the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 17 Apr 2019
| 17 Apr 2019
Evaluating the performance of five different chemical ionization techniques for detecting gaseous oxygenated organic species
Matthieu Riva
Pekka Rantala
Jordan E. Krechmer
Otso Peräkylä
Yanjun Zhang
Liine Heikkinen
Olga Garmash
Markku Kulmala
Douglas Worsnop
The impact of aerosols on climate and air quality remains poorly understood due to multiple factors. One of the current limitations is the incomplete understanding of the contribution of oxygenated products, generated from the gas-phase oxidation of volatile organic compounds (VOCs), to aerosol formation. Indeed, atmospheric gaseous chemical processes yield thousands of (highly) oxygenated species, spanning a wide range of chemical formulas, functional groups and, consequently, volatilities. While recent mass spectrometric developments have allowed extensive on-line detection of a myriad of oxygenated organic species, playing a central role in atmospheric chemistry, the detailed quantification and characterization of this diverse group of compounds remains extremely challenging. To address this challenge, we evaluated the capability of current state-of-the-art mass spectrometers equipped with different chemical ionization sources to detect the oxidation products formed from α-Pinene ozonolysis under various conditions. Five different mass spectrometers were deployed simultaneously for a chamber study. Two chemical ionization atmospheric pressure interface time-of-flight mass spectrometers (CI-APi-TOF) with nitrate and amine reagent ion chemistries and an iodide chemical ionization time-of-flight mass spectrometer (TOF-CIMS) were used. Additionally, a proton transfer reaction time-of-flight mass spectrometer (PTR-TOF 8000) and a new “vocus” PTR-TOF were also deployed. In the current study, we compared around 1000 different compounds between each of the five instruments, with the aim of determining which oxygenated VOCs (OVOCs) the different methods were sensitive to and identifying regions where two or more instruments were able to detect species with similar molecular formulae. We utilized a large variability in conditions (including different VOCs, ozone, NOx and OH scavenger concentrations) in our newly constructed atmospheric simulation chamber for a comprehensive correlation analysis between all instruments. This analysis, combined with estimated concentrations for identified molecules in each instrument, yielded both expected and surprising results. As anticipated based on earlier studies, the PTR instruments were the only ones able to measure the precursor VOC, the iodide TOF-CIMS efficiently detected many semi-volatile organic compounds (SVOCs) with three to five oxygen atoms, and the nitrate CI-APi-TOF was mainly sensitive to highly oxygenated organic (O > 5) molecules (HOMs). In addition, the vocus showed good agreement with the iodide TOF-CIMS for the SVOC, including a range of organonitrates. The amine CI-APi-TOF agreed well with the nitrate CI-APi-TOF for HOM dimers. However, the loadings in our experiments caused the amine reagent ion to be considerably depleted, causing nonlinear responses for monomers. This study explores and highlights both benefits and limitations of currently available chemical ionization mass spectrometry instrumentation for characterizing the wide variety of OVOCs in the atmosphere. While specifically shown for the case of α-Pinene ozonolysis, we expect our general findings to also be valid for a wide range of other VOC–oxidant systems. As discussed in this study, no single instrument configuration can be deemed better or worse than the others, as the optimal instrument for a particular study ultimately depends on the specific target of the study.
Please read the corrigendum first before continuing.
-
Notice on corrigendum
The requested paper has a corresponding corrigendum published. Please read the corrigendum first before downloading the article.
-
Article
(2992 KB)
-
The requested paper has a corresponding corrigendum published. Please read the corrigendum first before downloading the article.
- Article
(2992 KB) - Full-text XML
- Corrigendum
- BibTeX
- EndNote





Atmospheric aerosols, a mixture of solid and liquid particles consisting of organic and inorganic substances suspended in the air, have a significant impact on climate (Albrecht, 1989; Hallquist et al., 2009; Intergovernmental Panel on Climate Change, 2014; Twomey, 1977). They are also recognized to adversely impact air quality and human health, nowadays representing the fifth-ranking human health risk factor globally (Gakidou et al., 2017). Depending on the region, organic aerosol contributes on average 20 %–90 % to the submicron aerosol mass (Jimenez et al., 2009), with secondary organic aerosol (SOA) as the largest source of atmospheric organic aerosol (Hallquist et al., 2009; Jimenez et al., 2009). SOA is predominantly formed through the gas-phase oxidation of volatile organic compounds (VOCs), producing oxygenated VOCs (OVOCs), which can subsequently condense onto pre-existing aerosol particles. Generally, the more oxidized the OVOCs, the lower its volatility is and the greater the probability of this compound to partition to the particle phase. Recently, studies have provided new insights into how highly oxygenated organic molecules (HOMs) can form faster than previously expected and at high enough yields to make them a major source of condensing or even nucleating compounds (Ehn et al., 2014; Jokinen et al., 2015; Kirkby et al., 2016; Wang et al., 2018).
The quantitative assessment of the impact of aerosol on climate remains poorly understood due to a number of factors, including an incomplete understanding of how VOC oxidation processes contribute to new particle and SOA formation (Glasius and Goldstein, 2016). Indeed, atmospheric oxidation processes can lead to the formation of thousands of oxidized products from a single precursor (Glasius and Goldstein, 2016; Goldstein and Galbally, 2007). As a result of these complex oxidation processes, atmospheric organic species span an extremely wide range of chemical formulas, structures and physicochemical properties. Volatilities range from volatile species present only in the gas phase, via low- and semi-volatile organic compounds (LVOCs and SVOCs), to extremely low volatility organic compounds (ELVOCs) present mainly in the particle phase (Donahue et al., 2012). The chemical complexity of OVOC poses a major challenge in detecting, quantifying and characterizing such a large number and wide variety of organic compounds.
Mass spectrometric techniques, which can detect a large range of species simultaneously, are well-suited to tackling these challenges. This is underlined by the major role of the mass spectrometers in improving our understanding of the atmospheric chemical composition over the last 20 years (Breitenlechner et al., 2017; Ehn et al., 2014; Jokinen et al., 2012; Krechmer et al., 2018; Lindinger et al., 1998; Yuan et al., 2017). Proton transfer reaction (PTR) has been one of the most used medium-pressure ionization techniques since the mid-1990s (Lindinger et al., 1998). Since then, the PTR-MS technique has been greatly improved in terms of sensitivity, detection limit and mass resolution by introducing the PTR-TOF-MS (Yuan et al., 2017). The latest version has detection limits as low as 107 molecules cm−3. While such techniques can characterize VOCs, the PTR-MS technique has not been able to measure more oxygenated organic species. This is mostly due to losses of these low volatile compounds in the sampling lines and on the walls of the inlet (caused, for example, by very low flow rates), as the instrument was designed to primarily measure volatile compounds.
Several different chemical ionization mass spectrometry (CIMS) methods have been developed, including medium-pressure systems like CF3O−-CIMS for specific detection of oxygenated VOCs and SVOCs including hydroperoxides (Crounse et al., 2006), acetate CIMS for selective detection of organic acids (Bertram et al., 2011) and the iodide adduct ionization CIMS for the detection of a wider range of OVOCs, including alcohols, hydroperoxides and peroxy acids (Lee et al., 2014; Riva et al., 2017). These instruments, based on negative ion chemistry, can detect oxygenated gas-phase compounds at concentrations as low as ∼106 molecules cm−3. Finally, the discovery of the HOMs was possible due to the development of a nitrate chemical ionization source connected to an atmospheric pressure interface time-of-flight mass spectrometer (CI-APi-TOF) (Ehn et al., 2014; Jokinen et al., 2012). The selectivity and high sensitivity for molecules containing many functional groups (detection limit below 105 molecules cm−3) of the nitrate CI-APi-TOF makes this instrument perfect for detecting HOMs and even certain radicals (e.g., peroxy radicals). As part of the rapid development in gas-phase mass spectrometry, several new reagent ion chemistries have been tested over the last few years. With improvements in sensitivity and/or selectivity, new methods are now able to detect a wide variety of oxygenated species, including radicals and stabilized Criegee intermediates (Berndt et al., 2015, 2017, 2018; Breitenlechner et al., 2017; Hansel et al., 2018; Krechmer et al., 2018).
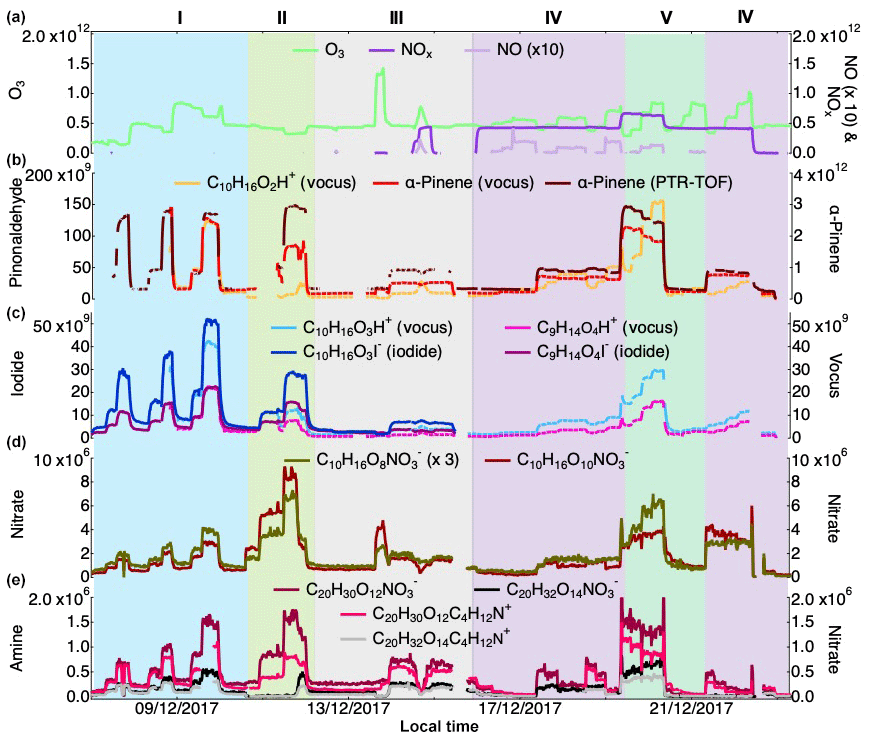
Figure 1Campaign overview, including the concentration of O3, NO, NOx (a) as well as α-Pinene measured by the vocus and the PTR-TOF and pinonaldehyde measured using the vocus (b). Concentrations of pinonic and pinic acids (vocus and iodide) are presented in (c), example of HOM monomers from nitrate (d) and example of HOM dimers from amine and nitrate (e). Concentrations for all the gaseous species are in molecules cm−3; see text for details on quantification. The experiments were separated into five types: I is α-Pinene + O3, II is α-Pinene + O3+CO (as an OH scavenger), III is tests (NO2 injection, H2O2 injection for generating HO2), IV is α-Pinene + O3+NO and V is α-Pinene + . Concentrations of NO and are scaled for clarity.
The selectivity and sensitivity of the different ionization chemistries makes it impossible for one mass spectrometer to be able to measure the full range of VOCs and OVOCs present in the atmosphere. Hence, only a simultaneous deployment of several mass spectrometry techniques can provide a comprehensive chemical characterization of the gaseous composition. While such a multi-instrument approach maximizes the fraction of organic species measured (Isaacman-VanWertz et al., 2017, 2018), a number of questions and limitations can arise in both laboratory and field measurements. For instance, the extent to which instruments can (i) measure species with identical molecular composition, (ii) cover the entire range of oxygenated species and (iii) provide constant sensitivity across different conditions, have to be determined. Most studies are typically limited to one, or perhaps two, mass spectrometers, and then it is also important to know which fraction of the OVOC distribution these instruments are sensitive to. To our knowledge, systematic comparisons of the most commonly used or recently developed gas-phase mass spectrometers are not yet available. In this work, we compared the suitability of five different chemical ionization methods (including iodide TOF-CIMS, nitrate and amine CI-APi-TOFs, a PTR-TOF and the newly developed vocus PTR-TOF) for the detection of OVOCs formed from α-Pinene ozonolysis during a comprehensive chamber study with varying VOCs, O3 and NOx concentrations. We characterized the time evolution of around 1000 compounds and explored the capability of these instruments to measure OVOCs of different oxygenation levels within different compound groups.
2.1 Chamber experiments
Experiments were performed at the University of Helsinki in a 2 m3 atmospheric simulation Teflon (FEP) chamber. The COALA chamber (named after the project for which it was constructed: Comprehensive molecular characterization of secondary Organic AerosoL formation in the Atmosphere) was operated under steady-state conditions, meaning that a constant flow of reactants and oxidants were continuously added to the chamber, while chamber air was sampled by the instruments. Under the conditions used in this study, the average residence time in the chamber was ∼30 min, and the majority of conditions were kept constant for 6 to 12 h before changing to new conditions. These experiments focused on the characterization of the oxidation products arising from the α-Pinene (C10H16) ozonolysis. α-Pinene was used for the generation of oxidation products because it is the most abundant monoterpene emitted by the boreal forests and is one of the most important SOA precursors on a global scale (Jokinen et al., 2015; Kelly et al., 2018).
