenes (e.g., monoterpenes and sesquiterpenes), coupled to the limited dependence on specific humidity, suggest that benzene cluster cation CI-ToFMS is suitable for field studies of biosphere–atmosphere interactions.
- Article
(7877 KB) - Full-text XML
- BibTeX
- EndNote





The annual global emission of biogenic volatile organic compounds (BVOCs) is estimated at 1000 TgC yr−1 and exceeds the total VOC emissions from anthropogenic activities (Guenther et al., 2012; IPCC). Foliage emissions account for 90 % of global BVOC emissions, of which isoprene (C5H8), monoterpenes (MTs; C10H16), and sesquiterpenes (SQTs; C15H24) are the primary constituents (Guenther et al., 1995). The emission rate and the chemical composition of emitted BVOCs is a complex function of the vegetation species and the wide array of stress factors that it is exposed to (Hallquist et al., 2009; Lang-Yona et al., 2010; Zhao et al., 2017). Atmospheric oxidation of BVOCs results in the formation of low-volatility compounds that can lead to new particle formation (Jokinen et al., 2015; Kirkby et al., 2016) and particle growth through secondary organic aerosol formation (Allan et al., 2006; Wiedensohler et al., 2009). Both of these processes impact Earth's radiative budget by scattering solar radiation and/or altering cloud formation and precipitation (Chung et al., 2012). The contribution of different types of BVOCs (e.g., isoprene, MTs and SQTs) to secondary organic aerosols (SOA) differ significantly (Zhao et al., 2017). Therefore, uncertainties in BVOCs emissions present significant issues in estimating net climate forcing (Kerminen et al., 2005; Kulmala et al., 2004). Identification of the chemical composition of the emitted BVOCs and quantification of the surface exchange rates of these compounds are essential for understanding complex and nonlinear biosphere–atmosphere interactions.
Chemical ionization mass spectrometry (CIMS) is a commonly utilized selective and sensitive method for in situ detection of trace gases (Huey, 2007). The sensitivity and selectivity towards a specific compound or class of compounds having similar functional groups rely on the selection of an appropriate ion (i.e., reagent ion) that reacts with and ionizes the analyte via an ion–molecule reaction. For example, iodide ions have been used to measure reactive nitrogen compounds, halogen-containing species, and oxygenated VOCs (Lopez-Hilfiker et al., 2015; Riedel et al., 2012; Thornton et al., 2010); CF3O− has been used for the detection of peroxides and organic nitrates (Crounse et al., 2006); NO+ has been used for the selective detection of primary alcohols and alkenes (Hunt and Harvey, 1975; Hunt et al., 1982; Karl et al., 2012; Koss et al., 2016; Mochalski et al., 2014); H3O+ has been used for VOCs and their oxygenated products (Lindinger et al., 1998); and benzene cluster cations have been used for dimethyl sulfide (DMS), isoprene, and terpenes (Kim et al., 2016; Leibrock and Huey, 2000).
The benzene cation clusters spontaneously with neutral benzene via attractive, non-covalent interactions (Chipot et al., 1996; Grover et al., 1987). Leibrock and Huey (2000) and recently Kim et al. (2016) demonstrated that select VOCs including isoprene, MTs, SQTs and aromatic compounds can be ionized by benzene cation clusters. Kim et al. studied the parameters that control the benzene cation cluster distribution (C6H6)+ ⋅ (C6H6)n at the operational conditions of the CI-ToFMS, concluding that, for the specific operating conditions used, the reagent ion within the ion–molecule reaction chamber was primarily in the form of the benzene dimer or larger clusters (Kim et al., 2016). This conclusion is in agreement with studies showing that the dissociation energy of the benzene cation dimer is significantly higher than that of the trimer or larger benzene cation clusters (Krause et al., 1991), suggesting that ionization in the CI-ToFMS by benzene cluster cations proceeds primarily through clusters that are at least the size of the benzene cation dimer.
The ionization mechanism for a given analyte (M) with the benzene cation dimer depends on the ionization energy (IE) of the analyte. Charge transfer (R1) is expected to be the dominant reaction for analytes having ionization energies smaller than the benzene dimer (8.69 eV) (Grover et al., 1987). In cases when the analyte IE is higher than that of benzene cation dimer, charge transfer is thermodynamically unfavored and adduct formation (R2) or ligand exchange (R3) are the sole modes of ionization. The ligand exchange product (R3) was previously reported for isoprene, dimethyl sulfide and select alkenes; however, the reaction pathway is not known (Kim et al., 2016; Leibrock and Huey, 2000).
The low IE of benzene clusters (8.69 eV for the dimer and even smaller for larger benzene cation clusters) (Grover et al., 1987; Shinohara and Nishi, 1989) is a major advantage in the quantification of monoterpenes or larger volatile organic compounds such as sesquiterpenes. The IE of these compounds is slightly smaller than that of the benzene dimer (e.g., 8.3 eV for β-caryophyllene; Novak et al., 2001) and the minimal excess energy in charge transfer reactions results in limited fragmentation. For example, approximately 60 % of β-caryophyllene was detected in its molecular ionic form (M+) in comparison to significant fragmentation observed by proton transfer reaction mass spectrometry (PTR-MS) (Kim et al., 2014, 2009).
The field-deployable CIMS that utilizes a time-of-flight mass analyzer (ToFMS), previously described by Kim et al., combines the efficient production and transmission of ions at high pressure (e.g., 75 mbar) with the high ion duty cycle of orthogonal extraction ToFMS (Bertram et al., 2011). This instrument configuration is highly sensitive and capable of measuring and logging mass spectra (10–800 m∕Q) at rates higher than 10 Hz (Bertram et al., 2011). These benefits make CI-ToFMS highly applicable for studying atmospheric exchange processes of trace gases at the air–ocean interface that require fast response rates (Kim et al., 2014). However, at these pressures, the distribution of benzene clusters and their associated ion–molecule reactions times are not well constrained. Unlike PTR-MS, it is not possible to directly derive the analyte mixing ratio from laboratory studies of the ion–molecule kinetics (reaction rates) that are conducted at lower pressure in which both the reaction times and cluster distribution have been previously determined. As such, quantitative analysis of atmospheric trace gases using high pressure CIMS necessitates either a direct or empirical calibration for each analyte as a function of the atmospheric conditions (e.g., humidity or temperature).
In what follows, we build on earlier studies in our group (Kim et al., 2016), which described the use of benzene cluster cations as a reagent ion for the detection and quantification of dimethyl sulfide, isoprene, and α-pinene. At the time of Kim et al. (2016), it was not known if benzene ion chemistry was equally sensitive to all monoterpene compounds, to what extent CI-ToFMS sensitivity was dependent on ambient specific humidity for a broad range of monoterpenes and sesquiterpenes, and the source of organic impurities in the reagent ion delivery. Here, we address each of these topics.
In this paper, we describe a high-purity liquid benzene source, which permits operation of the CI-ToFMS at higher reagent ion concentrations. We discuss the sensitivity of benzene cluster cation chemistry to a select number of terpenes (Fig. 1) at atmospherically relevant mixing ratios (< 500 pptv). We report on the effect of atmospheric water vapor and the neutral benzene reagent ion precursor concentration on CI-ToFMS sensitivity to select terpenes (isoprene, α- and β-pinene, D-limonene, β-myrcene, ocimene, farnesene, isolongifolene, α-humulene, and β-caryophyllene). We also examine the de-clustering power of the radio frequency (RF)-only quadrupole to better determine the cluster distribution present in the ion molecule reaction chamber.

2.1 Materials
The following analytes were purchased from Sigma-Aldrich and used with no further purification: isoprene, α-pinene, β-pinene, D-limonene (≥ 99 %), β-myrcene (96.2 %), ocimene (97.0 %, as a mixture of isomers), farnesene (> 90.0 %, as a mixture of isomers) α-humulene (> 96.5 %), β-caryophyllene (≥ 98.5 %), isolongifolene (≥ 98.0 %, as a mixture of isomers), benzene (≥ 99.5 %), and chloroform-d (99.8 at. % D). A compressed gas cylinder of 0.184 ppm of DMS-d3 in N2 was purchased from Praxair, USA. Water was supplied from a Milli-Q system at 18.2 MΩ cm. Nitrogen was used from a UHP liquid N2 dewar (Airgas). UHP (99.999 %) oxygen cylinders were purchased from Airgas.
2.2 Chemical ionization mass spectrometer
The detailed description of the CI-ToFMS (Tofwerk AG, Switzerland, and Aerodyne Research Inc., USA) and its performance are discussed in Bertram et al. (2011). In brief, reagent ions are generated by passing 10 sccm of UHP N2 over the headspace of a liquid benzene reservoir contained in a stainless steel bottle. Benzene vapor is diluted with 2.2 slpm of N2, prior to delivery to the 210Po source. The benzene vapor mixing ratio is estimated from the dilution ratio and benzene vapor pressure. In the experiments discussed here, we varied the benzene concentration between 60 and 360 ppm. A combination of stainless steel and Teflon tubing was used to transfer benzene vapors to minimize extraction of organic compounds from the tubing. Following dilution, benzene vapor flows through a 10 mCi α emitting radioactive 210Po source (NRD 2021–1000). The collision of α-particles with N2 results in the formation of N ions that ionize the benzene clusters (Dondes et al., 1966). The analyte sample is mixed with the formed benzene cluster cations at the ion–molecule reactor (IMR) held at 75 mbar. At this pressure, the estimated analyte residence time in the IMR is 100 ms. The reagent and product ions are transmitted from the IMR chamber into a collisional dissociation chamber (CDC, P= 2 mbar) equipped with a RF-only ion-guide quadrupole, followed by a subsequent chamber (P= 1.4 × 10−2 mbar) in which a second RF-only quadrupole is used to focus the ion beam. The ion beam is then guided by a further set of ion optics to the entrance point of the extraction region of the compact time-of-flight mass analyzer (Tofwerk AG, Switzerland).
2.3 Liquid calibration unit
A custom liquid calibration system was developed to deliver known, atmospherically relevant mixing ratios (< 500 pptv) of gas-phase terpenes to the CI-ToFMS. The liquid calibration system uses a syringe pump to continuously evaporate known quantities of solution into a heated carrier gas flow, generating known mixing ratios of select terpenes. To produce trace concentrations of each analyte, the standard liquid material was diluted in series with chloroform-d using a set of calibrated auto pipettes. Chloroform-d was chosen due to its solvent properties and low boiling point (61 ∘C) that enhances the evaporation of the analyte. Due to its ionization energy (IE > 11 eV; Bieri et al., 1981), higher than that of benzene cation clusters, it was expected that chloroform would not be ionized and would have negligible impact on the benzene cluster cation ionization mechanisms. To assess this, mass spectra were recorded for solutions containing solely deuterated chloroform for a variety of different pump flows from 0 to 5 µL min−1. We did not observe the molecular cation of chloroform-d (CDCl, 120 m∕Q) and only very small signatures of the fragments (at 48, 84, or 86 m∕Q) were observed (Fig. 2), consistent with the IE of chloroform-d being higher than that of the reagent ions (11.37 ± 0.02 eV compared with 8.69 eV) (Grover et al., 1987) (Werner et al., 1974). It was also determined that concentration of deuterated chloroform did not interfere with reagent ion or water cluster signal intensities.
