the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 14 Jul 2026
| 14 Jul 2026
Voltage scanning to calibrate Multi-Reagent Chemical Ionization Mass Spectrometers (MR-CIMS): from signal to sensitivity
Yuwei Wang
Aristeidis Voliotis
Emily Matthews
Rongrong Wu
Milan Roska
Max Gerrit Adam
René Dubus
Lukas Kesper
Franz Rohrer
Robert Wegener
Benjamin Winter
Kelvin H. Bates
Quanfu He
Thorsten Hohaus
Achim Grasse
Ralf Tillmann
Andreas Wahner
Christian Wesolek
Sergej Wedel
Yizhen Wu
Sören R. Zorn
Manjula Canagaratna
Douglas Worsnop
Felipe Lopez-Hilfiker
Georgios I. Gkatzelis
Thomas J. Bannan
Chemical ionization mass spectrometry (CIMS) offers high time-resolution measurements for diverse compounds, but atmospheric quantification remains challenging. Here, we combine a recently published method for determining the collision limit using a single reagent ion with a voltage scanning approach for assessing the relative sensitivities of diverse adduct ions. We used voltage scanning in a Multi-Reagent Chemical Ionization Mass Spectrometer (MR-CIMS) to assess ion–molecule adduct strength. The sensitivities to most detectable compounds were calculated based on this relationship using a collision-limit sensitivity of 13.87 ± 0.69 ncps pptv−1 determined for α-pinene using the benzene channel. Following previously published work, the collision limit sensitivity of the other reagent ions used was assumed to be equal to that of the benzene channel and was further examined using the binding energy and measured sensitivity of nitrophenol in the bromide channel. Calibration of 13 molecules, including nitric acid, formic acid, and oxygenated VOCs, was performed to obtain a universal relationship between the sensitivities and the voltage at which the adduct signal halves (dV50). Quantification uncertainties stayed below 20 % for compounds with sensitivities above 5.69 and 5.30 ncps pptv−1 in bromide and iodide channels, respectively. High-level quantum chemical calculations indicated that the detected compounds predominantly form hydrogen-bonded clusters with bromide and iodide. In a large photochemical chamber, estimated sensitivities of more than 260 compounds were determined. Based on this, we achieved their quantification with multiple negative reagent ions. The quantification was validated by comparing nitrous acid concentrations measured using MR-CIMS with those obtained from a calibrated iterative cavity-enhanced differential optical absorption spectroscopy (ICAD) (R2 = 0.891, slope = 1.24). Six of the organic compounds were taken as examples to show the results of this method for a daytime oxidation chamber experiment. The measurement uncertainties for these pptv-level compounds ranged from 8.9 % to 39.0 %, depending on their sensitivities and concentrations. Further theoretical and experimental investigations showed that halogen compounds can form intermolecular halogen bonds with strength comparable to their intramolecular bonds, preventing this approach from being applied to determine their sensitivities. This work highlights that voltage scanning is a useful approach to the determination ion–molecule adduct sensitivity in CIMS.
- Article
(2379 KB) - Full-text XML
-
Supplement
(1663 KB) - BibTeX
- EndNote




Chemical ionization mass spectrometry (CIMS) is a powerful analytical method providing high time resolution measurements for a wide range of compounds (Bertram et al., 2011). It uses soft ionization to charge analytes and has the advantage of allowing in situ measurements that preserve the structure of the analytes (Riva et al., 2019; Zhang et al., 2023). Among its many applications, the measurement of organics, including volatile organic compounds (VOCs) and their less volatile oxidation products, is one of the most prominent and critical. This capability meets the evolving demands of atmospheric chemistry research, addressing important questions regarding the formation of secondary organic aerosol (SOA), the cycling of atmospheric reactive nitrogen, and the sinks and sources of atmospheric radicals (Ehn et al., 2014; Lee et al., 2018; Yuan et al., 2017).