The experiments were conducted at room temperature (27±2 ∘C) and under dry conditions (RH < 1 %). An overview of the measurements, as well as the experimental conditions, are presented in Fig. 1. α-Pinene was introduced to the chamber from a gas cylinder, and steady-state concentrations of α-Pinene were varied from 20 to 100 ppb. As alkene ozonolysis yields OH radicals (Atkinson et al., 1997), in some experiments, ∼1500 ppm of carbon monoxide (CO) was injected to serve as the OH scavenger. Also, 10 to 50 ppb of O3 was generated by injecting purified air through an ozone generator (Dasibi 1008-PC) and monitored over the process of the campaign using a UV photometric analyzer (Model 49P, Thermo-Environmental). In the experiments performed in the presence of NOx, 400 nm LED lights were used to generate NO in the chamber from the photolysis of the injected NO2. The purified air ([O3∕NOx] and [VOC] reduced to less than 1 ppb and 5 ppb, respectively), generated by an air purification system (AADCO, 737 Series, Ohio, USA) running on compressed air, was used as a bath gas. Temperature, relative humidity (RH) and pressure were monitored by a Vaisala Humidity and Temperature Probe (INTERCAP® HMP60) and a differential pressure sensor (Sensirion SDP1000-L025).
Table 1Overview and characteristics of the mass spectrometers deployed during the campaign at the COALA chamber.
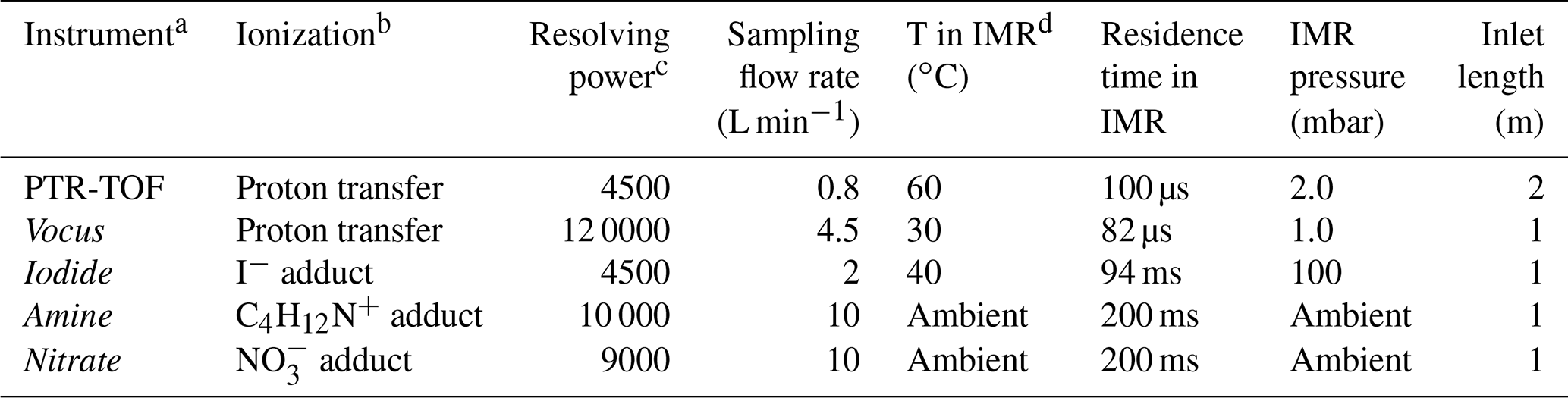
a The reagent ion is used to denote the instrument name. b Type of ionization method used for each instrument. c Corresponds to the mass resolution of the instruments under the conditions used in this study. d IMR is the ion–molecule reaction chamber, i.e., the region where sample molecules are mixed with reagent ions. The IMR has a different design in each of the instruments, except for the nitrate and amine, which are identical.
2.2 Mass spectrometers
We deployed five chemical ionization schemes to the COALA chamber in order to characterize the chemical composition of the gas-phase oxidation products formed from α-Pinene ozonolysis. In this section, we briefly present each instrument, summarized in Table 1. As each mass spectrometer has slightly different working principles, references to more detailed descriptions are provided. Specific benefits and limitations, which were not often discussed in earlier studies, are reviewed in Sect. 2.4. Each of the mass spectrometers were equipped with a mass analyzer manufactured by Tofwerk AG, either an HTOF (mass resolving power ∼5000) or long TOF (LTOF, mass resolving power ∼10 000) version.
In the analysis, we focused primarily on the relative behavior of the ions measured by the different mass spectrometers. An absolute comparison was also performed, but this approach has a larger uncertainty, as the sensitivity towards every molecule is different in each of the mass spectrometers, depending on molecular size, functionality, proton affinity, polarizability, etc. We attempted a rough estimate of absolute concentrations for each instrument, despite the fact that, with around a thousand ions analyzed, it is evident that we make no claim for them to all be accurate. As will be shown, the concentrations of gas-phase VOCs and OVOCs vary up to 7 orders of magnitude, and therefore useful information can still be obtained even in cases where concentration estimates could be off by an order of magnitude. Details about instruments used in this study as well as calibrations and instrumental limitations are discussed in the following sections.
2.2.1 PTR-TOF
α-Pinene concentration was measured in the COALA chamber by a proton transfer reaction time-of-flight mass spectrometer (PTR-TOF 8000, Ionicon Analytik Gmbh) – later referred to as PTR-TOF. The technical details have been described in detail elsewhere (Graus et al., 2010; Jordan et al., 2009). The sample air from the COALA chamber was drawn to the instrument using 2 m long PTFE tubing (6 mm o.d, 4 mm i.d.) and a piece of 20 cm capillary PEEK tubing (1.6 mm o.d., 1 mm i.d.), with a sampling flow of 0.8 L min−1 (liters per minute). The instrument was operated using a drift tube at a pressure of around 2 mbar and a drift tube at a temperature of 60∘ (∘C). Drift tube voltage was kept at 600 V, leading to Td, where E is the electrical field strength and N is the gas number density. With these settings, the primary ion isotope (, 21.0221 Th) level stayed at 4500 cps (counts per second), and the mass resolving power of the HTOF mass analyzer was ∼4500. Data were recorded using a time resolution of 10 s. The background of the instrument was measured approximately every day with VOC-free air generated using a custom-made catalytic converter heated to 350 ∘C (Schallhart et al., 2016).
2.2.2 Vocus
The vocus PTR-TOF (proton transfer reaction time-of-flight mass spectrometer, Tofwerk AG/Aerodyne Research, Inc.), later referred to as a vocus, is based on a new PTR-inlet design (i.e., focusing ion–molecule reactor, FIMR) with sub-ppt detection limits (Krechmer et al., 2018). Sample air was drawn to the instrument using 1 m long PTFE tubing (6 mm o.d, 4 mm i.d.), with a flow rate of 4.5 L min−1. Most of the sample air was directed to the exhaust, while the actual flow to the vocus was around 0.15 L min−1. The instrument was operated with 1.0 mbar drift tube pressure, the voltages being 350 and 400 V for axial and radial voltages, respectively and Td. The vocus was operated at a higher water flow than in Krechmer et al. (2018), resulting in a decrease in the OVOC (e.g., HOMs) fragmentation but also in a lower sensitivity. The signal level of the instrument had some instability during the campaign, thus the primary ion signal (H3O+, 19.0178 Th) varied from a few hundred to few thousand cps and the isotope of the second water cluster (, 57.0432 Th) was around 104–105 cps. The much lower signal at H3O+ was due to a high-pass band filter that removes most of the ions < 35 Th (Krechmer et al., 2018). The mass resolving power of the LTOF mass analyzer was 12 000–13 000 for the whole campaign. Data were recorded using a time resolution of 10 s. Zero air was produced with a built-in active carbon filter and background was measured hourly except during 15–17 December due the malfunctioning of the zero-air pump.
2.2.3 Iodide
Another deployed instrument was a time-of-flight chemical ionization mass spectrometer (TOF-CIMS, Tofwerk AG/Aerodyne Research, Inc.), equipped with iodide (I−) reagent ion chemistry – later referred to as iodide. While the molecules could be detected as deprotonated species or as adducts with I−, we restricted the analysis in this work to ions containing only an iodide adduct, which guarantees detection of the parent organic compounds without substantial fragmentation. Iodide TOF-CIMS has been described previously and has high sensitivity towards (multifunctional) oxygenated organic compounds (Iyer et al., 2017; Lee et al., 2014). The instrument was operated at 1 L min−1 reagent flow rate into the ion–molecule reaction (IMR) chamber of the instrument. Iodide ions were generated from methyl iodide (CH3I) using a polonium (Po-210) source. Sample air was drawn to the instrument using 1 m long PTFE tubing (6 mm o.d, 4 mm i.d.) with a flow rate of 2 L min−1. The IMR was temperature controlled at 40 ∘C and operated at a nominal pressure of 100 mbar. The instrument, equipped with an HTOF mass analyzer, was configured to measure singularly charged ions from 1 to 1000 Th with a mass resolving power and time resolution of 4000–5000 and 10 s, respectively.
2.2.4 Amine and nitrate
Two chemical ionization atmospheric pressure interface time-of-flight mass spectrometers (CI-APi-TOF, Tofwerk AG/Aerodyne Research, Inc.) were also deployed (Ehn et al., 2014; Jokinen et al., 2012). The inlet was designed to minimize wall losses through the use of coaxial sample (10 L min−1) and sheath flows (∼30 L min−1) in order to sample (extremely) low-volatile species which are easily lost to the walls. Two types of ionization schemes were utilized: the promising new amine reagent ion chemistry (Berndt et al., 2017, 2018) and the more commonly used nitrate chemistry – later referred to as amine and nitrate, respectively. The amine has been shown to be sensitive towards a very wide range of OVOCs and both closed-shell species and peroxy radicals, from molecules with a few oxygen atoms all the way to HOMs (Berndt et al., 2018). Previous work have shown that protonated amines are effective reagent ions, forming stable clusters with OVOCs (Berndt et al., 2018). The nitrate, on the other hand, has mainly been used for detection of HOMs (Ehn et al., 2014).
Sample air was drawn to the instruments using a common 1 m long PTFE inlet line (19.05 mm o.d, 16 mm i.d.) with the flow rate being ∼20 L min−1 (∼10 L min−1 for each mass spectrometer). Nitrate () ions were formed from nitric acid (HNO3) using an X-ray source, while protonated butylamine (C4H12N+) ions were produced using butylamine with a 7.5 MBq Am-241 source. or C4H12N+ ions enter the ion reaction zone together with a clean sheath airflow, concentric with the sample flow, and the two do not mix turbulently. The ions are then guided into the sample flow by an electrical field. The residence time in the IMR was ∼200 ms. The main reagent ions were (mass to charge of 62 Th), (125 Th) and (HNO3)2 188 Th) for the nitrate and C4H12N+ (74 Th) for the amine. Both instruments were equipped with LTOF mass analyzers, providing a mass resolving power of 9000–10 000.
2.3 Calibration of the mass spectrometers
In order to estimate absolute concentrations of all detected molecules, each instrument's signals, using an averaging period of 15 min, were normalized to the reagent ion signals (to eliminate the influence of changes affecting all signals in the instruments, e.g., due to a degrading response from the detector), followed by multiplication with a scaling factor. The reagent ion quantity used for normalization is described below, separately for each instrument. Normalized ion count rates are reported as normalized cps and normalized counts per second (ncps). The scaling factors were derived differently for each instrument (details provided below). For iodide, nitrate, and amine, the same factor was used for all ions in the spectrum, while for the PTR instruments the factors were different depending on the type of molecule (e.g., VOC or OVOC). For the PTR instruments and the iodide, a duty cycle correction was applied to compensate for mass-dependent transmission due to the orthogonal extraction of the mass analyzers. The amine and nitrate were calibrated by scaling a wide range of mass to charge based on earlier studies, where duty cycle corrections had not been performed. Therefore, we did not apply such a correction for the atmospheric pressure ionization mass spectrometers. Finally, we emphasize that the scaling factors should not be compared between instruments as a measure of sensitivity, since multiple factors impact these values, including, for example, the specific normalization approach and the chosen extraction frequency of the mass analyzers.
The PTR-TOF was calibrated twice using a calibration unit consisting of a calibration gas mixture of 16 different VOCs (Apel-Riemer Environmental Inc., USA) that was diluted with clean air purified by a catalytic converter (1.2 L min−1 of zero air and 8 sccm of standard gas), producing VOC mixing ratios of around 7 ppb (parts per billion) (Schallhart et al., 2016). Sensitivities were calculated to be 12.31, 27.92, and 30.51 ncps ppb−1 based on the concentrations of monoterpenes, MVK (methyl vinyl ketone) and m-/o-xylenes. PTR-TOF signals were normalized using the sum of the first primary ion isotope at 21.0221 Th and the first water cluster isotope at 39.0327 Th (e.g., Schallhart et al., 2016). According to common practice, the sensitivities above were scaled to correspond to a situation where the total reagent ion signal equaled 106 cps.
The vocus was calibrated four times during the campaign using the same calibration gas mixture as used for the PTR-TOF. There was variability in the sensitivity during the campaign and therefore the uncertainty in the vocus results are slightly larger than normal. Sensitivities were highest for acetone, at maximum around 1800 and around 650 cps ppb−1 for monoterpenes. α-Pinene concentration was retrieved using the authentic standard, while the concentrations of the OVOC and C10H14H+ were estimated using the calibration factor of the MVK and sum of m-/o-xylenes, respectively. MVK and m-/o-xylene sensitivities was around 1700 and 700 cps ppb−1, respectively. Vocus signals were normalized using the primary ion signal at 19.0178 Th only, as the water clusters have a negligible effect on the ion chemistry inside the FIMR (Krechmer et al., 2018). Due to the high-pass filter that removes almost all the signal at 19.0178 Th, we do not report the normalized sensitivities (i.e., in ncps ppb−1) for the vocus in order to avoid direct comparisons with the PTR-TOF. Instead, the sensitivities above are given without normalization, although a normalization was used for the final data. For the uncertainty estimates, the same applies as listed above for the PTR-TOF.