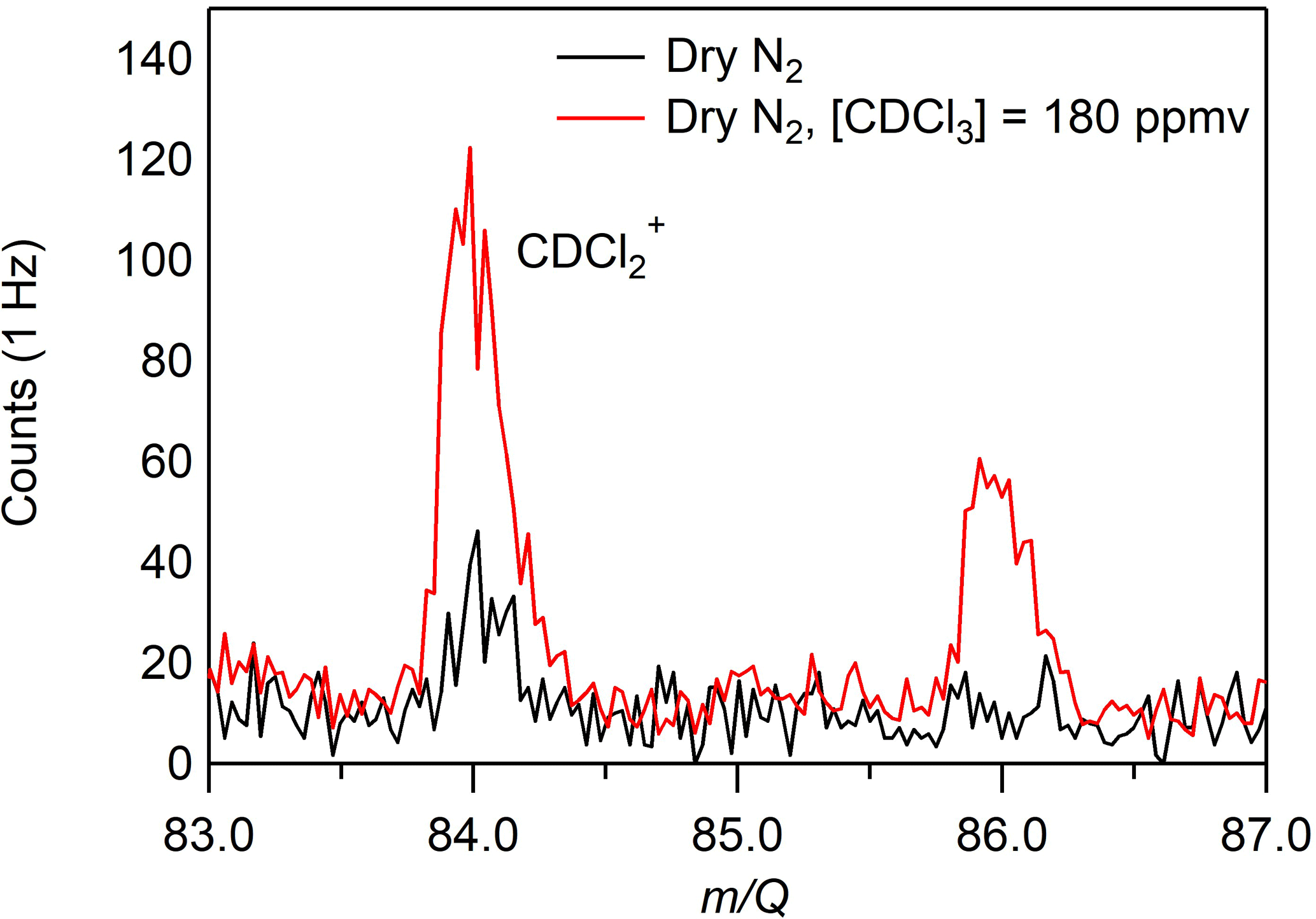
Figure 2CI-ToFMS mass spectrum acquired when overflowing the inlet with excess nitrogen (black) and for a nebulized solution of chloroform-d at a flow rate of 3 µL min−1 in a nitrogen carrier gas (red), where the resulting [CDCl3] = 180 ppmv. No signal was observed above the baseline for any other fragments or the parent (CDCl, 120 m∕Q).
To evaporate the analyte solution, a controlled amount (0–5 µL min−1) of the analyte solution was delivered by a syringe pump (Harvard Apparatus, model 11) via PEEK tubing (Upchurch Scientific) into a heated carrier stream resulting in CDCl3 mixing ratios from 60 to 300 ppmv. A synthetic 80:20 N2 : O2 mixture was used as zero air and heated by an in-line gas heater (Omega, AHP-3741). The temperature of the zero air flow at the point of intersection with the PEEK tubing was kept at 80 ∘C via a PID temperature controller (Omega, CN9300). Excess zero air flow was used to ensure an overflow of the CIMS inlet. The trace concentration of the evaporated analytes and the elevated temperature in front of the inlet (ca. 50 ∘C) helped to prevent re-condensation of the analyte on the inlet tubing. Humidified zero air was generated by passing a fraction of the total flow through the head space of a water reservoir. The relative humidity (RH) of the total air flow was measured using a relative humidity sensor (Vaisala, HMP110), calibrated using the procedure described in Greenspan (1977).
The sensitivities reported in this paper are presented in normalized counts per second per pptv (ncps pptv−1). We normalized the analyte ion count rates by the sum of the benzene cation monomer (78 m∕Q) and dimer (156 m∕Q) count rates to a reference of 1 × 106 counts per second of total reagent ion signal in order to account for changes in ion transmission and generation over time. Sensitivities are calculated as the slope of the linear fit of each calibration curve of 5–7 steps (Fig. 3). Error bars are the standard deviation of repeated triplicate measurements. The performance of the liquid evaporation technique was validated by comparing the sensitivity to dimethyl-1,1,1-d3 sulfide (Praxair certified compressed gas standard, 0.184 ppm ± 10 %) diluted by zero air to a desired mixing ratio, with that of a diluted nebulized solution of DMS. The slope of the linear fit for calibration measurements from the pressurized cylinder (DMS-d3, 65 m∕Q) and the solution (DMS, 62 m∕Q) agreed to better than 10 %.
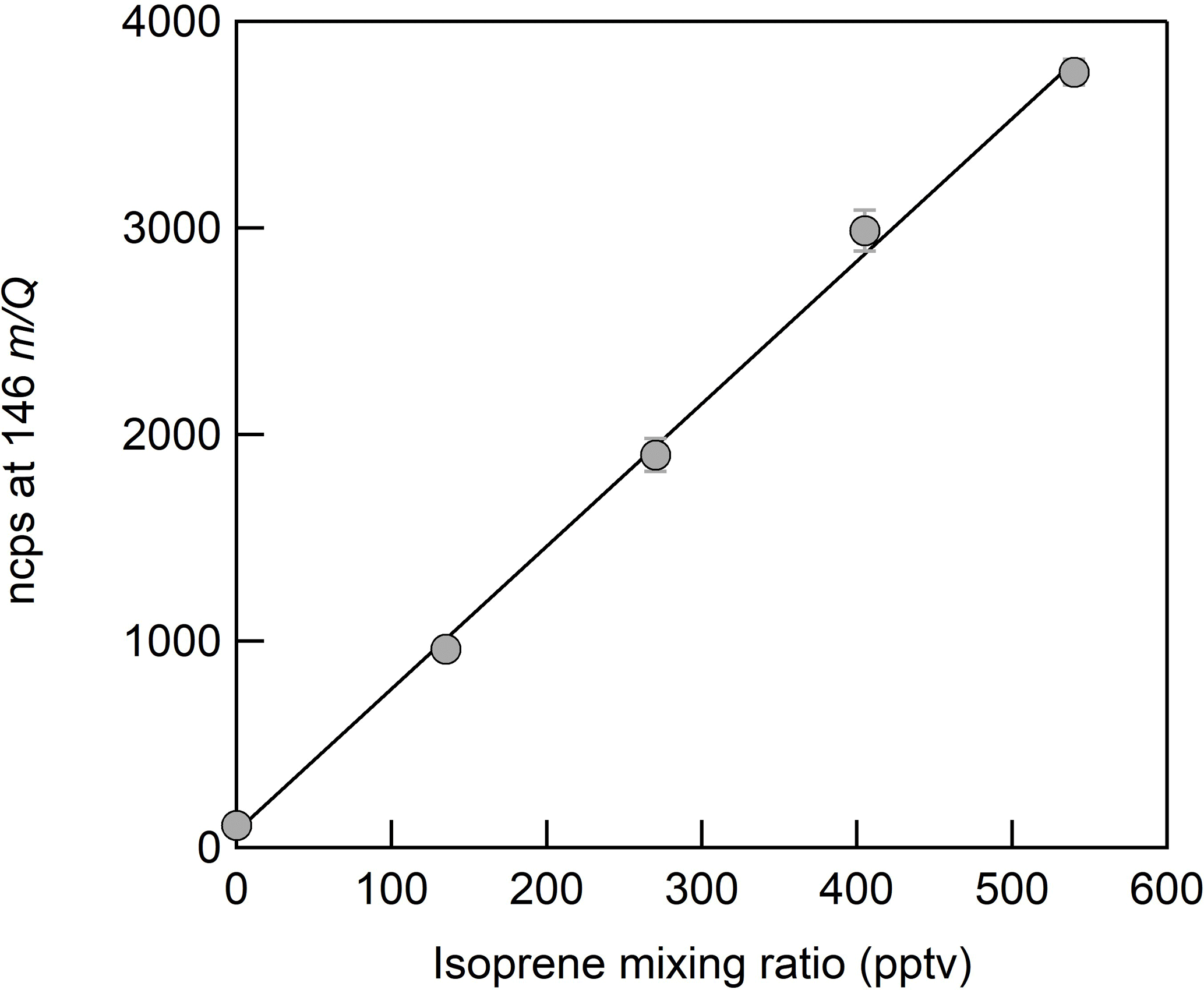
Figure 3CI-ToFMS calibration curve for isoprene, detected as ⋅ C5H8 at 146 m∕Q. The sensitivity (slope) is 7 ncps, R2= 0.99. Error bars represent the standard deviation of the triplicate calibrations.