Quantifying detected compounds is fundamental to CIMS but is challenging particularly in clustering ion chemistry. This is due to two main factors: first, certain reagent ions, e.g., iodide, and bromide, are more sensitive to oxygenated compounds, for which commercially available and atmospherically relevant standards are often lacking; second, the sensitivities of some reagent ions, e.g., ammonium and iodide, span a wide range, complicating accurate quantification (Bianchi et al., 2019; Lee et al., 2014; Rissanen et al., 2019; Xu et al., 2022; Zaytsev et al., 2019). Over the past decades, several investigations have sought to characterize the sensitivity of CIMS to compounds for which commercial standards are unavailable. Huey et al. (1995) measured accurate reaction rate constants for SF and I− with various atmospheric analytes, thereby firstly validating that the detected ions represent the target analytes rather than interferences and establishing a quantitative link between ion-molecule rate constants and signal responses in CIMS (Huey et al., 1995). In addition, the community often adopts the strategy of synthesizing target analytes for the direct calibration of CIMS, including peroxyacetyl nitrate (Slusher et al., 2004; Veres and Roberts, 2015), organic hydroperoxides (Crounse et al., 2006), nitryl chloride (ClNO2) and dinitrogen pentoxide (N2O5) (Kercher et al., 2009), peroxynitric acid (Veres et al., 2015), chloramines (Chen et al., 2025; Wang et al., 2023), tertiary isoprene nitrates (Vasquez et al., 2020), and biogenic hydroxy nitrates (Robinson et al., 2024).
For analytes that are difficult to synthesize or whose structures and functionalities are uncertain, using semi-quantitative calibration or quantum model estimation provides a practical alternative for estimating sensitivities (Iyer et al., 2016; Lee et al., 2014; Lopez-Hilfiker et al., 2016). Very recently, Aggarwal et al. (2025) conducted calibrations on 39 Tofwerk CIMS, statistically demonstrating that the collision limit can be used to put constraints on sensitivity from one ion mode polarity to another, as long as the critical parameters are held constant, with the discrepancy between the two polarities being within 5 %. This enables the determination of the collision limit of negative reagent ions to be made from the positive channels, which are often easier to complete, simplifying calibration requirements across various reagent ion chemistries (Aggarwal et al., 2025). Under this condition, once the distributions of relative sensitivities to detectable compounds are obtained, the true sensitivities can be readily calculated. To achieve that, voltage scanning is one of the most frequently used semi-quantitative calibration methods (Lopez-Hilfiker et al., 2016; Song et al., 2024; Xu et al., 2022; Zaytsev et al., 2019). It is a procedure to determine the estimated sensitivity of reagent ion adducts by imparting energy to the ions via electric fields. The binding energy is inferred from the change in applied voltage at which the adduct signal drops to half of its original intensity. This voltage change is referred to as dV50. Compounds' sensitivities can be estimated once the empirical relationship between binding energies and sensitivities is established. Lopez-Hilfiker et al. (2016) was the first to report the utilization of the voltage scanning method to constrain the relative sensitivities of I-CIMS to multifunctional organic molecules. Song et al. (2024) established a comprehensive characterization on the relative sensitivity of I-CIMS to multifunctional compounds. An increase in sensitivity was observed with the addition of polar functional groups to the compound, such as keto, hydroxyl, and carboxylic acid groups. Semi-quantitative equations were established for monophenols, monoacids, polyphenol or diacid species, and species with multiple functional groups in I-CIMS. Despite the useful implementation of the voltage scanning technique in some works, it is still not widely adopted with CIMS users.