The uncertainties for the compounds that were directly calibrated are estimated to be ±20 % for PTR-TOF and vocus. For other compounds, the uncertainties are much higher due to uncertain ionization efficiencies and potential fragmentation of the compounds with unknown structures. For example, we used sensitivity of MVK for all oxygenated monoterpenes, even though all those compounds may have very different fragmentation patterns, transmission rates and/or proton transfer reaction rates. Therefore, we refrain from quantitative estimates of the uncertainties for these species.
The iodide was calibrated twice during the campaign (15 and 23 December) by injecting known amounts of formic acid into the instrument. Due to unknown reasons, the response of the iodide decayed throughout the campaign, and therefore only data measured before 17 December, when a stronger drop occurred, were included for the direct comparison of the nonnitrate OVOCs. While normalization should compensate for this type of behavior, this particular instrument utilized a time-to-digital converter (TDC) acquisition card, which meant the primary ion peak was heavily saturated. Lacking any isotopic signatures for I−, we found that utilizing a region of the rising edge of the I− signal (126.5–126.65 Th) provided a reasonable correction to our data. The sensitivity without normalization was 1.0 cps ppt−1 for formic acid, and following the normalization, this sensitivity was applied for all ions throughout the period where iodide data were included in the analysis. We acknowledge that this brings with it a large uncertainty, as the iodide has sensitivities ranging over a few orders of magnitude depending on the specific molecule (Lee et al., 2014), and refrain from quantitative uncertainty estimates, as in the case for the PTR instruments above.
Standards for OVOC compounds measurable by the nitrate are still lacking, and this instrument was therefore not directly calibrated during the campaign. However, to be able to roughly estimate concentrations, a calibration was inferred by assuming that the molar yield of HOMs, i.e., molecules with six or more oxygen atoms, was 5 % during α-Pinene ozonolysis experiments. Different values have been reported for the HOM yield in this system, ranging from slightly above to slightly below 5 % (Ehn et al., 2014; Jokinen et al., 2014, 2015). Clearly such an approach yields large uncertainties, and we estimated it here to roughly ±70 %. Earlier work with more direct calibrations reported an uncertainty of ±50 % (Ehn et al., 2014) and the added 20 p.p. in this work reflects the increased uncertainty in scaling the sensitivity based on expected HOM yields. This method requires knowledge of the wall loss rate of HOMs in the COALA chamber, which was estimated to be 1∕300 s−1 in our study. This estimate is based on a rough scaling to a slightly smaller chamber (1.5 m3) with active mixing by a fan, where the loss rate was measured to be 0.01 s−1 (Ehn et al., 2014). As our chamber is larger, and our mixing fan was only spinning at a moderate speed, we estimated the loss rates to roughly 3 times lower. The resulting calibration coefficient was 2×1010 molecules cm−3 ncps−1, which is similar to that in previous studies (Ehn et al., 2014; Jokinen et al., 2012). As for nitrate, the amine was also not calibrated directly, and in order to achieve an estimate of the concentrations measured by this instrument, we scaled the sensitivity of the amine to match that of the nitrate for specific HOM dimers ( and ), which were found to correlate very well between the two instruments (as described in more detail in the Results section). This approach gave a calibration factor of 6×108 molecules cm−3 ncps−1, with similar uncertainty estimates to the nitrate. In the CI-APi-TOFs, the calibration factor is generally close to 1010 molecules cm−3 ncps−1, but as discussed later in Sect. 3.1, the amine reagent ion was considerably depleted during the experiments, which led to the relatively low calibration factor. As mentioned earlier, the scaling factors should not be compared directly between instruments. The lower value for the amine is a result of the normalization rather than an indication of higher sensitivity. This reagent ion depletion also means that the most abundant species were most likely no longer responding linearly to concentration changes, and therefore their concentrations can be off by an order of magnitude or more.
2.4 Instrumental limitations and considerations
In this section we aim to highlight some of the limitations involved when characterizing and quantifying OVOCs measured by online mass spectrometers. The list below is not exhaustive but addresses several issues that are relevant for the interpretation of our results.
2.4.1 Mass resolving power
One major limitation for all of the mass spectrometers described above is the mass resolving power, ranging from 4000 to 14 000. Even though the new generation of LTOF mass analyzers with higher resolving power can enhance the separation of measured ions, it remains challenging to accurately identify and deconvolve the elemental composition of many ions. Indeed, it is common for one CIMS mass spectrum to include more than 1000 different ions. For high-resolution (HR) peak identification and separation, firstly one needs to generate a list of ions, i.e., a peak list. Its construction can be time consuming, even if only based on one single spectrum, and once conditions change, different ions may appear. For measurements lasting weeks or months, it is nearly impossible to ensure that all ions are correctly identified and fitted. If the peak list contains too few ions compared to reality, signals from nonfitted ions will assign the adjacent ions with artificially high signals. On the contrary, if too many closely lying ions are included in the peak list, even small errors in the mass axis determination can cause the signal to be fitted to specific ions even though their signals are nonexistent. In such extreme cases, with closely overlapping ions, traditional HR analysis becomes impossible.
While less selective detection techniques can sound more useful for monitoring and characterizing OVOCs, spectra acquired using such ionization techniques (e.g., PTR, iodide or amine) pose a significant challenge for data analysis and may ultimately provide even less useful information. Statistical analysis techniques can be used in order to better constrain the uncertainties associated with peak fitting, as recently proposed (Cubison and Jimenez, 2015; Stark et al., 2015). These previous studies pointed out that the uncertainties related to the peak fitting can become significant if the overlapping peaks are separated by less than a full-width at half maximum (Cubison and Jimenez, 2015). This is very often the case for CIMS instruments, and the more the ions overlap, the larger the uncertainty is. Peak fitting becomes increasingly problematic as molecular masses increase, since the number of potential ions increases dramatically with mass.
2.4.2 Ionization, declustering and fragmentation
The response of a mass spectrometer to a certain compound is to first approximation a result of two factors: the ionization probability of the neutral molecule and the detection probability of the formed ion. The ionization process is largely controlled by the stability of the products compared to the primary ions, whether a question of adduct formation or (de)protonation processes. Different reagent ion chemistries have been studied computationally in recent years, successfully reproducing several observations (Berndt et al., 2017; Hyttinen et al., 2015, 2018; Iyer et al., 2016). While a neutral molecule can bind to a reagent ion at the collision limit, the adduct can undergo collision-induced dissociation (i.e., declustering) during transport through interfacing with the high vacuum in the mass analyzer. Ultimately, the binding strength of the adduct and the energy of the collisions in the mass spectrometer will define the survival probability of the ions. To address this issue, procedures have been proposed, for example to probe the response of adducts to different collision energies (Isaacman-VanWertz et al., 2018; Lopez-Hilfiker et al., 2016), providing critical information on the sensitivity of the instrument.
Similarly to declustering, (de)protonated compounds can undergo fragmentation reactions where molecular bonds are broken. For example, the detection of monoterpenes (C10H16) using PTR instruments often shows equally large signals at the parent ion () and at a fragment ion (). Also, iodide adducts have been shown to cause molecules to fragment, as in the case of peroxy acids decomposing to carboxylate anions (Lee et al., 2014). Both declustering and fragmentation processes are associated with the optimization of the voltages of each instrument, which is performed by the instrument operator (Breitenlechner et al., 2017; Krechmer et al., 2018; Lopez-Hilfiker et al., 2016). While using voltage scans to probe such processes is possible, and even desirable, performing, interpreting and utilizing the results across the mass spectrum and across different conditions remains challenging and has only been utilized in a few studies to date (Isaacman-VanWertz et al., 2018; Lopez-Hilfiker et al., 2016).
2.4.3 Quantification
For quantification, the instrument sensitivity is generally determined via calibration standards, while a background level was measured by zero air. The challenges involved in these procedures are highly dependent on the type of compounds to be quantified. As an example, we discuss three kinds of molecules with different volatilities: VOCs, SVOCs and ELVOCs.
- a.
VOCs: volatile species are relatively easy to quantify since they can be contained in gas bottles or easily evaporated from standard samples in known quantities. Their responses are also fast due to negligible adsorption and evaporation from the walls.
- b.
SVOCs: many semi-volatile organic compounds (SVOCs) are commercially available and can be evaporated in known amounts from liquid standards into the gas phase. However, the nature of SVOCs results in both condensed and gas phases for these species, meaning that once clean air is introduced, the signal of SVOCs will often show a gradual decay over minutes or even hours due to evaporation of the “leftovers” from surfaces in the inlet lines and the inlet itself (Pagonis et al., 2017). The procedure used to determine the “correct” blank is not trivial, and the blank will look different depending on whether it is done at the entrance of the instrument or at the sampling inlet and depending on the duration of the blank measurement itself. Another related challenge for SVOC quantification is that temperature fluctuations of a few degrees may cause net evaporation (temperature increasing) or condensation (temperature decreasing) of SVOCs from sampling lines and the inlet.
- c.
ELVOCs: for ELVOCs, finding standard compounds for calibration remains extremely difficult. Most organic compounds, including hydroperoxide or acid, with low volatility are likely to decompose before evaporating. Thus, their quantification is often inferred from other similar compounds. For the nitrate CI-APi-TOF, sulfuric acid is often used for calibration by being formed in situ from SO2 (Kürten et al., 2012). This is, to some extent, a similar approach to the one we took for the nitrate in this work and scaled to the estimated HOM yield, as both methods require knowledge of formation rates from the initial precursors and loss rates of the formed compound of interest. Other studies have used permeation sources of perfluorinated carboxylic acids, which are semi-volatile yet found to bind strongly to nitrate ions (Ehn et al., 2014; Heinritzi et al., 2016). However, while the calibration is complicated, the blank measurements are often not even needed for exactly the same reasons. Whatever contaminants might be present in the system, most are irreversibly lost to instrument surfaces and unable to evaporate into the gas phase due to the extremely low vapor pressures. Potential oxidation processes occurring inside the mass spectrometer may be an exception, but to our knowledge, this has not been reported to be a large concern for ELVOCs.
In addition to the list above, the response of an instrument to specific molecules may vary according to the conditions at which they were sampled. Temperature (change) was listed as one consideration and water vapor, or relative humidity (RH), is another important limitation for several mass spectrometers described above (Breitenlechner et al., 2017; Krechmer et al., 2018; Kürten et al., 2012; Lee et al., 2014; Li et al., 2019). For chemical ionization techniques, the water vapor can either compete with the OVOC ionization, leading to a decrease in sensitivity, or stabilize the adduct, resulting in an increase in the sensitivity. Alternatively, if a compound forms a very stable complex, it may have an adduct formation efficiency that is independent of water vapor. If the sensitivity is RH dependent, calibrations and blanks should optimally be performed at the same RH as the sampling in order to be representative. This, in turn, may cause considerable practical challenges for both RH control and calibration and blank cleanliness.
In summary, recent computational and experimental work has shown that many approaches exist for optimizing the ability of CIMS instruments to quantify OVOCs, including different blanks, calibration methods, voltage scans, etc. However, all these approaches are very rarely utilized in a single study, simply due to the immense time and effort required, both during the experiments and during the data analysis, where the results of all steps need to be incorporated. Ultimately, each study needs to prioritize producing larger amounts of data (i.e., performing more measurements) with less capability for detailed quantification or producing a smaller amount of data with more accurate quantification.
We applied our five CIMS instruments at the COALA chamber over a period of nearly 1 month, where we tried to provide different types of atmospherically relevant oxidation conditions for α-Pinene. With such high variability in the conditions, we compared signals between the mass spectrometers more robustly, even though certain limitations were inevitable. For example, it is often the case that mass spectra will show some signal at almost every mass, which can be due to multiple reasons, and it is important to separate when the signal is truly from the sampled air and not from some internal background or contamination. Similarly, one needs to assess whether the instrument is measuring the majority of the species with the same elemental composition or only detecting a small subset of those compounds due to specific selectivity for one isomer. In addition, an instrument may be able to detect a certain molecule, but the resulting signal remains unreliable. This may be the case if the sensitivity is extremely low for the molecule or if the peak is close to a much larger unrelated signal, which will create large interferences when performing HR fitting. In both cases the signal is likely to be influenced by different types of noise.
First, we performed correlation analyses in order to identify signals which were physically meaningful. We conducted the analysis with the whole data set (a total of ∼1000 ions in each instrument) rather than selectively focusing on individual ions. This comprehensive approach utilized more data but also resulted in larger uncertainties as not all fitted ions could be validated for all CIMS. From the correlation analysis we identified when two instruments agree, i.e., observing identical elemental compositions and having a similar temporal behavior, concerning some group of compounds. From a subsequent absolute comparison, we estimated which chemical ionization method was likely to be detecting a certain group of compounds more efficiently.
3.1 Instrument comparisons: correlations
3.1.1 Medium pressure ionization mass spectrometers
Peak fitting was performed by utilizing the Igor-based Tofware or Matlab-based TofTools software (Junninen et al., 2010) for ion mass to charge up to ∼600 Th, depending on the mass spectrometers. To select which ions to fit (i.e., include in the peak lists), both the exact masses and the isotopic distributions were used as criteria. A Pearson correlation coefficient R was calculated between molecules with the same elemental composition measured by different instruments. As a practical example, the time series of C10H16O3 measured by vocus and iodide are shown in Fig. 1c, and the time series correlation for this compound between the two instruments was R=0.85. For later comparisons we will use R squared and, in this case, R2=0.73. For iodide, the data set covered only the first half of the campaign, but the other instruments covered nearly the whole period. This includes a wide variety of conditions, with and without NOx, and therefore high correlations are very suggestive of two instruments measuring the exact same compound(s) at that specific elemental composition. However, as an increase in α-Pinene is likely to increase almost all measured OVOC signals to some extent, low positive correlations can arise artificially and should not be overinterpreted. Due to the selectivity and the sensitivity of the ionization methods, not all ions were observed in all the different instruments, and thus only a certain fraction of the identified compounds can be compared between mass spectrometers.