3.1 Benzene cluster cation mass spectra
The CI-ToFMS mass spectra, obtained while overflowing the inlet with nominally dry zero air, are shown in Fig. 4a. To maximize the transmission of weakly bound ion–molecule adducts, we operated the instrument in all of the experiments described here with a minimal applied electric field between the instrument inlet and the entrance of the second RF-only quadrupole ion guide. The two primary peaks in the mass spectrum correspond to the benzene cation (; 78 m∕Q) and the benzene cation clustered to a single, neutral benzene ( ⋅ (C6H6); 156 m∕Q), where and ⋅ (C6H6) combined account over 90 % of the total ion current (TIC) for a benzene neutral concentration of 300 ppm. Benzene cation clusters larger than the dimer were not observed, as expected from their dissociation enthalpy, which is significantly smaller than that of the benzene cation clustered with a single neutral benzene molecule (Krause et al., 1991). The observed mass spectrum indicates significant ion intensity at 39, 50, 51, and 52 m∕Q that are attributed to the dissociation of the molecular () ion into its fragments , , , and , accounting for ca. 5 % of TIC. The fragmentation may result from the interaction of , α-particles, or electrons with benzene clusters in the ion molecule reaction region (Lifshitz and Reuben, 1969; Talebpour et al., 2000). For comparison, a similar spectrum is shown in Fig. 4b, using the same benzene neutral concentration and operating voltages, but without the RF and voltage bias applied to the first quadrupole ion guide. In this mode of operation, the total ion current is reduced by over 95 %, and and ⋅ (C6H6) are nearly equal in intensity, highlighting that benzene cluster collisional dissociation is occurring within this region. Even with the first RF-only quadrupole electronics turned off, the n= 2 cluster ( ⋅ (C6H6)2; 234 m∕Q) was not observed. Of notable absence (< 1 % TIC) in both Fig. 4a and b are the organic contaminants (92, 106, and 120 m∕Q) previously attributed to alkyl substituted benzene and protonated water clusters (H3O+ ⋅ (H2O)n; 19, 37, 55, and 73 m∕Q) that were present at high abundance (> 10 % of TIC) in Kim et al. (2016). It was postulated in Kim et al. that the source of the organic contaminants was the benzene compressed gas cylinder, as their combined contribution to TIC scaled with the neutral benzene concentration. It was also noted that low benzene neutral concentrations led to elevated water cluster abundance. This resulted in an optimum benzene neutral concentration of 10 ppm to balance the contributions from organic contaminants and water clusters. Here, we eliminate the organic contaminants through the use of a high-purity benzene liquid source permitting operation at higher neutral benzene concentrations (> 300 ppm). As discussed in Sect. 3.2, this has critical advantages for the detection of analytes such as isoprene and effectively eliminates competing ion chemistry stemming from protonated water clusters.
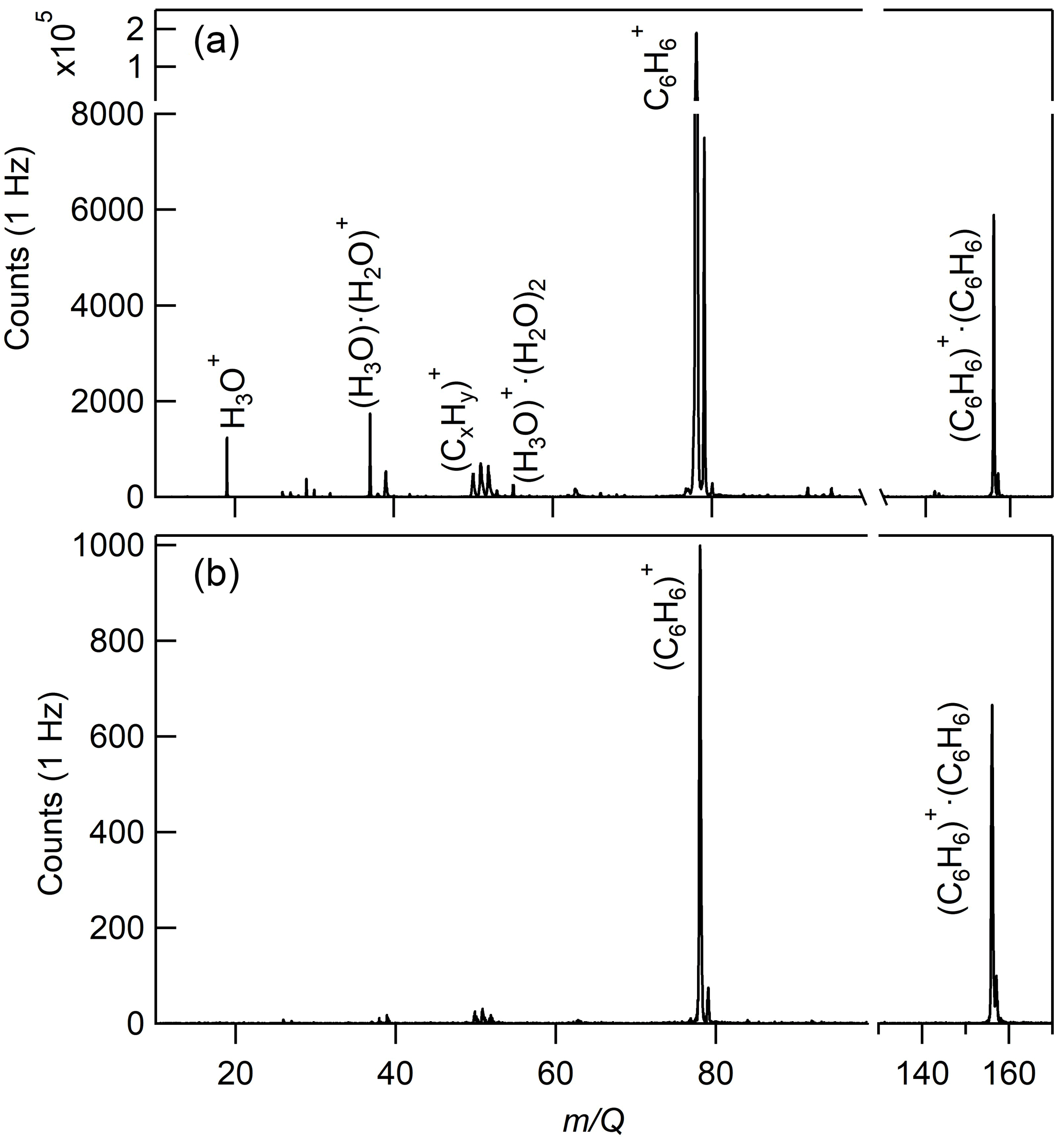
Figure 4(a) CI-ToFMS mass spectrum acquired when overflowing the inlet with nominally dry zero air for a benzene neutral concentration of 300 ppm using a liquid reagent ion delivery and (b) same as in (a), but with the first RF-only octupole ion guide turned off, resulting in a much weaker electric field strength.
In what follows we assess the CI-ToFMS sensitivity to a series of terpenes, including isoprene, α-pinene, β-pinene, D-limonene, ocimene, β-myrcene, farnesene, α-humulene, β-caryophyllene, and isolongifolene at atmospherically relevant mixing ratios (< 100 pptv) and determine the dependence of CI-ToFMS sensitivity on the reagent ion neutral delivery concentration (Sect. 3.2) and water vapor concentration (Sect. 3.3).
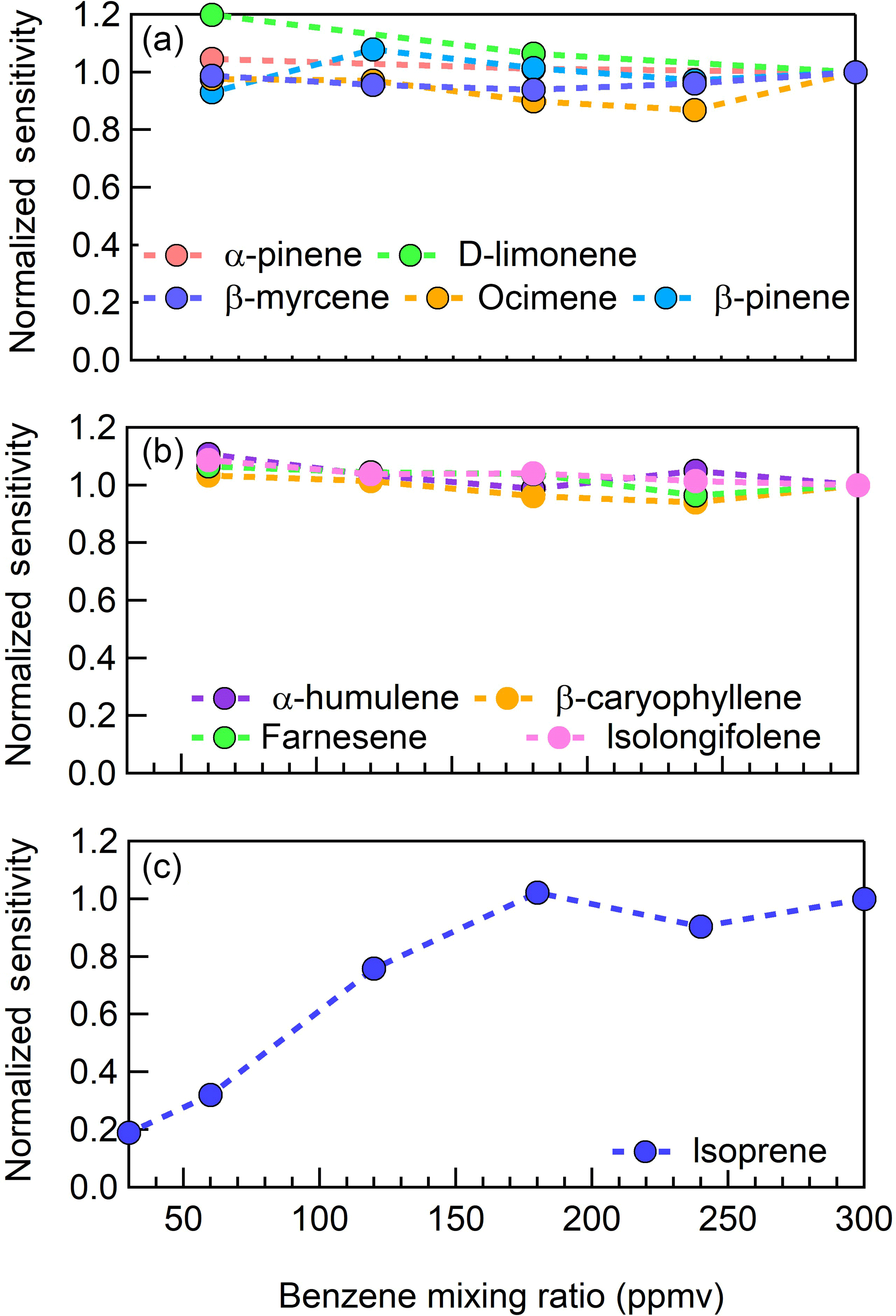
Figure 5CI-ToFMS sensitivity to (a) monoterpenes (; 136 m∕Q), (b) sesquiterpenes (; 204 m∕Q), and (c) isoprene ( ⋅ C5H8; 146 m∕Q) as a function of benzene neutral concentration normalized to the sensitivity at 300 ppmv neutral benzene. Measurements were conducted in nominally dry zero air.