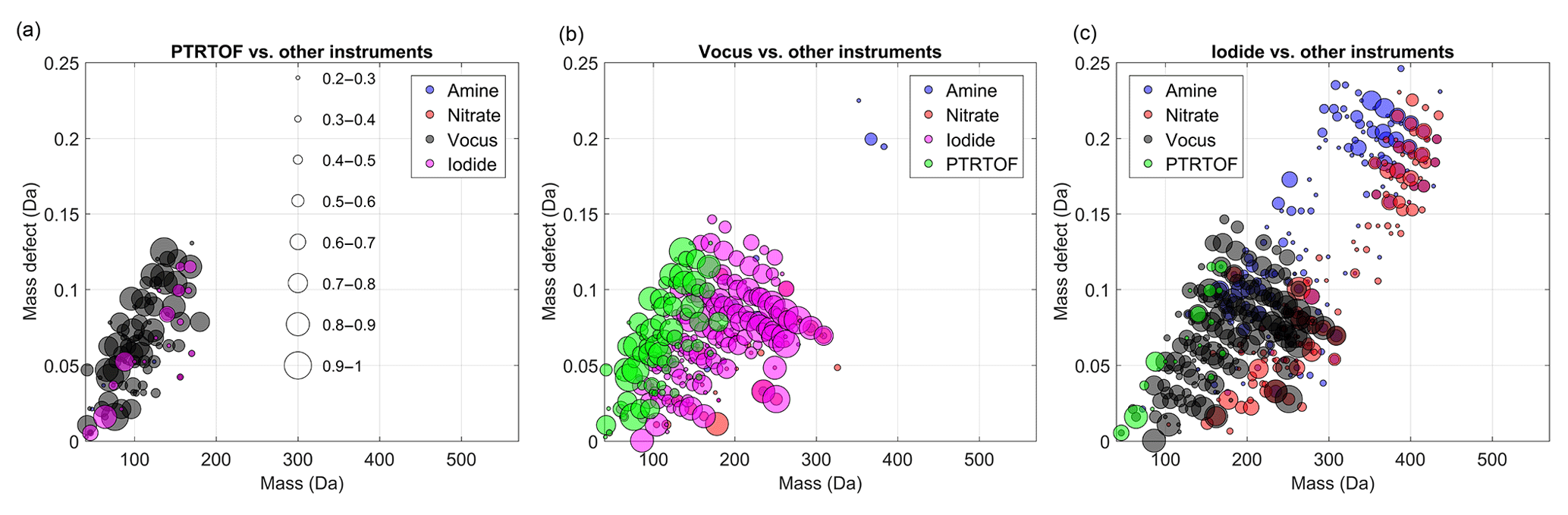
Figure 2Mass-defect plots showing the compounds for which a time series correlation (R2 > 0.2) was observed by the medium-pressure chemical ionization mass spectrometers, (a) PTR-TOF, (b) vocus and (c) iodide. Each circle represents a distinct molecular composition and the marker area represents the correlation (R2, legend shown in a) of the time series of that molecule between two different CIMS instruments. The color of each marker depicts the instrument against which the correlation is calculated.
Figure 2 shows the correlation analysis for the medium-pressure chemical ionization mass spectrometers, with marker size scaled by R2. In those figures, the abscissa represents the measured mass-to-charge ratio of the compounds and the y axis their mass defect, which is calculated as the exact mass of the compound minus the mass rounded to the closest integer (Schobesberger et al., 2013). For example, the mass of C10H16O3 is 184.110 Da, and the mass defect is +0.110 Da. The contribution of the reagent ions has been removed in the different figures. A mass-defect diagram helps to separate the molecules into two dimensions and allows some degree of identification of the plotted markers.
As expected, the PTR-TOF and the vocus are strongly correlated for compounds with low (0–3) oxygen number (Fig. 2a). Contrariwise, only a few compounds were identified by the PTR-TOF and the iodide with a fairly good correlation (i.e., R2 > 0.5). The correlating compounds included small acids such as formic and acetic acid. As discussed earlier, the inlet of the PTR-TOF is not well enough designed to sample OVOCs with low volatility, which explained the lack of correlations for larger and more oxidized products between the PTR-TOF and the nitrate CI-APi-TOF. The molecules with the lowest correlations (R2 < 0.2) were not included in the plots, as the intention is to show regions where instruments agree. If an ion is included in a peak list, it will always be fit, and thereby a value of R2 > 0 is always expected, filling markers throughout the MD-mass space.
In addition to VOCs, the vocus was able to measure a large range of OVOCs (150–300 Th) as revealed in Fig. 2b, displaying a very good correlation with species identified by the iodide. Indeed, most of the identified compounds have R2 > 0.7. As noted earlier, several different experimental conditions were tested (Fig. 1), and these high correlations indicate that both instruments were likely sensitive to the same compounds. In other words, a good correlation was seen in this mass range for nearly all compositions, the iodide and the vocus did not seem to be strongly impacted by the exact chemical conformation of the organic compounds. Interestingly no dimers (mass to charge > 300 Th) were observed with the vocus, which suggests some potential limitation of the instrument or the used settings. As a result, a very limited correlation was observed between compounds measured by the vocus and the amine or nitrate CI-APi-TOFs. The two main exceptions were C5H6O7 (178.011 Da) and C7H9NO8 (235.033 Da). Note that the latter is less clear, as the correlation is nearly identical between three instruments (nitrate, vocus, and iodide). The lack of correlation was not only due to lack of ion transmission at higher masses in the vocus, since the instrument was able to detect some ions up to 400 Th, including C10H30O5Si5H+ and C19H29O6NH+. One possibility was that since the compounds above ∼300 Th were likely to contain hydroperoxides, or in the case of dimers, organic peroxides, the ions may have fragmented before detection in the vocus, either during the protonation or due to the strong electric fields in the vocus FIMR. In the case of HOM monomers with more than seven oxygen atoms, an additional limitation comes from more abundant and closely overlapping ions in the spectra, impacting the accurate fitting of these ion signals in the vocus. From our data set, it was not possible to determine the exact cause(s) for this lack of sensitivity for larger molecules in the vocus, but it is possible that changes in instrument operating conditions can extend the range of molecules detectable using the vocus in future studies.
As shown in Fig. 2c, the iodide was capable of measuring ions with larger masses (i.e., above 300 Th), indicating the detection of more complex (e.g., dimers) and oxygenated compounds than the vocus. This was the case in spite of the lower flow rate for the iodide than the vocus and thus less optimal for sampling of low-volatile species (Table 1). The iodide seemed to have the widest detection range of the mass spectrometers deployed in this study, showing high correlation with other instruments for organic molecules, from C1 (like formic acid) to C20, as long as the molecules had at least two oxygen atoms. This is in line with earlier findings that the iodide is sensitive to most species that are polar or have polarizable functional groups (Iyer et al., 2017; Lee et al., 2014). However, the correlation with the CI-APi-TOFs was still somewhat limited (R2 < 0.7) for HOM monomers and dimers. One reason may have been that these HOMs contain peroxy acid functionalities, which have been shown to undergo reactions in the iodide TOF-CIMS (Lee et al., 2014). In this work, we only analyzed the ions containing I−, as these were believed to be the ones where the parent molecule remained intact. Another reason for lower correlation was the fact that I− is less selective than other ionization methods, resulting in many overlapping peaks at the same integer mass and ambiguous peak fitting (Lee et al., 2014; Stark et al., 2015, 2017), similar to the case in the vocus. This means that, although the iodide and/or the vocus might be able to charge a specific molecule, and it would not fragment before detection, the ion may remain unquantifiable due to a highly ambiguous peak fitting as a result of multiple overlapping signals.
3.1.2 Atmospheric pressure interface mass spectrometers
Figure 3 shows similar comparisons to those in Fig. 2 for the nitrate (Fig. 3a) and the amine (Fig. 3b). Interestingly, these two instruments show excellent correlation (R2 > 0.9) for dimeric products (molecules within 350–500 Th) but showed mostly low correlations (R2 < 0.6) with other instruments in the monomer range. The nitrate had some agreement with the iodide for certain monomer compounds, but in the HOM-monomer range where the nitrate generally saw its largest signals (C10 molecules with 7 to 11 oxygen atoms; Ehn et al., 2014), none of the other instruments showed strongly correlating signatures.

Figure 3Mass-defect plots showing the compounds for which time series correlation (R2 > 0.2) was observed by the atmospheric-pressure chemical ionization mass spectrometers, (a) nitrate and (b) amine. Each circle represents a distinct molecular composition and the marker area represents the correlation (R2, legend shown in Fig. 2a) of the time series of that molecule between two different CIMS instruments. The color of each marker depicts the instrument against which the correlation is calculated.
Despite showing signals at almost all OVOCs, the amine presented low correlations for all OVOCs except the dimers. In the amine the reagent ion was greatly depleted due to the relatively high signals (Fig. 4), likely leading to a nonlinear response for most of the OVOCs, apparently with the exception of the HOM dimers. It may be that the amine reagent ion formed extremely stable clusters with these dimers, and thus any collision involving these dimers with the reagent ion (regardless of whether already clustered with an OVOC) in the IMR led to an amine–dimer cluster. While the amine showed very low correlation with the other instruments for most molecules, it has been demonstrated to be an extremely useful detector of both radicals and closed-shell OVOCs under very clean, low-loading flow tube experiments (Berndt et al., 2017, 2018). In other words, it can provide information on a wide variety of OVOCs, but to obtain quantitative information, the amine CI-APi-TOF has to be used in a very diluted system (with very clean air) and at low loadings. Determining the limitations more explicitly requires further studies, but as a rough approximation, the typical CI-APi-TOF sensitivity of ∼1010 molecules cm−3 ncps−1 means that when sampling detectable molecules at 1010 molecules cm−3 (∼0.4 ppb), these molecules will have ion signals of equal abundance to the reagent ions. Consequently, once the concentration of measurable molecules exceeds roughly 100 ppt, the CI-APi-TOF may no longer be an optimal choice. For the nitrate CI-APi-TOF, which mainly detects HOMs with short lifetimes due to their low volatilities, this has rarely been a limitation, but for less selective reagent ions, like amines, this can be an important consideration.
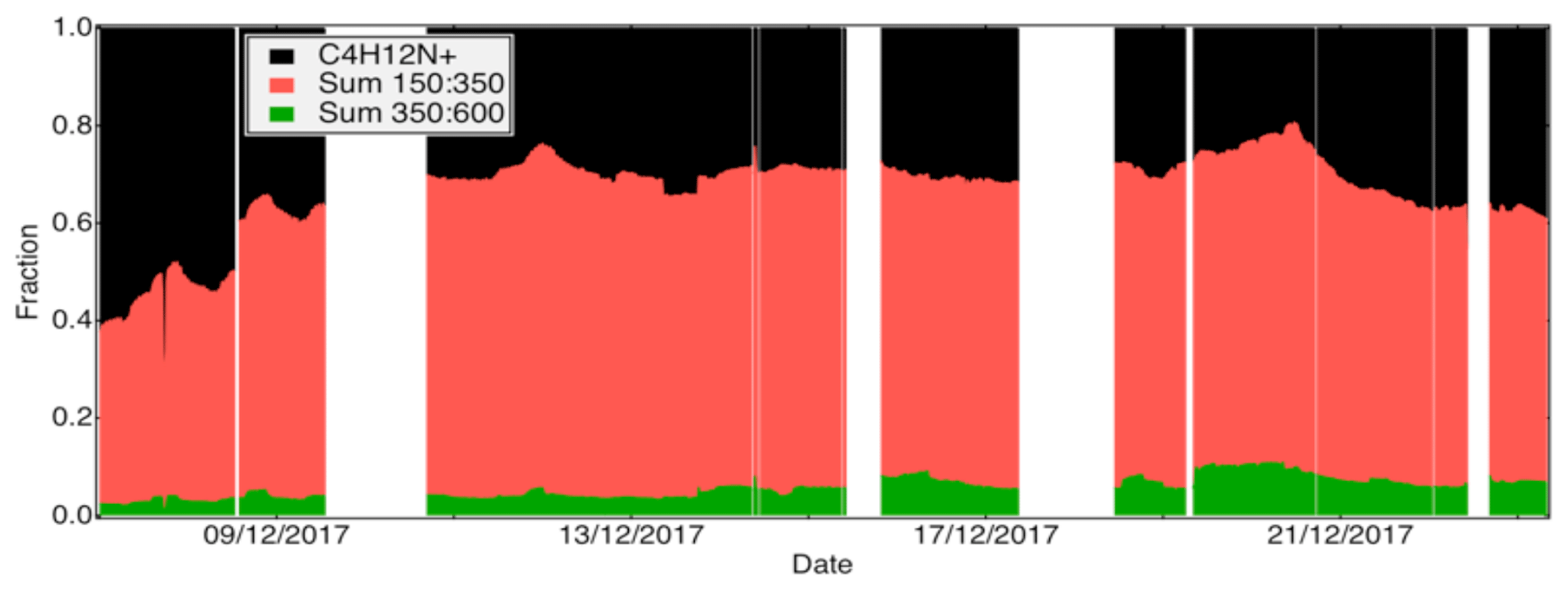
Figure 4Contribution of the reagent ion, sum of ions from 150 to 350 Th and sum of ions from 350 to 600 Th to total ion count throughout the campaign for the amine CI-APi-TOF. Only a negligible fraction of the signal was found below 150 Th (excluding C4H12N+).