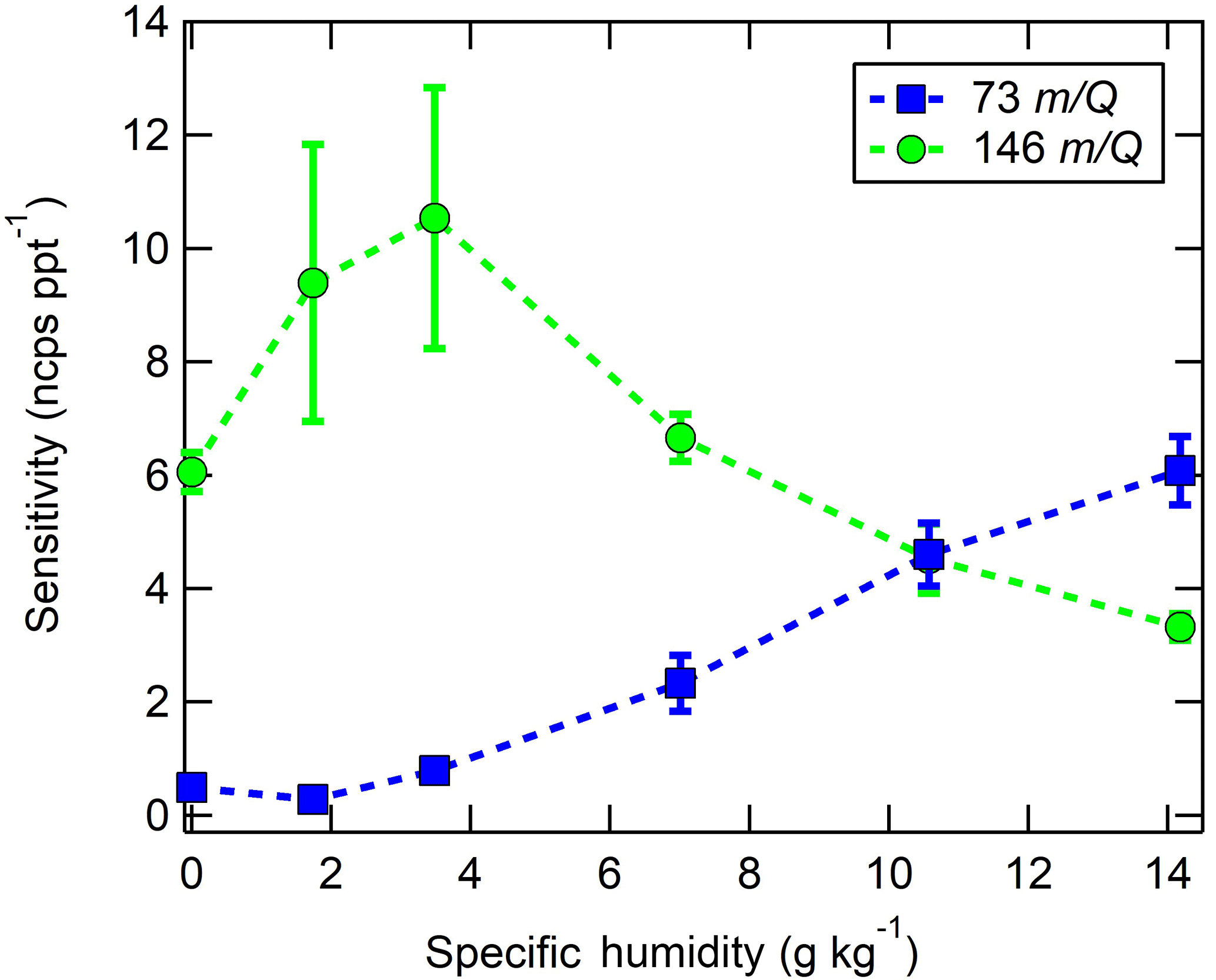
Figure 6Humidity-dependent CI-ToFMS sensitivities to isoprene (green circles, ⋅ C5H8, 146 m∕Q), and the protonated water tetramer (blue squares, H3O+ ⋅ (H2O)3, 73 m∕Q ), derived from calibration curves such as those shown in Fig. 6. The reported sensitivities are the average of triplicate calibration curves with all linear best fits having R2 > 0.98. Error bars represent the standard deviation of the triplicate calibrations. All calibrations were performed in zero air.
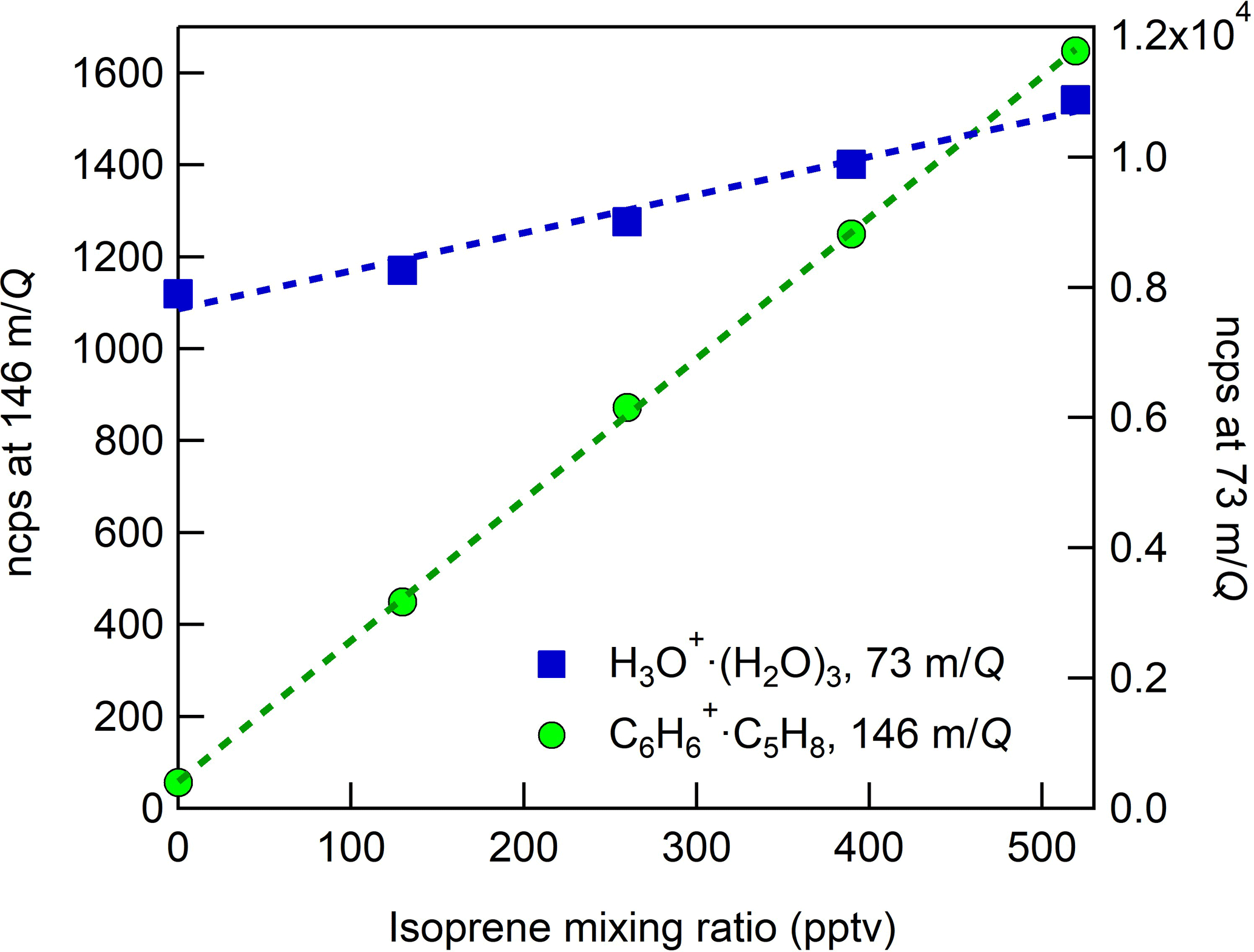
Figure 7CI-ToFMS sensitivity to isoprene, observed as the isoprene–benzene cluster (green circles, ⋅ C5H8, 146 m∕Q) and water protonated tetramer (blue squares, H3O+ ⋅ (H2O)3, 73 m∕Q). Dashed lines are the least square best-fit lines (R2 > 0.98). Calibration was performed at SH of 14 g kg−1 in zero air.

Figure 8Humidity-dependent sensitivities to select MTs detected as M+ (, 136 m∕Q). Error bars indicate the standard deviation of triplicate measurements. All calibrations were conducted in zero air. Error bars represent the standard deviation of the triplicate calibrations. All calibrations were performed in zero air.

Figure 9Normalized calibration of D-limonene for all major product ions (, 136 m∕Q, green circles), (, 135 m∕Q, orange squares), and (C10H16O, 168 m∕Q, purple triangles). Calibration was performed in zero air at 14 g kg−1 specific humidity (80 % RH at 23 ∘C). Dashed lines are least squares best-fit lines (all R2 > 0.99).
3.2 Impact of benzene neutral concentration on terpene sensitivity
We examined the impact of the benzene reagent ion precursor concentration on terpene sensitivity in nominally dry zero air for benzene neutral concentrations between 60 and 300 ppm. For the selection of monoterpenes and sesquiterpenes studied here, there was no indication that instrument sensitivity was dependent on the neutral benzene reagent ion precursor concentration between 60 and 300 ppm (Fig. 5a–b). In Fig. 5a–c, the reported sensitivity for each terpene is normalized to that measured at a benzene neutral concentration of 300 ppm. Unlike MTs and SQTs, the sensitivity of the isoprene–benzene adduct ( ⋅ C5H8; 146 m∕Q) strongly depends on the benzene concentration below 200 ppm (Fig. 5c) and therefore all the measurements in this study, were conducted at 300 ppm benzene. The cause for this dependence in benzene concentration is unclear as the exact mechanism for ⋅ C5H8 formation is unknown. It should also be noted that the sensitivity to DMS is independent of benzene concentration. Based on these analyses, we suggest that future studies utilizing benzene ion chemistry operate at neutral benzene reagent ion precursor concentrations of 300 ppm, generated from a high-purity liquid source.
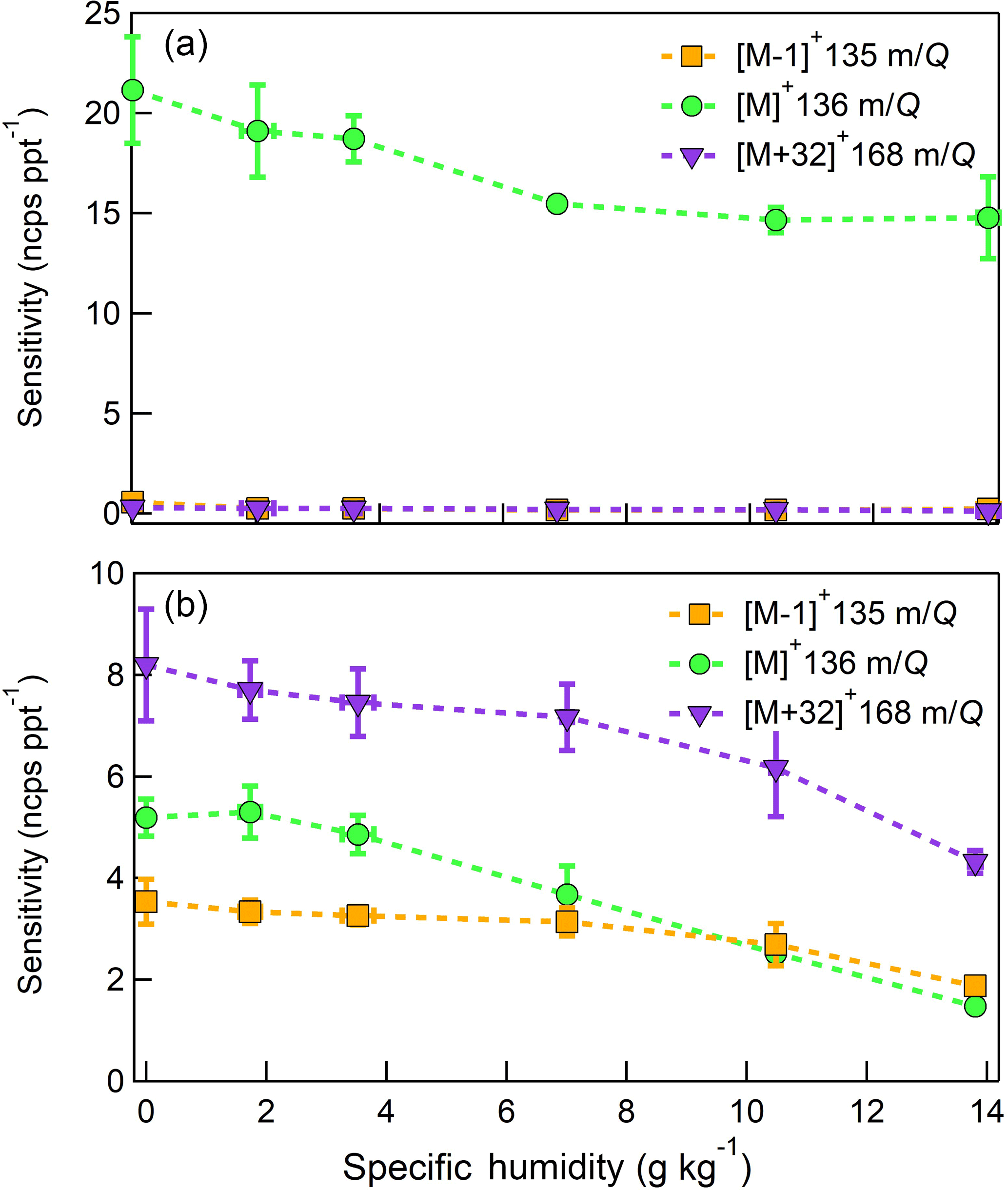
Figure 10Humidity-dependent, normalized sensitivities to (a) α-pinene (b) D-limonene for all major product ions (, 136 m∕Q, green circles), (, 135 m∕Q, orange squares), and (C10H16O, 168 m∕Q, purple triangles). Error bars represent the standard deviation of the triplicate calibrations. All calibrations were performed in zero air.
3.3 Impact of specific humidity on sensitivity
3.3.1 Isoprene
In these experiments, the specific humidity (SH) was varied between 0 and 14 g kg−1, equivalent to 0–80 % RH at 23 ∘C, to assess its effect on the sensitivity. Our reported “nominally dry” cases correspond to 0.7 % RH or ca. 0.01 g kg−1 SH. As shown in Fig. 6, the sensitivity of the CI-ToFMS to isoprene ( ⋅ C5H8; 146 m∕Q) displays a strong, nonlinear dependence on SH. Instrument sensitivity increases with increasing SH, reaching a maximum value of 10 ncps ppt−1 at 4 g kg−1 (25 % RH at 23 ∘C), then decreases significantly at higher humidity. Surprisingly, we observed a linear correlation (R2 > 0.95) between the protonated water tetramer signal (73 m∕Q) and the delivered isoprene mixing ratio at constant SH that was not observed for smaller protonated water clusters (Fig. 7). The apparent sensitivity, derived from the slope of the linear least squares fit of the observed water tetramer signal vs. delivered isoprene concentration, increases with increasing specific humidity above 2 g kg−1 (Fig. 6). We reiterate that Fig. 6 does not show the protonated tetramer signal as a function of SH but the sensitivity of the 73 m∕Q signal to the delivered isoprene mixing ratio as shown in Fig. 7. The decreased sensitivity to isoprene adduct and increase in water tetramer signal with isoprene mixing ratio are unlikely the result of the formation of water protonated clusters via charge transfer reaction with benzene cations since the IE of water is significantly higher than that of the benzene dimer (12.62 and 8.69 eV respectively) (Chan et al., 1993; Grover et al., 1987). Since the formation of water tetramer clusters increases with isoprene mixing ratio and humidity, it is suggested that the interaction between water clusters and isoprene–benzene adducts in the IMR results in a charge exchange from the isoprene adduct to the water tetramer in a similar way that was previously described between benzene cation and water clusters. For example, Miyazaki et al. (2004) showed that the IR spectra of benzene–water ion clusters, with more than four water molecules, resemble those of protonated water clusters and suggested that the charge is held by the water molecules; such clusters that are likely to be formed in the IMR are expected to be broken apart in the ion optics. It is likely that the observed trends of the humidity-dependent sensitivity of isoprene and water tetramer signal also results from a similar formation and de-clustering in our CI-ToFMS.
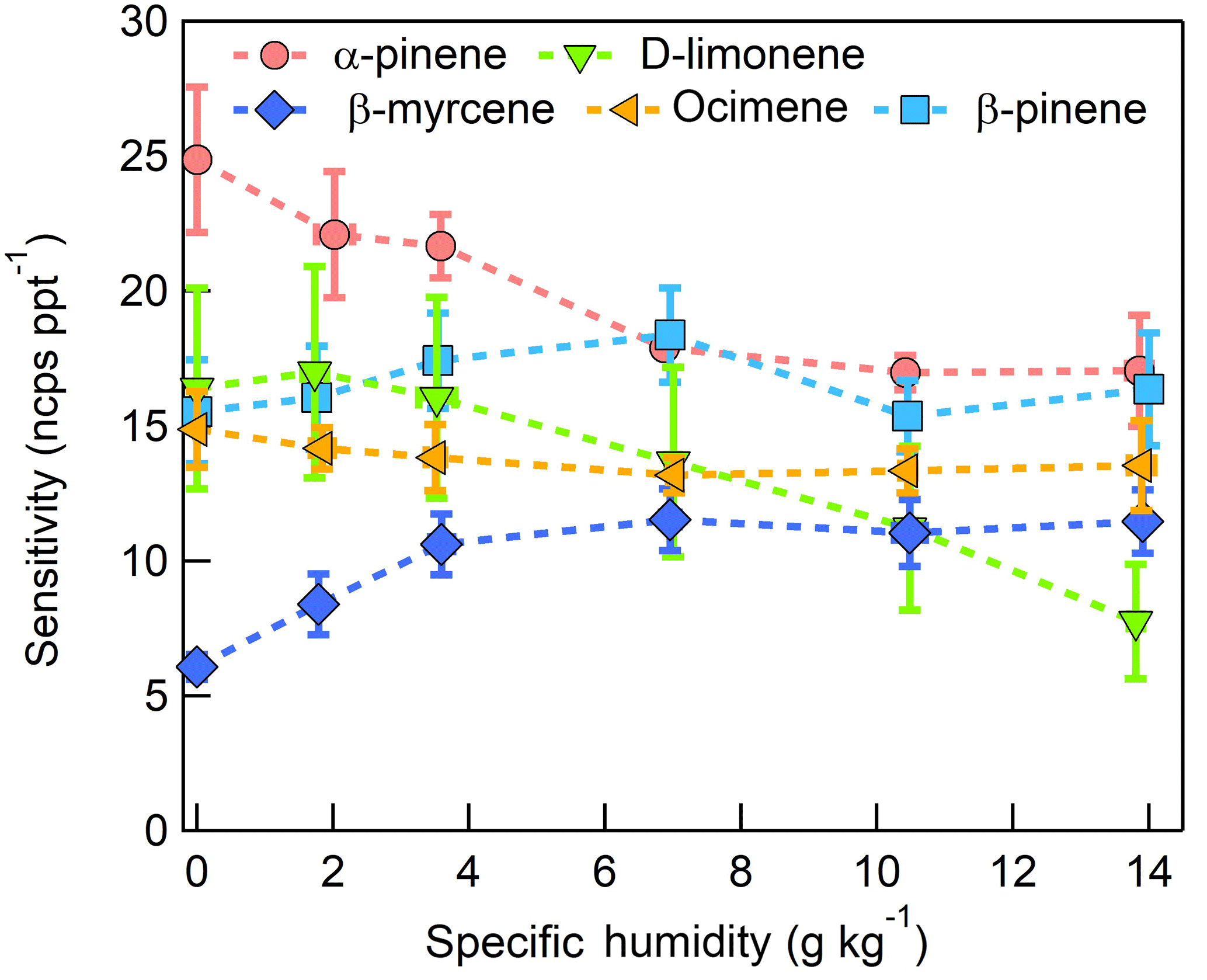
Figure 11Humidity-dependent, CI-ToFMS monoterpene sensitivities reported as the sum of all detected masses (135, 136, and 168 m∕Q). Error bars represent the standard deviation of the triplicate calibrations. All calibrations were performed in zero air.