3.2 Instrument comparisons: concentration estimates
Concentrations of the identified compounds were estimated for all the different instruments, as described in Sect. 2.6. It should be noted that no separate inlet loss corrections were applied. The estimations for the results of PTR-TOF and the vocus are the most reliable as both instruments were calibrated using authentic standards with a proven method, while larger uncertainties in the total measured concentrations are expected for the iodide and the CI-APi-TOFs.
With around 1000 identified ions for each instrument, except for the PTR-TOF, we decided to focus our attention in this section on a few particular compound groups: the most abundant C10 monomers (i.e., C10H14∕16On), C10 organonitrates (C10H15NOn) and dimers (C20H32On). For the nonnitrate compounds, the concentrations were measured during steady-state conditions on 9 December from 15:30 to 23:00 with [O3] = 25 ppb and [α-Pinene] =100 ppb) during period I (Fig. 1 in blue). The organonitrate concentrations were compared using steady-state conditions from 20 December, from 02:45 to 07:45 with [O3] =35, [α-Pinene] =100 and NO =0.5 ppb, during period IV (Fig. 1 in purple). Figure 5a–d show the concentrations of the selected species as a function of oxygen number in the molecules. While we again emphasize that all the concentrations were only rough estimates, these plots painted a similar picture to the correlation analysis, as described in more detail in the next paragraphs.
Focusing first on the nonnitrate monomers (Fig. 5a–b), for compounds with zero or one oxygen atoms, the PTR-TOF agreed well with the concentration estimated by the vocus, while molecules with more than two oxygen atoms were already close to, or below, the noise level of the PTR-TOF. In contrast, as the number of oxygen atoms in the molecule reached two or more, the iodide signal increased and for most compounds showed concentrations similar to the vocus. These two instruments agreed on concentration estimates fairly well all the way up to an oxygen content of around nine oxygen atoms, where the measured signals were close to the instruments' noise levels. However, when comparing to the nitrate, which is assumed to have good sensitivity for HOMs with seven or more oxygen atoms, the concentrations suggested by the vocus and iodide for the O7 and O8 monomers were very high. We preliminarily attributed this to an overestimation of the concentrations of HOMs by these two instruments, possibly due to higher sensitivities towards these molecules compared to the compounds used for calibration (i.e., MVK). We also did not correct for potential backgrounds using the blanks for the iodide, although they were measured, since the variability in the blank concentrations (see also discussion in Sect. 2.4) was large enough to cause artificially high fluctuations in the final signals. Therefore, we opted to not include such a correction but also note that, even if half the signal at a given ion was attributable to background in the iodide, then it would only have a small impact on the logarithmic scales used in Fig. 5. Other possible reasons for this discrepancy was that the iodide and vocus were able to detect isomers that the nitrate was not, or that the nitrate sensitivity was underestimated. However, considering that the nitrate HOM signal was scaled to match a 5 % molar HOM yield, it was unlikely that the HOM concentrations can be considerably higher than this. Other estimated parameters involved in the formation and loss rates of HOMs also had uncertainties, but we did not expect any of them to be off by more than 50 %. This concentration discrepancy thus remained unresolved and will require more dedicated future studies.
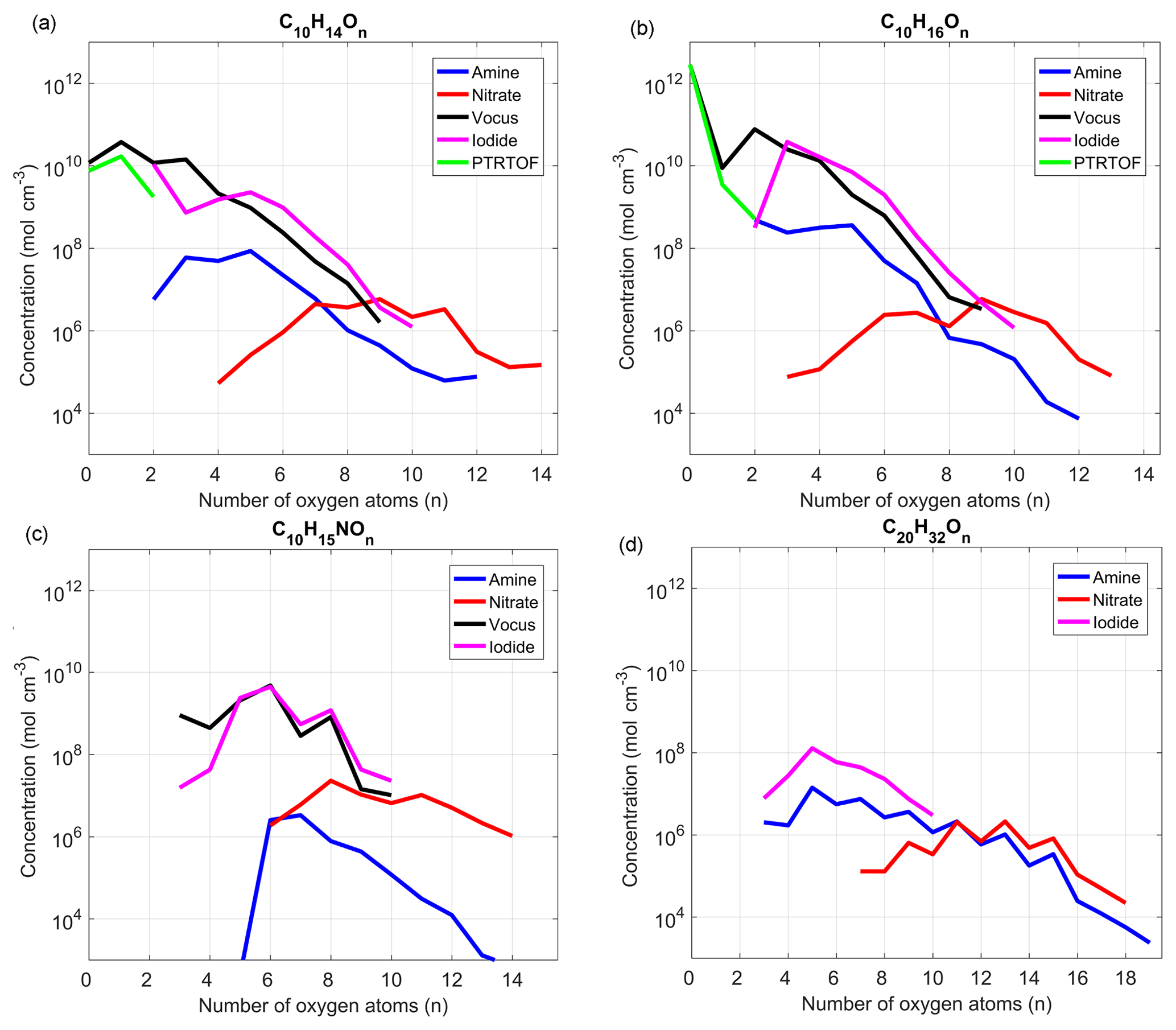
Figure 5Estimated concentrations of the main α-Pinene C10-monomer oxidation products (a, b), C10-monomer organonitrates (c) and α-Pinene dimers (d) by the different mass spectrometers deployed in this study. The average concentrations were estimated when the system reached steady state in two experiments: without NO (a, b, d), 9 December (15:30–23:00), and with NO (c), 20 December (02:45 to 07:45). See text for more details. Data are plotted only for ions for which the average concentrations were higher than 3 times the standard deviation during the campaign.
Finally, the quantities estimated using the amine are significantly lower (1–2 orders of magnitude) for all monomers when compared to the other instruments. This was presumably related to the titration of the reagent ion, which meant that the majority of charged OVOCs will undergo multiple subsequent collisions with other OVOCs, potentially losing their charge in the process. The nitrate had, as expected, very low sensitivity towards less oxygenated compounds and its highest detection efficiency for HOMs (i.e., molecules with at least six oxygen atoms).
The organonitrate comparison in Fig. 5c suggested that both the vocus and the iodide were efficient at detecting these compounds, as both instruments agreed well (R2 > 0.7) for C10 organonitrates with 5 to 10 oxygen atoms. While organonitrates have been detected before using the iodide (Lee et al., 2016), this was the first observation in which the vocus also detected such compounds efficiently. However, we cannot exclude such compounds undergoing fragmentation within the drift tube as commonly observed in other PTR instruments (Yuan et al., 2017). For larger oxygen content, the nitrate again seemed to be most sensitive, showing clear signals above 10 oxygen atoms, where the previous instruments were already close to noise levels. The amine seemed worse at detecting organonitrates compared to nonnitrate monomers.
Neither of the PTR instruments were able to detect any dimers in this study within their measurement ranges (up to 320 Th for PTR-TOF). The amine and the nitrate were able to quantify the widest range of HOM dimers, while the iodide was able to detect less oxidized dimers (Fig. 5d). Based on the concentration estimates, the amine detection range also extended to less oxidized dimers than the nitrate, as has already been shown by Berndt et al. (2018). Dimers measured by the iodide were more abundant than the ones detected by the amine, but from the monomer comparisons we speculated that the amine might be underestimating concentrations, while the iodide might be overestimating them. With the data available to us, we can only speculate on the relative sensitivities of the instruments able to detect dimers, especially with the vocus providing no support to the comparison.
One aspect lending credibility to the amine dimer data, in addition to the good time series correlation with the nitrate, was the odd–even oxygen atom patterns visible both in the amine and nitrate data. Such a pattern is to be expected, since the 32 hydrogen atoms in the selected dimers indicate that they have been formed from RO2 radicals, one of which had 15 hydrogen atoms (which is what ozonolysis will yield, following OH loss) (Docherty et al., 2005; Lee et al., 2006; Ziemann and Atkinson, 2012), while the second RO2 had 17 hydrogen atoms (which is the number expected from OH oxidation of an alkene where OH adds to the double bond). The first RO2 from ozonolysis had four oxygen atoms, and further autoxidation will keep an even number of oxygen atoms, while the opposite was true for the OH-derived RO2, which started from three oxygen atoms. In other words, the major dimers from this pathway should contain an odd number of oxygen atoms after they are combined. In the case of C20H30On dimers, mainly formed from two ozonolysis RO2, the pattern was expected to show peaks at even numbers, which is also the case (not shown).
Odd–even patterns for the oxygen content were not visible in the iodide, but the reason remained unknown. It was possible that the dimers detected by the iodide might be formed via other pathways, where such a selectivity did not occur. This topic should be explored further in future studies, since dimers formed from the oxidation of biogenic compounds are important for new-particle formation, and it is therefore critical to accurately identify and quantify the formation and evolution of different types of dimers. To date, both dimers measured by iodide (Mohr et al., 2017) and nitrate (Tröstl et al., 2016) have been found to be important for particle formation from monoterpenes.
3.3 Performance in detecting oxygenated species
Figure 6 summarizes our results and depicts the performance of each mass spectrometer in detecting monomer and dimer monoterpene oxidation products. Molecules of C10H16On, C10H15NOn and C20H30On were provided as examples. We emphasized that the oxygen content alone was not the determining factor for whether a certain type of mass spectrometer will detect a compound, but we utilized this simplified representation in order to provide an overview of the performances of the different chemical ionization schemes. The results were primarily based on the correlation analysis from Sect. 3.1, and as apparent from the y axis, this comparison was only qualitative. However, our aim was to provide an easy-to-interpret starting point, especially for new CIMS users wanting to compare different available techniques.