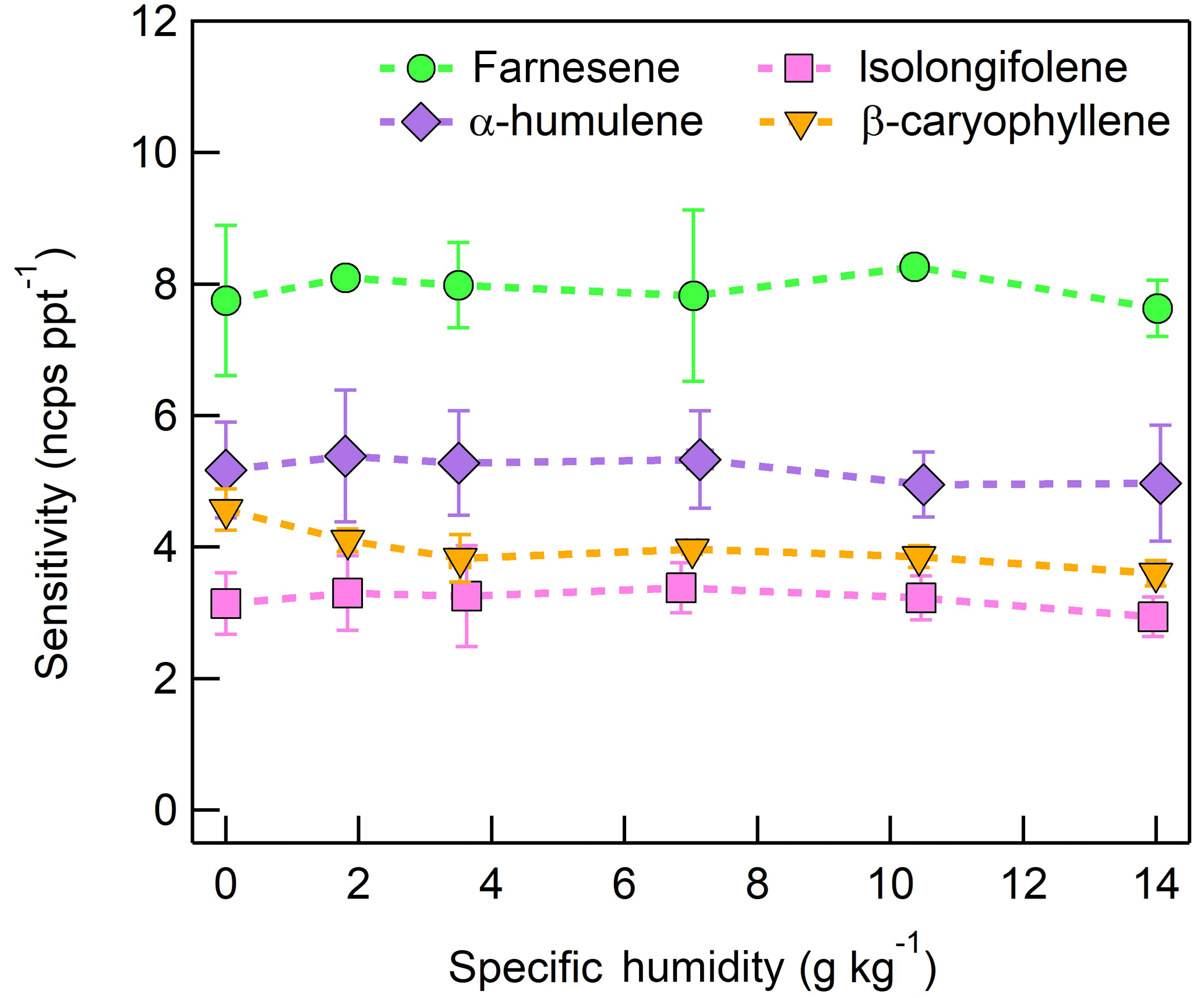
Figure 12Humidity-dependent sensitivities of SQTs detected as C15H24 (204 m∕Q). Error bars represent the standard deviation of triplicate measurements. All calibrations were performed in zero air.
3.3.2 Monoterpenes
The dependence of monoterpene sensitivity on SH is shown in Fig. 8 for the molecular ion (; 136 m∕Q). Instrument sensitivity under nominally dry conditions displays a wide range of sensitivities that are species dependent (4.8 to 21.0 ncps ppt−1). At high specific humidity, sensitivities converge significantly (9.5 to 15.0 ncps ppt−1). The observed dependence in the α-pinene sensitivity on SH reported here is counter to that previously reported by our group in Kim et al. (2016). This is attributed to the different instrument operational configuration used here (e.g., high concentration and purity benzene reagent ion precursor and low electric field strengths).
Table 1Monoterpene sensitivities and dependence on operating and sampling conditions.
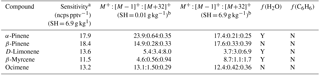
a SH = 6.9 g kg−1 corresponds to 65 % RH at 15 ∘C, representative of boreal regions. The reported sensitivity includes the contributions from the M+, , and M+32+ ions. b Sensitivities (ncps pptv−1) at M+, , and M+32+, are reported for SH = 0.01 and 6.9 g kg−1.
The humidity-dependent sensitivity of D-limonene is anomalous compared with the other monoterpenes studied, where the CI-ToFMS sensitivity to D-limonene decreases by a factor of 4 over the studied humidity range. The gradual and systematic decrease in the sensitivity suggests that the ionization of D-limonene by charge transfer is not the only ionization mechanism and/or that the D-limonene cation is subjected to subsequent reactions which results in the formation of other detectable ions. We calculated the calibration curves of each of the recorded mass-to-charge ratios to identify product ions that showed (1) high correlation with the delivered D-limonene mixing ratio (R2 > 0.98) and (2) the contribution to the total sensitivity (i.e., slope) was higher than 1 ncps ppt −1. A representative normalized calibration curve of the three ions (135, 136, and 168 m∕Q) that met these criteria is presented in Fig. 9. The peak at 168 m∕Q (C10H16O) is attributed to either a D–limonene-O2 adduct or a D-limonene oxidation product (e.g., limonene epoxide). The peak at 135 m∕Q(C10H15)+ represents the product. We speculate that this product could be formed following the oxidation of an [M+1]+ ion, formed via proton transfer, and the subsequent departure of HOOH (Karlberg et al., 1994). The purity of the primary standard was confirmed via GC-MS, and comparable peak ratios were measured when sampling the standard directly, ruling out the potential for the nebulization process to alter the MS peak ratios. Finally, the [M+32]+ and peak intensities are reduced to baseline by sampling the terpene in nitrogen, suggesting that these peaks are a result of secondary ion chemistry involving O2. The normalized sensitivity of each of these three peaks decreases with increasing SH (Fig. 10), suggesting that water clusters compete or suppress the charge transfer to the contributing ions. The humidity-dependent sensitivity of all the studied MTs, calculated as the sum of all their contributing ions, shows lower variability, mostly due to the higher sensitivity to D-limonene when all product ions are accounted for (Fig. 11). The variations in the sensitivities between different monoterpenes is small (14 ± 3 ncps ppt−1) and instrumental response is largely independent on SH from 4 to 14 g kg−1. This range is typical at boreal forests during the summer (Suni et al., 2003). The reported sensitivities, product ions, and dependence on ambient water concentrations and neutral benzene concentration for select monoterpenes are shown in Table 1.
Table 2Sesquiterpene sensitivities and dependence on operating and sampling conditions.
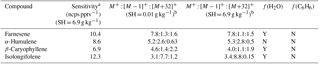
a SH = 6.9 g kg−1 corresponds to 65 % RH at 15 ∘C, representative of Boreal regions. The reported sensitivity includes the contributions from the M+, , and M+32+ ions. b Sensitivities (ncps pptv−1) at M+, , and M+32+, is reported for SH = 0.01 and 6.9 g kg−1.
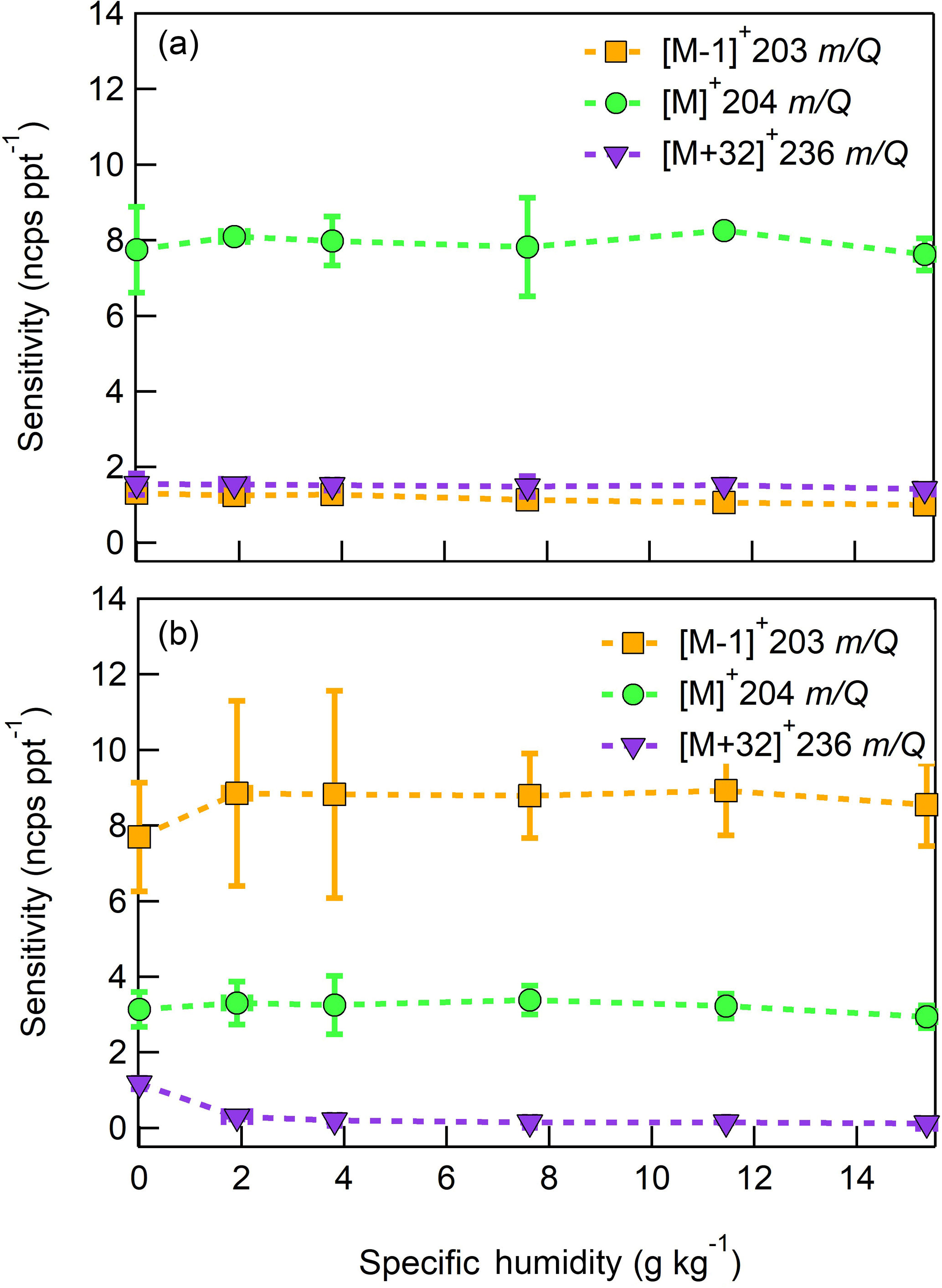
Figure 13Humidity-dependent, normalized sensitivities to (a) farnesene and (b) isolongifolene for all major product ions (, 203 m∕Q, orange squares), (, 204 m∕Q, green circles), and (, 236 m∕Q, purple triangles). Error bars represent the standard deviation of the triplicate measurement.
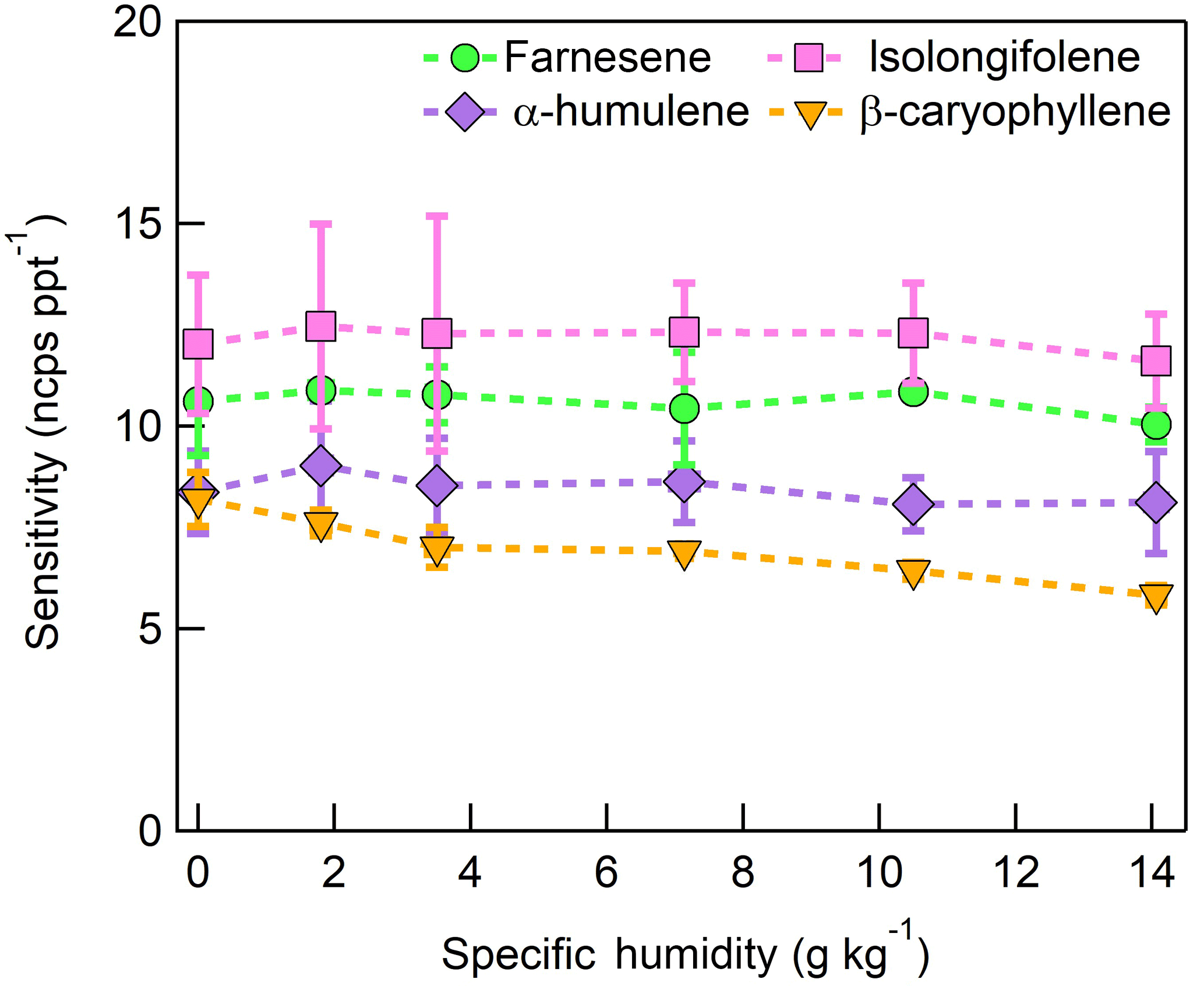
Figure 14Humidity-dependent, normalized sensitivities to sesquiterpenes, reported as the sum of the major product ions (, 203 m∕Q), (, 204 m∕Q), and (, 236 m∕Q). Error bars represent the standard deviation of triplicate measurements.
3.3.3 Sesquiterpenes
The sensitivities of the CI-ToFMS toward SQTs, detected as the charge transfer product at 204 m∕Q, show minimal dependence on SH between nominally dry conditions and 14 g kg−1 (Fig. 12). Using the same process discussed in Sect. 3.3.2 for identifying other product ions, it was found that 203 and 236 m∕Q ( and ) also contributed to product ion intensity.
The response of the farnesene and isolongifolene molecular ions and their related contributing ions are presented as examples of SQTs dependence on SH (Fig. 13). All three major ions were observed at all measured SHs, and in the case of isolongifolene, the normalized response of 203 m∕Q () was higher than the molecular ion (204 m∕Q, ) over the entire SH range, including at nominally dry conditions (Fig. 13). At present, we do not have a definitive mechanism for the product ion distribution, but the presence of similar products (i.e., ( and ([M+32]+) and their humidity dependence suggest that the molecular ions of sesquiterpenes are subjected to similar reactions to MTs which results in a lower signal of the molecular ion. Similar to MTs, the humidity-dependent sensitivities of sesquiterpenes calculated as the sum of all contributing ions lower the variability in calculated sensitivities (Fig. 14). Since the sensitivity is independent of the humidity, a general sensitivity to all SQTs of 9.6 ± 2.3 ncps pptv−1 can be further used for quantification of ambient SQTs. The reported sensitivities, product ions, and dependence on ambient water concentrations and neutral benzene concentration for select sesquiterpenes are shown in Table 2.
We show that benzene cluster cations are a sensitive reagent ion for chemical ionization of select biogenic volatile organic compounds. We demonstrate that isoprene is primarily detected as an adduct (C5H8 ⋅ ) with a sensitivity ranging between 4 and 10 ncps ppt−1, which depends strongly on the reagent ion precursor concentration and specific humidity (SH). This highlights the importance of continuous infield calibrations for isoprene concentration measurements. We show that monoterpenes are primarily detected as the molecular ion () with an average sensitivity, across the five measured compounds, of 14 ± 3 ncps ppt−1 for SH between 7 and 14 g kg−1, typical of the boreal forest during summer. Sesquiterpenes are detected primarily as the molecular ion () with an average sensitivity, across the four measured compounds, of 9.6 ± 2.3 ncps ppt−1 that is also independent of specific humidity. Given that signal intensity was observed at ( and ([M+32]+ for a few select terpenes (e.g., D-limonene) we recommend that future measurements of total monoterpenes utilize all three product ions. We suggest that future studies that utilize benzene cluster cation chemistry use high-purity liquid reservoirs and benzene neutral concentrations at or above 300 ppmv.
Calibration data generated in this study can be found online at https://minds.wisconsin.edu/handle/1793/76304 (last access: 6 June 2018).
The authors declare that they have no conflict of interest.
This work was supported by a National Science Foundation (NSF) CAREER Award
(grant no. AGS-1151430) and the Office of Science (Office of Biological and
Environmental Research), U.S. Department of Energy (grant no. DE-SC0006431).
Avi Lavi gratefully acknowledges support from the Dreyfus Foundation
Environmental Chemistry Postdoctoral Fellowship Program.
Edited by: Mingjin Tang
Reviewed by: three anonymous referees
Allan, J. D., Alfarra, M. R., Bower, K. N., Coe, H., Jayne, J. T., Worsnop, D. R., Aalto, P. P., Kulmala, M., Hyötyläinen, T., Cavalli, F., and Laaksonen, A.: Size and composition measurements of background aerosol and new particle growth in a Finnish forest during QUEST 2 using an Aerodyne Aerosol Mass Spectrometer, Atmos. Chem. Phys., 6, 315–327, https://doi.org/10.5194/acp-6-315-2006, 2006.
Bertram, T. H., Kimmel, J. R., Crisp, T. A., Ryder, O. S., Yatavelli, R. L. N., Thornton, J. A., Cubison, M. J., Gonin, M., and Worsnop, D. R.: A field-deployable, chemical ionization time-of-flight mass spectrometer, Atmos. Meas. Tech., 4, 1471–1479, https://doi.org/10.5194/amt-4-1471-2011, 2011.
Bieri, G., Asbrink, L., and Vonniessen, W.: 30.4-Nm He(Ii) Photoelectron-Spectra of Organic-Molecules 4. Fluoro-Compounds (C,H,F), J. Electron Spectrosc., 23, 281–322, 1981.
Chan, W. F., Cooper, G., and Brion, C. E.: The Electronic-Spectrum of Water in the Discrete and Continuum Regions – Absolute Optical Oscillator-Strengths for Photoabsorption (6–200 Ev), Chem. Phys., 178, 387–400, 1993.
Chipot, C., Jaffe, R., Maigret, B., Pearlman, D. A., and Kollman, P. A.: Benzene dimer: A good model for pi-pi interactions in proteins? A comparison between the benzene and the toluene dimers in the cas phase and in an aqueous solution, J. Am. Chem. Soc., 118, 11217–11224, 1996.
Chung, C. E., Ramanathan, V., and Decremer, D.: Observationally constrained estimates of carbonaceous aerosol radiative forcing, P. Natl. Acad. Sci. USA, 109, 11624–11629, 2012.
Crounse, J. D., McKinney, K. A., Kwan, A. J., and Wennberg, P. O.: Measurement of gas-phase hydroperoxides by chemical ionization mass spectrometry, Anal. Chem., 78, 6726–6732, 2006.
Dondes, S., Harteck, P., and Kunz, C.: A Spectroscopic Study of Alpha-Ray-Induced Luminescence in Gases 1, Radiat. Res., 27, 174–210, 1966.
Greenspan, L.: Humidity Fixed-Points of Binary Saturated Aqueous-Solutions, J. Res. NBS A Phys. Ch., 81, 89–96, 1977.
Grover, J. R., Walters, E. A., and Hui, E. T.: Dissociation-Energies of the Benzene Dimer and Dimer Cation, J. Phys. Chem.-US, 91, 3233–3237, 1987.
Guenther, A., Hewitt, C. N., Erickson, D., Fall, R., Geron, C., Graedel, T., Harley, P., Klinger, L., Lerdau, M., Mckay, W. A., Pierce, T., Scholes, B., Steinbrecher, R., Tallamraju, R., Taylor, J., and Zimmerman, P.: A Global-Model of Natural Volatile Organic-Compound Emissions, J. Geophys. Res.-Atmos., 100, 8873–8892, 1995.
Guenther, A. B., Jiang, X., Heald, C. L., Sakulyanontvittaya, T., Duhl, T., Emmons, L. K., and Wang, X.: The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 5, 1471–1492, https://doi.org/10.5194/gmd-5-1471-2012, 2012.
Hallquist, M., Wenger, J. C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N. M., George, C., Goldstein, A. H., Hamilton, J. F., Herrmann, H., Hoffmann, T., Iinuma, Y., Jang, M., Jenkin, M. E., Jimenez, J. L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, Th. F., Monod, A., Prévôt, A. S. H., Seinfeld, J. H., Surratt, J. D., Szmigielski, R., and Wildt, J.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009.