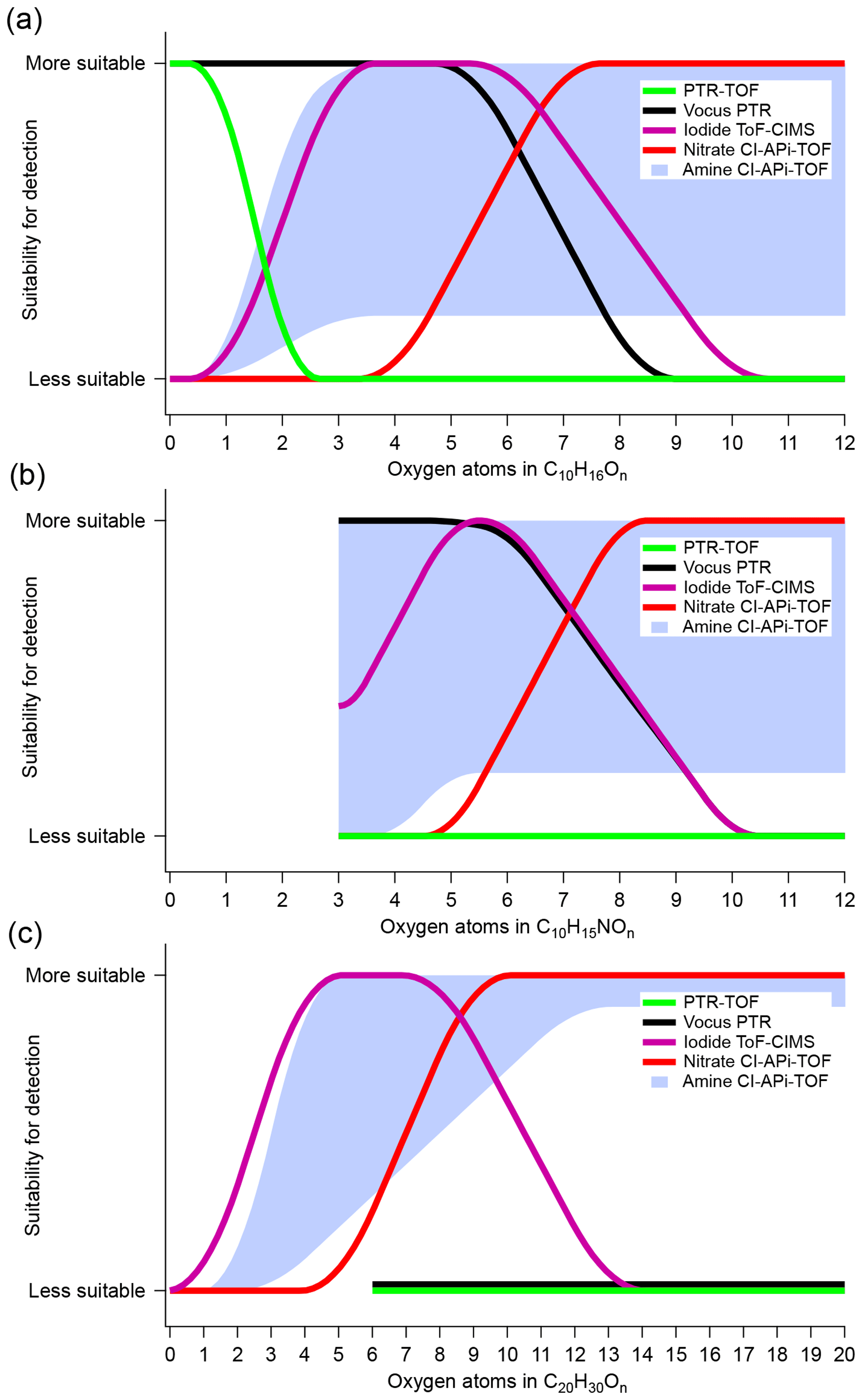
Figure 6Estimated detection suitability of the different CIMS techniques for α-Pinene and its oxidation products, plotted as a function of the number of oxygen atoms. Each panel symbolizes a compound group: monomers (a), organonitrate monomers (b) and dimers (c). The figures are indicative only, as none of the reagent ion chemistries are direct functions of the oxygen atom content in the molecules. See text for more details.
For monomer compounds without N atoms, shown in Fig. 6a, the PTR-TOF was limited to the detection of VOCs, while the vocus was additionally able to measure a large range of OVOCs, up to at least five to six oxygen atoms. The iodide detected OVOCs with oxygen content starting from ∼3 atoms but did not seem to efficiently observe HOM monomers (i.e., C10HxO>7). While being a very promising instrument for a broad detection of OVOCs, the performance of the amine was limited in our study due to a significant drop in the reagent ion to ∼40 % of the total signal. Therefore, the amine was marked with a shaded region rather than a line, with the lower limit based roughly on its usefulness under the conditions we probed, while the upper limit was an estimate based on findings in a cleaner system with low loadings (Berndt et al., 2018). Finally, the nitrate was mainly selective towards HOMs. The detection and quantification of monomeric OVOCs containing five to eight oxygen atoms remained the most uncertain, since there were inconsistencies in both concentration and correlation between the nitrate, measuring the more oxygenated species, and the vocus and iodide, which detected the less oxidized compounds.
In Fig. 6b, the suitability for the different instruments was plotted for organonitrate monomers. The vocus efficiently detected the less oxidized organonitrates, while the iodide displayed good sensitivity for the same compounds, with the exception of the least oxygenated ones. For larger number of oxygens, the nitrate again seemed the most suitable method. For dimers (Fig. 6c), neither of the PTR techniques showed any ability to detect these compounds in our study. We did not extend the lines all the way down to n=0 for the compounds, as it was still possible that these methods can be able to detect the least oxidized and most volatile C20 compounds, which might not have been present during our experiments. The iodide showed some correlation with the nitrate but had good signals mainly in the range of dimers with four to eight oxygen atoms. The amine and nitrate correlated well for the most oxidized dimers, suggesting good suitability for dimer detection of HOM dimers. The amine concentrations stayed high, with the expected odd–even pattern in oxygen number, even at lower oxygen content than the nitrate, and therefore the suitability extended further towards lower O-atom contents. Again, the shaded area was based on a combination of our findings and those of Berndt et al. (2018).
The results in Fig. 6 are based on the α-Pinene ozonolysis system. While we will not speculate too much about the extent to which these findings can be extrapolated to other systems, certain features will remain similar for other atmospherically relevant reactions. For example, the most oxidized gaseous HOM species will likely have been formed through autoxidation processes, which means that they will contain hydroperoxide functionalities and could thus be detectable by the nitrate. Likewise, the HOMs, and in particular the dimers, will very likely have low volatilities, requiring high sample flows with minimal wall contact, as in the case of the Eisele-type CI inlets used in the nitrate and amine. Several other key features are also expected to be valid in different VOC–oxidant systems, and therefore we believe that our findings are also relevant for many other reaction partners.
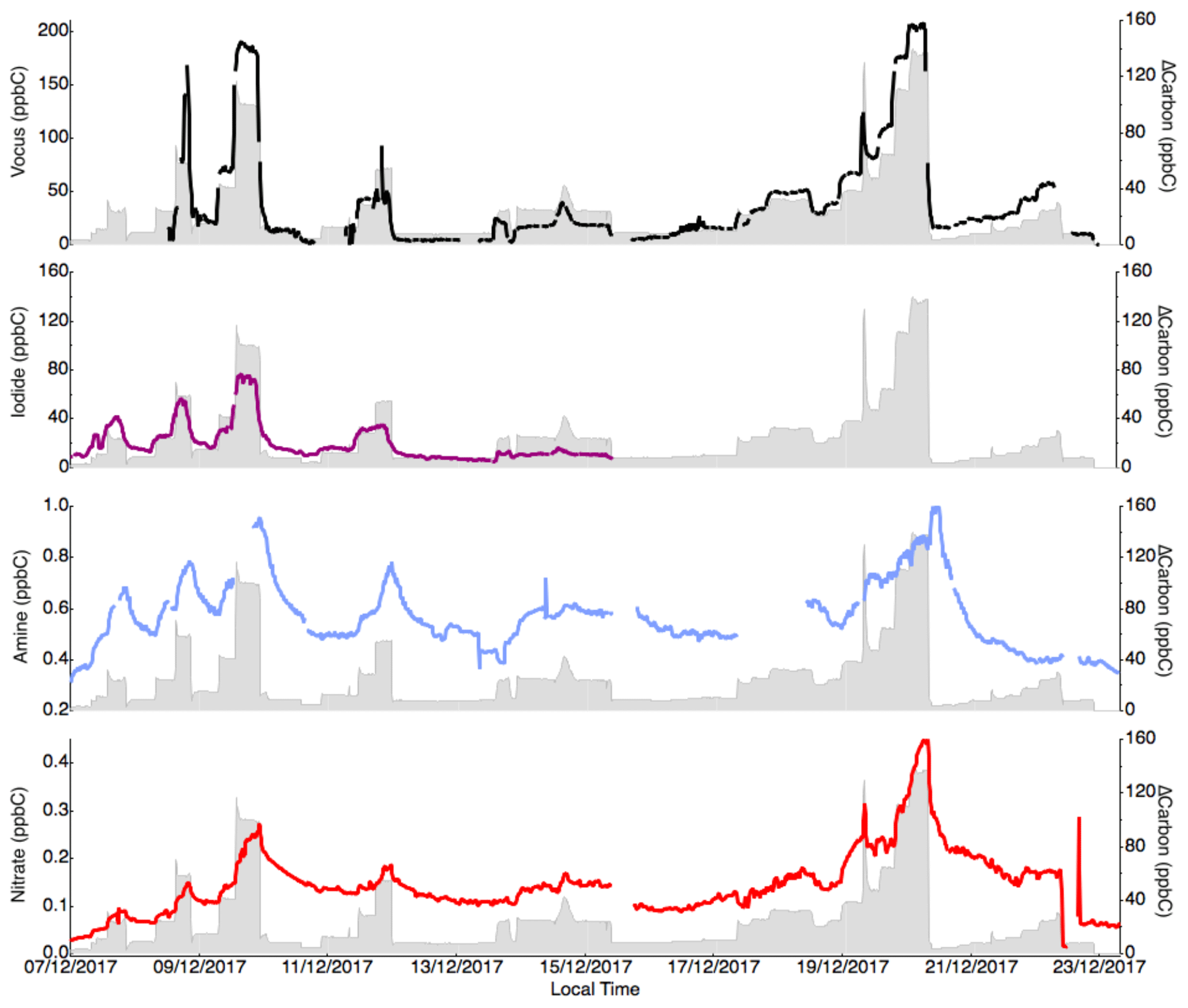
Figure 7Concentration (in ppbC) of the sum of the compounds measured by each instrument (vocus, iodide, amine and nitrate) throughout the campaign compared to the amount of reacted carbon through α-Pinene oxidation. Large uncertainties remain in the quantification of the OVOCs for all instruments, but it is clear that the iodide and vocus are able to measure a large fraction of the reacted carbon in the gas phase.
As a final test for each instrument, we estimated how much of the reacted carbon (in ppbC) the different mass spectrometers can explain. As shown in Fig. 7, both the iodide and vocus seemed to capture most of the reacted carbon within uncertainties. The concentration determined using the vocus was overestimated, explaining more carbon than was reacted. Out of the largest contributors to the reacted carbon, pinonaldehyde (C10H16O2) was not efficiently detected by iodide, but otherwise most of the abundant molecules were quantified by both vocus and iodide. Any carbon lost by condensation to walls or particles would not have been quantifiable by any of the instruments in this study. While the nitrate was calibrated with an assumption that it can measure 5 % of the reacted α-Pinene, it only detected less than 0.1 of that amount. The reason was that the HOMs it can detect were quickly lost to walls (or particles), and thus the gas-phase concentration was not equivalent to the branching ratio of the VOC oxidation reaction. In fact, and as revealed by the slow changes in the times series in Fig. 7d, most of the carbon ultimately measured by the nitrate was semi-volatile, as such compounds accumulated and reached higher concentration in the chamber, unlike HOMs. Thus, while the nitrate was able to detect a critical group of OVOCs from an aerosol formation perspective, i.e., HOMs, for carbon closure studies (Isaacman-VanWertz et al., 2017, 2018), it will be of limited use. This again highlights the need to first determine the target of a study before deciding which CIMS technique is the most useful. For the closure comparison in our study, the overestimations emphasized the need to perform calibration with an extensive set of OVOCs, ideally with monoterpene-oxidation products, in order to better constrain the sensitivity of the products of interest. The study by Isaacman-VanWertz et al. (2018), as the only study to achieve full carbon closure during chamber oxidation of α-Pinene by OH, also successfully utilized voltage scanning to determine sensitivities of each compound.
The primary goal of this work was to evaluate the performance of five chemical ionization mass spectrometers (PTR-TOF, vocus PTR, iodide TOF-CIMS, amine CI-APi-TOF and nitrate CI-APi-TOF) in the identification and quantification of a wide variety of products formed in the ozonolysis of α-Pinene. In addition, we wanted to estimate the capabilities of the newly developed vocus PTR in measuring OVOC species. By comparing the regions of coverage of the instruments across multiple experimental conditions (i.e., in different O3, VOC, NO and OH radical concentrations), we demonstrated that the current instrumentation captures nearly the entire range of OVOCs, spanning from VOCs to ELVOCs. The PTR-TOF was only able to measure the most volatile compounds, while the vocus appeared to be able to measure both VOCs and most of the OVOCs up to five to six oxygen atoms. In combination with the iodide and nitrate, most of the OVOC range can be measured. The iodide showed good overlap with the vocus for most SVOCs with three to five oxygen atoms, while the nitrate mainly detected products with six or more oxygen atoms. No dimer species were observed with either of the PTR instruments, which might be due to wall losses (likely at least for the PTR-TOF) and/or potential fragmentation in the instruments. The amine CI-APi-TOF is a promising technique, as shown in earlier studies, but it likely requires low loadings in order to not titrate the reagent ion, limiting its utility for many chamber experiments and, potentially, atmospheric observations. The large uncertainties in attempting a quantification of the wide variety of species measurable with these mass spectrometers underline the urgent need to develop robust, simple and complete calibration methods in order to obtain a better estimation of the concentrations. Finally, it is important to underline that the experimental and analytical procedures performed by the user will ultimately impact the sensitivity, the selectivity and the interpretability of the results attainable from each instrument.
Mass spectrometry data are available upon request to the corresponding authors.
MR and ME designed the experiments. Instrument deployment, operation, and data analysis were carried out by MR, PR, JEK, OP, YZ, LH, OG, CY and ME; MR, PR, OP and ME interpreted the compiled data set. MR, PR and ME wrote the paper. All co-authors discussed the results and commented the manuscript. The authors declare that they have no conflict of interest.
JEK and DW both work for Aerodyne.
This work was supported by the European Research Council (ERC-StG COALA, grant no. 638703). We gratefully acknowledge Pasi Aalto, Petri Keronen, Frans Korhonen, and Erkki Siivola for technical support. Olga Garmash thanks Doctoral Programme in Atmospheric Sciences (ATM-DP) at the University of Helsinki for financial support. Otso Peräkylä would like to thank the Vilho, Yrjö and Kalle Väisälä Foundation. We thank the TofTools team for providing tools for mass spectrometry data analysis.
This paper was edited by Keding Lu and reviewed by three anonymous referees.
Albrecht, B. A.: Aerosols, Cloud Microphysics, and Fractional Cloudiness, Science, 245, 1227–1230, https://doi.org/10.1126/science.245.4923.1227, 1989.