Huey, L. G.: Measurement of trace atmospheric species by chemical ionization mass spectrometry: Speciation of reactive nitrogen and future directions, Mass Spectrom. Rev., 26, 166–184, 2007.
Hunt, D. F. and Harvey, T. M.: Nitric oxide chemical ionization mass spectra of alkanes, Anal. Chem., 47, 1965–1969, 1975.
Hunt, D. F., Harvey, T. M., Brumley, W. C., Ryan, J. F., and Russell, J. W.: Nitric oxide chemical ionization mass spectrometry of alcohols, Anal. Chem., 54, 492–496, 1982.
Ibrahim, Y. M., Mautner, M. M. N., Alshraeh, E. H., El-Shall, M. S., and Scheiner, S.: Stepwise hydration of ionized aromatics. Energies, structures of the hydrated benzene cation, and the mechanism of deprotonation reactions, J. Am. Chem. Soc., 127, 7053–7064, 2005.
Jokinen, T., Berndt, T., Makkonen, R., Kerminen, V. M., Junninen, H., Paasonen, P., Stratmann, F., Herrmann, H., Guenther, A. B., Worsnop, D. R., Kulmala, M., Ehn, M., and Sipila, M.: Production of extremely low volatile organic compounds from biogenic emissions: Measured yields and atmospheric implications, P. Natl. Acad. Sci. USA, 112, 7123–7128, 2015.
Karl, T., Hansel, A., Cappellin, L., Kaser, L., Herdlinger-Blatt, I., and Jud, W.: Selective measurements of isoprene and 2-methyl-3-buten-2-ol based on NO+ ionization mass spectrometry, Atmos. Chem. Phys., 12, 11877–11884, https://doi.org/10.5194/acp-12-11877-2012, 2012.
Karlberg, A. T., Shao, L. P., Nilsson, U., Gafvert, E., and Nilsson, J. L. G.: Hydroperoxides in Oxidized D-Limonene Identified as Potent Contact Allergens, Arch. Dermatol. Res., 286, 97–103, 1994.
Kerminen, V. M., Lihavainen, H., Komppula, M., Viisanen, Y., and Kulmala, M.: Direct observational evidence linking atmospheric aerosol formation and cloud droplet activation, Geophys. Res. Lett., 32, https://doi.org/10.1029/2005GL023130, 2005.
Kim, M. J., Farmer, D. K., and Bertram, T. H.: A controlling role for the air-sea interface in the chemical processing of reactive nitrogen in the coastal marine boundary layer, P. Natl. Acad. Sci. USA, 111, 3943–3948, 2014.
Kim, M. J., Zoerb, M. C., Campbell, N. R., Zimmermann, K. J., Blomquist, B. W., Huebert, B. J., and Bertram, T. H.: Revisiting benzene cluster cations for the chemical ionization of dimethyl sulfide and select volatile organic compounds, Atmos. Meas. Tech., 9, 1473–1484, https://doi.org/10.5194/amt-9-1473-2016, 2016.
Kim, S., Karl, T., Helmig, D., Daly, R., Rasmussen, R., and Guenther, A.: Measurement of atmospheric sesquiterpenes by proton transfer reaction-mass spectrometry (PTR-MS), Atmos. Meas. Tech., 2, 99–112, https://doi.org/10.5194/amt-2-99-2009, 2009.
Kirkby, J., Duplissy, J., Sengupta, K., Frege, C., Gordon, H., Williamson, C., Heinritzi, M., Simon, M., Yan, C., Almeida, J., Trostl, J., Nieminen, T., Ortega, I. K., Wagner, R., Adamov, A., Amorim, A., Bernhammer, A. K., Bianchi, F., Breitenlechner, M., Brilke, S., Chen, X., Craven, J., Dias, A., Ehrhart, S., Flagan, R. C., Franchin, A., Fuchs, C., Guida, R., Hakala, J., Hoyle, C. R., Jokinen, T., Junninen, H., Kangasluoma, J., Kim, J., Krapf, M., Kurten, A., Laaksonen, A., Lehtipalo, K., Makhmutov, V., Mathot, S., Molteni, U., Onnela, A., Perakyla, O., Piel, F., Petaja, T., Praplan, A. P., Pringle, K., Rap, A., Richards, N. A., Riipinen, I., Rissanen, M. P., Rondo, L., Sarnela, N., Schobesberger, S., Scott, C. E., Seinfeld, J. H., Sipila, M., Steiner, G., Stozhkov, Y., Stratmann, F., Tome, A., Virtanen, A., Vogel, A. L., Wagner, A. C., Wagner, P. E., Weingartner, E., Wimmer, D., Winkler, P. M., Ye, P., Zhang, X., Hansel, A., Dommen, J., Donahue, N. M., Worsnop, D. R., Baltensperger, U., Kulmala, M., Carslaw, K. S., and Curtius, J.: Ion-induced nucleation of pure biogenic particles, Nature, 533, 521–526, 2016.
Koss, A. R., Warneke, C., Yuan, B., Coggon, M. M., Veres, P. R., and de Gouw, J. A.: Evaluation of NO+ reagent ion chemistry for online measurements of atmospheric volatile organic compounds, Atmos. Meas. Tech., 9, 2909–2925, https://doi.org/10.5194/amt-9-2909-2016, 2016.
Krause, H., Ernstberger, B., and Neusser, H. J.: Binding-Energies of Small Benzene Clusters, Chem. Phys. Lett., 184, 411–417, 1991.
Kulmala, M., Suni, T., Lehtinen, K. E. J., Dal Maso, M., Boy, M., Reissell, A., Rannik, Ü., Aalto, P., Keronen, P., Hakola, H., Bäck, J., Hoffmann, T., Vesala, T., and Hari, P.: A new feedback mechanism linking forests, aerosols, and climate, Atmos. Chem. Phys., 4, 557–562, https://doi.org/10.5194/acp-4-557-2004, 2004.
Lang-Yona, N., Rudich, Y., Mentel, Th. F., Bohne, A., Buchholz, A., Kiendler-Scharr, A., Kleist, E., Spindler, C., Tillmann, R., and Wildt, J.: The chemical and microphysical properties of secondary organic aerosols from Holm Oak emissions, Atmos. Chem. Phys., 10, 7253–7265, https://doi.org/10.5194/acp-10-7253-2010, 2010.
Leibrock, E. and Huey, L. G.: Ion chemistry for the detection of isoprene and other volatile organic compounds in ambient air, Geophys. Res. Lett., 27, 1719–1722, 2000.
Lifshitz, C. and Reuben, B. G.: Ion-Molecule Reactions in Aromatic Systems I. Secondary Ions and Reaction Rates in Benzene, J. Chem. Phys., 50, 951–960, 1969.
Lindinger, W., Hansel, A., and Jordan, A.: On-line monitoring of volatile organic compounds at pptv levels by means of proton-transfer-reaction mass spectrometry (PTR-MS) – Medical applications, food control and environmental research, Int. J. Mass Spectrom., 173, 191–241, 1998.
Lopez-Hilfiker, F. D., Mohr, C., Ehn, M., Rubach, F., Kleist, E., Wildt, J., Mentel, Th. F., Carrasquillo, A. J., Daumit, K. E., Hunter, J. F., Kroll, J. H., Worsnop, D. R., and Thornton, J. A.: Phase partitioning and volatility of secondary organic aerosol components formed from α-pinene ozonolysis and OH oxidation: the importance of accretion products and other low volatility compounds, Atmos. Chem. Phys., 15, 7765–7776, https://doi.org/10.5194/acp-15-7765-2015, 2015.
Miyazaki, M., Fujii, A., Ebata, T., and Mikami, N.: Infrared spectroscopy of size-selected benzene-water cluster cations [C6H6-(H2O)(n)](+) (n= 1–23): Hydrogen bond network evolution and microscopic hydrophobicity, J. Phys. Chem. A, 108, 10656–10660, 2004.
Mochalski, P., Unterkofler, K., Spanel, P., Smith, D., and Amann, A.: Product ion distributions for the reactions of NO+ with some physiologically significant aldehydes obtained using a SRI-TOF-MS instrument, Int. J. Mass Spectrom., 363, 23–31, 2014.
Novak, I., Kovac, B., and Kovacevic, G.: Electronic structure of terpenoids, J. Org. Chem., 66, 4728–4731, 2001.
Riedel, T. P., Bertram, T. H., Crisp, T. A., Williams, E. J., Lerner, B. M., Vlasenko, A., Li, S. M., Gilman, J., de Gouw, J., Bon, D. M., Wagner, N. L., Brown, S. S., and Thornton, J. A.: Nitryl Chloride and Molecular Chlorine in the Coastal Marine Boundary Layer, Environ. Sci. Technol., 46, 10463–10470, 2012.
Shinohara, H. and Nishi, N.: Excited-State Lifetimes and Appearance Potentials of Benzene Dimer and Trimer, J. Chem. Phys., 91, 6743–6751, 1989.
Suni, T., Rinne, J., Reissell, A., Altimir, N., Keronen, P., Rannik, U., Dal Maso, M., Kulmala, M., and Vesala, T.: Long-term measurements of surface fluxes above a Scots pine forest in Hyytiala, southern Finland, 1996–2001, Boreal Environ. Res., 8, 287–301, 2003.
Talebpour, A., Bandrauk, A. D., Vijayalakshmi, K., and Chin, S. L.: Dissociative ionization of benzene in intense ultra-fast laser pulses, J. Phys. B-At. Mol. Opt., 33, 4615–4626, 2000.
Thornton, J. A., Kercher, J. P., Riedel, T. P., Wagner, N. L., Cozic, J., Holloway, J. S., Dube, W. P., Wolfe, G. M., Quinn, P. K., Middlebrook, A. M., Alexander, B., and Brown, S. S.: A large atomic chlorine source inferred from mid-continental reactive nitrogen chemistry, Nature, 464, 271–274, 2010.
Werner, A. S., Tsai, B. P., and Baer, T.: Photoionization study of the ionization potentials and fragmentation paths of the chlorinated methanes and carbon tetrabromide, J. Chem. Phys., 60, 3650–3657, 1974.
Wiedensohler, A., Cheng, Y. F., Nowak, A., Wehner, B., Achtert, P., Berghof, M., Birmili, W., Wu, Z. J., Hu, M., Zhu, T., Takegawa, N., Kita, K., Kondo, Y., Lou, S. R., Hofzumahaus, A., Holland, F., Wahner, A., Gunthe, S. S., Rose, D., Su, H., and Pöschl, U.: Rapid aerosol particle growth and increase of cloud condensation nucleus activity by secondary aerosol formation and condensation: A case study for regional air pollution in northeastern China, J. Geophys. Res.-Atmos., 114, https://doi.org/10.1029/2008JD010884, 2009.
Zhao, D. F., Buchholz, A., Tillmann, R., Kleist, E., Wu, C., Rubach, F., Kiendler-Scharr, A., Rudich, Y., Wildt, J., and Mentel, T. F.: Environmental conditions regulate the impact of plants on cloud formation, Nat. Commun., 8, https://doi.org/10.1038/ncomms14067, 2017.