Atkinson, R., Baulch, D. L., Cox, R. A., Hampson, R. F., Kerr, J. A., Rossi, M. J., and Troe, J.: Evaluated Kinetic and Photochemical Data for Atmospheric Chemistry: Supplement VI. IUPAC Subcommittee on Gas Kinetic Data Evaluation for Atmospheric Chemistry, J. Phys. Chem. Ref. Data, 26, 1329–1499, https://doi.org/10.1063/1.556010, 1997.
Berndt, T., Richters, S., Kaethner, R., Voigtländer, J., Stratmann, F., Sipilä, M., Kulmala, M., and Herrmann, H.: Gas-Phase Ozonolysis of Cycloalkenes: Formation of Highly Oxidized RO2 Radicals and Their Reactions with NO, NO2, SO2 and Other RO2 Radicals, J. Phys. Chem. A, 119, 10336–10348, https://doi.org/10.1021/acs.jpca.5b07295, 2015.
Berndt, T., Herrmann, H., and Kurtén, T.: Direct Probing of Criegee Intermediates from Gas-Phase Ozonolysis Using Chemical Ionization Mass Spectrometry, J. Am. Chem. Soc., 139, 13387–13392, https://doi.org/10.1021/jacs.7b05849, 2017.
Berndt, T., Scholz, W., Mentler, B., Fischer, L., Herrmann, H., Kulmala, M., and Hansel, A.: Accretion Product Formation from Self- and Cross-Reactions of RO2 Radicals in the Atmosphere, Angew. Chem. Int. Edit., 57, 3820–3824, https://doi.org/10.1002/anie.201710989, 2018.
Bertram, T. H., Kimmel, J. R., Crisp, T. A., Ryder, O. S., Yatavelli, R. L. N., Thornton, J. A., Cubison, M. J., Gonin, M., and Worsnop, D. R.: A field-deployable, chemical ionization time-of-flight mass spectrometer, Atmos. Meas. Tech., 4, 1471–1479, https://doi.org/10.5194/amt-4-1471-2011, 2011.
Breitenlechner, M., Fischer, L., Hainer, M., Heinritzi, M., Curtius, J., and Hansel, A.: PTR3: An Instrument for Studying the Lifecycle of Reactive Organic Carbon in the Atmosphere, Anal. Chem., 89, 5824–5831, https://doi.org/10.1021/acs.analchem.6b05110, 2017.
Crounse, J. D., McKinney, K. A., Kwan, A. J., and Wennberg, P. O.: Measurement of Gas-Phase Hydroperoxides by Chemical Ionization Mass Spectrometry, Anal. Chem., 78, 6726–6732, https://doi.org/10.1021/ac0604235, 2006.
Cubison, M. J. and Jimenez, J. L.: Statistical precision of the intensities retrieved from constrained fitting of overlapping peaks in high-resolution mass spectra, Atmos. Meas. Tech., 8, 2333–2345, https://doi.org/10.5194/amt-8-2333-2015, 2015.
Docherty, K. S., Wu, W., Lim, Y. B., and Ziemann, P. J.: Contributions of Organic Peroxides to Secondary Aerosol Formed from Reactions of Monoterpenes with O3, Environ. Sci. Technol., 39, 4049–4059, https://doi.org/10.1021/es050228s, 2005.
Donahue, N. M., Robinson, A. L., Trump, E. R., Riipinen, I., and Kroll, J. H.: Volatility and Aging of Atmospheric Organic Aerosol, in: Atmospheric and Aerosol Chemistry, vol. 339, edited by: McNeill, V. F. and Ariya, P. A., Springer Berlin Heidelberg, Berlin, Heidelberg, 97–143, 2012.
Ehn, M., Thornton, J. A., Kleist, E., Sipilä, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I.-H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurtén, T., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., Canagaratna, M., Maso, M. D., Berndt, T., Petäjä, T., Wahner, A., Kerminen, V.-M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476–479, https://doi.org/10.1038/nature13032, 2014.
Gakidou, E., Afshin, A., Abajobir, A. A., Abate, K. H., Abbafati, C., Abbas, K. M., Abd-Allah, F., Abdulle, A. M., Abera, S. F., Aboyans, V., Abu-Raddad, L. J., Abu-Rmeileh, N. M. E., Abyu, G. Y., Adedeji, I. A., Adetokunboh, O., Afarideh, M., Agrawal, A., Agrawal, S., Ahmadieh, H., Ahmed, M. B., Aichour, M. T. E., Aichour, A. N., Aichour, I., Akinyemi, R. O., Akseer, N., Alahdab, F., Al-Aly, Z., Alam, K., Alam, N., Alam, T., Alasfoor, D., Alene, K. A., Ali, K., Alizadeh-Navaei, R., Alkerwi, A., Alla, F., Allebeck, P., Al-Raddadi, R., Alsharif, U., Altirkawi, K. A., Alvis-Guzman, N., Amare, A. T., Amini, E., Ammar, W., Amoako, Y. A., Ansari, H., Antó, J. M., Antonio, C. A. T., Anwari, P., Arian, N., Ärnlöv, J., Artaman, A., Aryal, K. K., Asayesh, H., Asgedom, S. W., Atey, T. M., Avila-Burgos, L., Avokpaho, E. F. G. A., Awasthi, A., Azzopardi, P., Bacha, U., Badawi, A., Balakrishnan, K., Ballew, S. H., Barac, A., Barber, R. M., Barker-Collo, S. L., Bärnighausen, T., Barquera, S., Barregard, L., Barrero, L. H., Batis, C., Battle, K. E., Baumgarner, B. R., Baune, B. T., Beardsley, J., Bedi, N., Beghi, E., Bell, M. L., Bennett, D. A., Bennett, J. R., Bensenor, I. M., Berhane, A., Berhe, D. F., Bernabé, E., Betsu, B. D., Beuran, M., Beyene, A. S., Bhansali, A., Bhutta, Z. A., Bicer, B. K., Bikbov, B., Birungi, C., Biryukov, S., Blosser, C. D., Boneya, D. J., Bou-Orm, I. R., Brauer, M., Breitborde, N. J. K., et al.: Global, regional, and national comparative risk assessment of 84 behavioural, environmental and occupational, and metabolic risks or clusters of risks, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016, Lancet, 390, 1345–1422, https://doi.org/10.1016/S0140-6736(17)32366-8, 2017.
Glasius, M. and Goldstein, A. H.: Recent Discoveries and Future Challenges in Atmospheric Organic Chemistry, Environ. Sci. Technol., 50, 2754–2764, https://doi.org/10.1021/acs.est.5b05105, 2016.
Goldstein, A. H. and Galbally, I. E.: Known and Unexplored Organic Constituents in the Earth's Atmosphere, Environ. Sci. Technol., 41, 1514–1521, https://doi.org/10.1021/es072476p, 2007.
Graus, M., Müller, M., and Hansel, A.: High Resolution PTR-TOF: Quantification and Formula Confirmation of VOC in Real Time, J. Am. Soc. Mass Spectrom., 21, 1037–1044, https://doi.org/10.1016/j.jasms.2010.02.006, 2010.
Hallquist, M., Wenger, J. C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N. M., George, C., Goldstein, A. H., Hamilton, J. F., Herrmann, H., Hoffmann, T., Iinuma, Y., Jang, M., Jenkin, M. E., Jimenez, J. L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, Th. F., Monod, A., Prévôt, A. S. H., Seinfeld, J. H., Surratt, J. D., Szmigielski, R., and Wildt, J.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009.
Hansel, A., Scholz, W., Mentler, B., Fischer, L., and Berndt, T.: Detection of RO2 radicals and other products from cyclohexene ozonolysis with and acetate chemical ionization mass spectrometry, Atmos. Environ., 186, 248–255, https://doi.org/10.1016/j.atmosenv.2018.04.023, 2018.
Heinritzi, M., Simon, M., Steiner, G., Wagner, A. C., Kürten, A., Hansel, A., and Curtius, J.: Characterization of the mass-dependent transmission efficiency of a CIMS, Atmos. Meas. Tech., 9, 1449–1460, https://doi.org/10.5194/amt-9-1449-2016, 2016.
Hyttinen, N., Kupiainen-Määttä, O., Rissanen, M. P., Muuronen, M., Ehn, M., and Kurtén, T.: Modeling the Charging of Highly Oxidized Cyclohexene Ozonolysis Products Using Nitrate-Based Chemical Ionization, J. Phys. Chem. A, 119, 6339–6345, https://doi.org/10.1021/acs.jpca.5b01818, 2015.
Hyttinen, N., Otkjær, R. V., Iyer, S., Kjaergaard, H. G., Rissanen, M. P., Wennberg, P. O., and Kurtén, T.: Computational Comparison of Different Reagent Ions in the Chemical Ionization of Oxidized Multifunctional Compounds, J. Phys. Chem. A, 122, 269–279, https://doi.org/10.1021/acs.jpca.7b10015, 2018.
Intergovernmental Panel on Climate Change: Climate Change 2013 – The Physical Science Basis: Working Group I Contribution to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge University Press, Cambridge, 2014.
Isaacman-VanWertz, G., Massoli, P., O'Brien, R. E., Nowak, J. B., Canagaratna, M. R., Jayne, J. T., Worsnop, D. R., Su, L., Knopf, D. A., Misztal, P. K., Arata, C., Goldstein, A. H., and Kroll, J. H.: Using advanced mass spectrometry techniques to fully characterize atmospheric organic carbon: current capabilities and remaining gaps, Faraday Discuss., 200, 579–598, https://doi.org/10.1039/C7FD00021A, 2017.
Isaacman-VanWertz, G., Massoli, P., O'Brien, R., Lim, C., Franklin, J. P., Moss, J. A., Hunter, J. F., Nowak, J. B., Canagaratna, M. R., Misztal, P. K., Arata, C., Roscioli, J. R., Herndon, S. T., Onasch, T. B., Lambe, A. T., Jayne, J. T., Su, L., Knopf, D. A., Goldstein, A. H., Worsnop, D. R., and Kroll, J. H.: Chemical evolution of atmospheric organic carbon over multiple generations of oxidation, Nat. Chem., 10, 462–468, https://doi.org/10.1038/s41557-018-0002-2, 2018.
Iyer, S., Lopez-Hilfiker, F., Lee, B. H., Thornton, J. A., and Kurtén, T.: Modeling the Detection of Organic and Inorganic Compounds Using Iodide-Based Chemical Ionization, J. Phys. Chem. A, 120, 576–587, https://doi.org/10.1021/acs.jpca.5b09837, 2016.
Iyer, S., He, X., Hyttinen, N., Kurtén, T., and Rissanen, M. P.: Computational and Experimental Investigation of the Detection of HO2 Radical and the Products of Its Reaction with Cyclohexene Ozonolysis Derived RO2 Radicals by an Iodide-Based Chemical Ionization Mass Spectrometer, J. Phys. Chem. A, 121, 6778–6789, https://doi.org/10.1021/acs.jpca.7b01588, 2017.
Jimenez, J. L., Canagaratna, M. R., Donahue, N. M., Prevot, A. S. H., Zhang, Q., Kroll, J. H., DeCarlo, P. F., Allan, J. D., Coe, H., Ng, N. L., Aiken, A. C., Docherty, K. S., Ulbrich, I. M., Grieshop, A. P., Robinson, A. L., Duplissy, J., Smith, J. D., Wilson, K. R., Lanz, V. A., Hueglin, C., Sun, Y. L., Tian, J., Laaksonen, A., Raatikainen, T., Rautiainen, J., Vaattovaara, P., Ehn, M., Kulmala, M., Tomlinson, J. M., Collins, D. R., Cubison, M. J., E., Dunlea, J., Huffman, J. A., Onasch, T. B., Alfarra, M. R., Williams, P. I., Bower, K., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Salcedo, D., Cottrell, L., Griffin, R., Takami, A., Miyoshi, T., Hatakeyama, S., Shimono, A., Sun, J. Y., Zhang, Y. M., Dzepina, K., Kimmel, J. R., Sueper, D., Jayne, J. T., Herndon, S. C., Trimborn, A. M., Williams, L. R., Wood, E. C., Middlebrook, A. M., Kolb, C. E., Baltensperger, U., and Worsnop, D. R.: Evolution of Organic Aerosols in the Atmosphere, Science, 326, 1525–1529, https://doi.org/10.1126/science.1180353, 2009.
Jokinen, T., Sipilä, M., Junninen, H., Ehn, M., Lönn, G., Hakala, J., Petäjä, T., Mauldin III, R. L., Kulmala, M., and Worsnop, D. R.: Atmospheric sulphuric acid and neutral cluster measurements using CI-APi-TOF, Atmos. Chem. Phys., 12, 4117–4125, https://doi.org/10.5194/acp-12-4117-2012, 2012.
Jokinen, T., Sipilä, M., Richters, S., Kerminen, V.-M., Paasonen, P., Stratmann, F., Worsnop, D., Kulmala, M., Ehn, M., Herrmann, H., and Berndt, T.: Rapid Autoxidation Forms Highly Oxidized RO2 Radicals in the Atmosphere, Angew. Chem. Int. Edit., 53, 14596–14600, https://doi.org/10.1002/anie.201408566, 2014.
Jokinen, T., Berndt, T., Makkonen, R., Kerminen, V.-M., Junninen, H., Paasonen, P., Stratmann, F., Herrmann, H., Guenther, A. B., Worsnop, D. R., Kulmala, M., Ehn, M., and Sipilä, M.: Production of extremely low volatile organic compounds from biogenic emissions: Measured yields and atmospheric implications, P. Natl. Acad. Sci. USA, 112, 7123–7128, https://doi.org/10.1073/pnas.1423977112, 2015.
Jordan, A., Haidacher, S., Hanel, G., Hartungen, E., Märk, L., Seehauser, H., Schottkowsky, R., Sulzer, P., and Märk, T. D.: A high resolution and high sensitivity proton-transfer-reaction time-of-flight mass spectrometer (PTR-TOF-MS), International J. Mass Spectrom., 286, 122–128, https://doi.org/10.1016/j.ijms.2009.07.005, 2009.
Junninen, H., Ehn, M., Petäjä, T., Luosujärvi, L., Kotiaho, T., Kostiainen, R., Rohner, U., Gonin, M., Fuhrer, K., Kulmala, M., and Worsnop, D. R.: A high-resolution mass spectrometer to measure atmospheric ion composition, Atmos. Meas. Tech., 3, 1039–1053, https://doi.org/10.5194/amt-3-1039-2010, 2010.
Kelly, J. M., Doherty, R. M., O'Connor, F. M., and Mann, G. W.: The impact of biogenic, anthropogenic, and biomass burning volatile organic compound emissions on regional and seasonal variations in secondary organic aerosol, Atmos. Chem. Phys., 18, 7393–7422, https://doi.org/10.5194/acp-18-7393-2018, 2018.
Kirkby, J., Duplissy, J., Sengupta, K., Frege, C., Gordon, H., Williamson, C., Heinritzi, M., Simon, M., Yan, C., Almeida, J., Tröstl, J., Nieminen, T., Ortega, I. K., Wagner, R., Adamov, A., Amorim, A., Bernhammer, A.-K., Bianchi, F., Breitenlechner, M., Brilke, S., Chen, X., Craven, J., Dias, A., Ehrhart, S., Flagan, R. C., Franchin, A., Fuchs, C., Guida, R., Hakala, J., Hoyle, C. R., Jokinen, T., Junninen, H., Kangasluoma, J., Kim, J., Krapf, M., Kürten, A., Laaksonen, A., Lehtipalo, K., Makhmutov, V., Mathot, S., Molteni, U., Onnela, A., Peräkylä, O., Piel, F., Petäjä, T., Praplan, A. P., Pringle, K., Rap, A., Richards, N. A. D., Riipinen, I., Rissanen, M. P., Rondo, L., Sarnela, N., Schobesberger, S., Scott, C. E., Seinfeld, J. H., Sipilä, M., Steiner, G., Stozhkov, Y., Stratmann, F., Tomé, A., Virtanen, A., Vogel, A. L., Wagner, A. C., Wagner, P. E., Weingartner, E., Wimmer, D., Winkler, P. M., Ye, P., Zhang, X., Hansel, A., Dommen, J., Donahue, N. M., Worsnop, D. R., Baltensperger, U., Kulmala, M., Carslaw, K. S., and Curtius, J.: Ion-induced nucleation of pure biogenic particles, Nature, 533, 521–526, https://doi.org/10.1038/nature17953, 2016.
Krechmer, J., Lopez-Hilfiker, F., Koss, A., Hutterli, M., Stoermer, C., Deming, B., Kimmel, J., Warneke, C., Holzinger, R., Jayne, J. T., Worsnop, D. R., Fuhrer, K., Gonin, M., and de Gouw, J. A.: Evaluation of a New Reagent-Ion Source and Focusing Ion-Molecule Reactor for use in Proton-Transfer-Reaction Mass Spectrometry, Anal. Chem., 90, 12011–12018, https://doi.org/10.1021/acs.analchem.8b02641, 2018.
Kürten, A., Rondo, L., Ehrhart, S., and Curtius, J.: Calibration of a Chemical Ionization Mass Spectrometer for the Measurement of Gaseous Sulfuric Acid, J. Phys. Chem. A, 116, 6375–6386, https://doi.org/10.1021/jp212123n, 2012.
Lee, A., Goldstein, A. H., Keywood, M. D., Gao, S., Varutbangkul, V., Bahreini, R., Ng, N. L., Flagan, R. C., and Seinfeld, J. H.: Gas-phase products and secondary aerosol yields from the ozonolysis of ten different terpenes, J. Geophys. Res., 111, D07302, https://doi.org/10.1029/2005JD006437, 2006.
Lee, B. H., Lopez-Hilfiker, F. D., Mohr, C., Kurtén, T., Worsnop, D. R., and Thornton, J. A.: An Iodide-Adduct High-Resolution Time-of-Flight Chemical-Ionization Mass Spectrometer: Application to Atmospheric Inorganic and Organic Compounds, Environ. Sci. Technol., 48, 6309–6317, https://doi.org/10.1021/es500362a, 2014.
Lee, B. H., Mohr, C., Lopez-Hilfiker, F. D., Lutz, A., Hallquist, M., Lee, L., Romer, P., Cohen, R. C., Iyer, S., Kurtén, T., Hu, W., Day, D. A., Campuzano-Jost, P., Jimenez, J. L., Xu, L., Ng, N. L., Guo, H., Weber, R. J., Wild, R. J., Brown, S. S., Koss, A., de Gouw, J., Olson, K., Goldstein, A. H., Seco, R., Kim, S., McAvey, K., Shepson, P. B., Starn, T., Baumann, K., Edgerton, E. S., Liu, J., Shilling, J. E., Miller, D. O., Brune, W., Schobesberger, S., D'Ambro, E. L., and Thornton, J. A.: Highly functionalized organic nitrates in the southeast United States: Contribution to secondary organic aerosol and reactive nitrogen budgets, P. Natl. Acad. Sci. USA, 113, 1516–1521, https://doi.org/10.1073/pnas.1508108113, 2016.
Li, X., Chee, S., Hao, J., Abbatt, J. P. D., Jiang, J., and Smith, J. N.: Relative humidity effect on the formation of highly oxidized molecules and new particles during monoterpene oxidation, Atmos. Chem. Phys., 19, 1555–1570, https://doi.org/10.5194/acp-19-1555-2019, 2019.
Lindinger, W., Hansel, A., and Jordan, A.: On-line monitoring of volatile organic compounds at pptv levels by means of proton-transfer-reaction mass spectrometry (PTR-MS) medical applications, food control and environmental research, Int. J. Mass Spectrom., 173, 191–241, https://doi.org/10.1016/S0168-1176(97)00281-4, 1998.
Lopez-Hilfiker, F. D., Iyer, S., Mohr, C., Lee, B. H., D'Ambro, E. L., Kurtén, T., and Thornton, J. A.: Constraining the sensitivity of iodide adduct chemical ionization mass spectrometry to multifunctional organic molecules using the collision limit and thermodynamic stability of iodide ion adducts, Atmos. Meas. Tech., 9, 1505–1512, https://doi.org/10.5194/amt-9-1505-2016, 2016.
Mohr, C., Lopez-Hilfiker, F. D., Yli-Juuti, T., Heitto, A., Lutz, A., Hallquist, M., D'Ambro, E. L., Rissanen, M. P., Hao, L., Schobesberger, S., Kulmala, M., Mauldin, R. L., Makkonen, U., Sipilä, M., Petäjä, T., and Thornton, J. A.: Ambient observations of dimers from terpene oxidation in the gas phase: Implications for new particle formation and growth: Ambient Observations of Gas-Phase Dimers, Geophys. Res. Lett., 44, 2958–2966, https://doi.org/10.1002/2017GL072718, 2017.
Pagonis, D., Krechmer, J. E., de Gouw, J., Jimenez, J. L., and Ziemann, P. J.: Effects of gas-wall partitioning in Teflon tubing and instrumentation on time-resolved measurements of gas-phase organic compounds, Atmos. Meas. Tech., 10, 4687–4696, https://doi.org/10.5194/amt-10-4687-2017, 2017.
Riva, M., Budisulistiorini, S. H., Zhang, Z., Gold, A., Thornton, J. A., Turpin, B. J., and Surratt, J. D.: Multiphase reactivity of gaseous hydroperoxide oligomers produced from isoprene ozonolysis in the presence of acidified aerosols, Atmos. Environ., 152, 314–322, https://doi.org/10.1016/j.atmosenv.2016.12.040, 2017.
Schallhart, S., Rantala, P., Nemitz, E., Taipale, D., Tillmann, R., Mentel, T. F., Loubet, B., Gerosa, G., Finco, A., Rinne, J., and Ruuskanen, T. M.: Characterization of total ecosystem-scale biogenic VOC exchange at a Mediterranean oak-hornbeam forest, Atmos. Chem. Phys., 16, 7171–7194, https://doi.org/10.5194/acp-16-7171-2016, 2016.
Schobesberger, S., Junninen, H., Bianchi, F., Lonn, G., Ehn, M., Lehtipalo, K., Dommen, J., Ehrhart, S., Ortega, I. K., Franchin, A., Nieminen, T., Riccobono, F., Hutterli, M., Duplissy, J., Almeida, J., Amorim, A., Breitenlechner, M., Downard, A. J., Dunne, E. M., Flagan, R. C., Kajos, M., Keskinen, H., Kirkby, J., Kupc, A., Kurten, A., Kurten, T., Laaksonen, A., Mathot, S., Onnela, A., Praplan, A. P., Rondo, L., Santos, F. D., Schallhart, S., Schnitzhofer, R., Sipila, M., Tome, A., Tsagkogeorgas, G., Vehkamaki, H., Wimmer, D., Baltensperger, U., Carslaw, K. S., Curtius, J., Hansel, A., Petaja, T., Kulmala, M., Donahue, N. M., and Worsnop, D. R.: Molecular understanding of atmospheric particle formation from sulfuric acid and large oxidized organic molecules, P. Natl. Acad. Sci. USA, 110, 17223–17228, https://doi.org/10.1073/pnas.1306973110, 2013.
Stark, H., Yatavelli, R. L. N., Thompson, S. L., Kimmel, J. R., Cubison, M. J., Chhabra, P. S., Canagaratna, M. R., Jayne, J. T., Worsnop, D. R., and Jimenez, J. L.: Methods to extract molecular and bulk chemical information from series of complex mass spectra with limited mass resolution, Int. J. Mass Spectrom., 389, 26–38, https://doi.org/10.1016/j.ijms.2015.08.011, 2015.
Stark, H., Yatavelli, R. L. N., Thompson, S. L., Kang, H., Krechmer, J. E., Kimmel, J. R., Palm, B. B., Hu, W., Hayes, P. L., Day, D. A., Campuzano-Jost, P., Canagaratna, M. R., Jayne, J. T., Worsnop, D. R., and Jimenez, J. L.: Impact of Thermal Decomposition on Thermal Desorption Instruments: Advantage of Thermogram Analysis for Quantifying Volatility Distributions of Organic Species, Environ. Sci. Technol., 51, 8491–8500, https://doi.org/10.1021/acs.est.7b00160, 2017.
Tröstl, J., Chuang, W. K., Gordon, H., Heinritzi, M., Yan, C., Molteni, U., Ahlm, L., Frege, C., Bianchi, F., Wagner, R., Simon, M., Lehtipalo, K., Williamson, C., Craven, J. S., Duplissy, J., Adamov, A., Almeida, J., Bernhammer, A.-K., Breitenlechner, M., Brilke, S., Dias, A., Ehrhart, S., Flagan, R. C., Franchin, A., Fuchs, C., Guida, R., Gysel, M., Hansel, A., Hoyle, C. R., Jokinen, T., Junninen, H., Kangasluoma, J., Keskinen, H., Kim, J., Krapf, M., Kürten, A., Laaksonen, A., Lawler, M., Leiminger, M., Mathot, S., Möhler, O., Nieminen, T., Onnela, A., Petäjä, T., Piel, F. M., Miettinen, P., Rissanen, M. P., Rondo, L., Sarnela, N., Schobesberger, S., Sengupta, K., Sipilä, M., Smith, J. N., Steiner, G., Tomè, A., Virtanen, A., Wagner, A. C., Weingartner, E., Wimmer, D., Winkler, P. M., Ye, P., Carslaw, K. S., Curtius, J., Dommen, J., Kirkby, J., Kulmala, M., Riipinen, I., Worsnop, D. R., Donahue, N. M., and Baltensperger, U.: The role of low-volatility organic compounds in initial particle growth in the atmosphere, Nature, 533, 527–531, https://doi.org/10.1038/nature18271, 2016.
Twomey, S.: The Influence of Pollution on the Shortwave Albedo of Clouds, J. Atmos. Sci., 34, 1149–1152, https://doi.org/10.1175/1520-0469(1977)034<1149:TIOPOT>2.0.CO;2, 1977.
Wang, S., Riva, M., Yan, C., Ehn, M., and Wang, L.: Primary Formation of Highly Oxidized Multifunctional Products in the OH-Initiated Oxidation of Isoprene. A Combined Theoretical and Experimental Study, Environ. Sci. Technol., 52, 12255–12264, https://doi.org/10.1021/acs.est.8b02783, 2018.
Yuan, B., Koss, A. R., Warneke, C., Coggon, M., Sekimoto, K., and de Gouw, J. A.: Proton-Transfer-Reaction Mass Spectrometry: Applications in Atmospheric Sciences, Chem. Rev., 117, 13187–13229, https://doi.org/10.1021/acs.chemrev.7b00325, 2017.
Ziemann, P. J. and Atkinson, R.: Kinetics, products, and mechanisms of secondary organic aerosol formation, Chem. Soc. Rev., 41, 6582, https://doi.org/10.1039/c2cs35122f, 2012.
The requested paper has a corresponding corrigendum published. Please read the corrigendum first before downloading the article.
- Article
(2992 KB) - Full-text XML