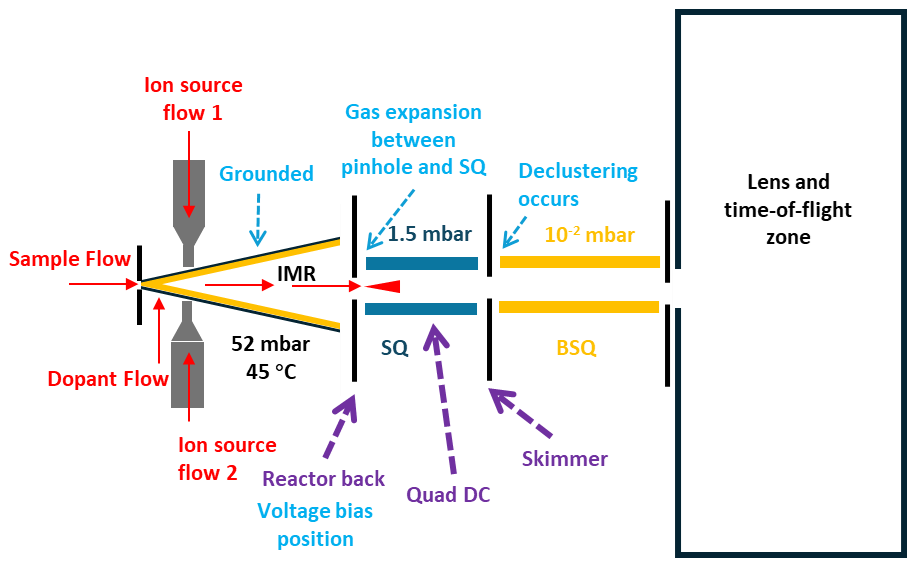
Figure 1Schematic of the MR-CIMS structure. Gas flow paths are indicated in red, and the electrodes/voltages adjusted during voltage scanning are highlighted by purple dashed arrows and texts. Explanatory annotations are denoted by blue dashed arrows and words. Pressures of the IMR, SQ (short quadrupole), and BSQ (big-segmented quadruple), as well as the temperature of the IMR, are labeled to facilitate understanding of the operating conditions in MR-CIMS.
Knowledge on the sensitivities of CIMS and the voltage scanning method is still insufficient. First, this method has not been evaluated for the many reagent ions that are used for characterizing the range of organics of interest. The distribution of Br-CIMS sensitivities to compounds with different functionalities, for example, is not well understood. Second, for the sake of safety and minimizing side reactions, some CIMS utilize a grounded flow tube ion–molecule reactor (IMR), such as the newly developed Vocus AIM (adduct ionization mechanism) IMR (Riva et al., 2024). The voltage scanning protocol needs to be refined for these reactors, including the additional tuning necessary to keep transmission from the IMR to the quadrupoles constant when applying voltage scanning methods. The schematic diagram of this process is shown in Fig. 1 and its details will be discussed more in the following sections. Third, as voltage scanning essentially determines the binding energy of the adducts through cluster dissociation, which significantly reduces the signals of the analytes, voltage scanning is only suitable for analytes with a sufficient signal-to-noise ratio (SNR) in the mass spectrum. The SNR threshold required to make voltage scanning applicable is currently unknown.
Here we present an approach to estimating sensitivities of a wide range of compounds using multiple reagent ion chemistries in the grounded Vocus AIM IMR. The collision limit sensitivity of a multi-reagent ion CIMS was determined based on the method reported by Aggarwal et al. (2025). Based on this, sensitivities were obtained with voltage scanning. We assessed the performance of the approach using a wide range of calibrants, chamber experiments and quantum chemical calculations. Calibration experiments were conducted on a Multi-Reagent Chemical Ionization time-of-flight Mass Spectrometer (MR-CIMS, Vocus B2, Tofwerk AG, Switzerland) (Aggarwal et al., 2025). The MR-CIMS is a newly developed fast-switching CIMS equipped with four reagent ions, i.e., bromide (Br−) and iodide (I−) anions, acetone dimers (C6H13O+) and benzene cations (C6H) that are each measured over a 2 s cycle. This allows measurements of a much wider range of compounds with a single instrument, making it ideal for mobile measurements, such as on an aircraft. We focus on the sensitivities and binding energies of adducts in the two negative channels, which are widely utilized in detecting oxygenated organic molecules, radicals, and halogen compounds in the atmosphere (Albrecht et al., 2019; Finkenzeller et al., 2023; Lee et al., 2014; Mohr et al., 2019; Tham et al., 2021).
Direct calibration experiments were performed to determine the relationship between the sensitivity to organics and dV50. This voltage scanning method was further demonstrated to be applicable to other CIMS using Vocus AIM IMR. To better investigate the sensitivities to compounds with different functionalities, quantum chemistry calculations were performed at the DLPNO-CCSD(T)/def2-QZVPPD level to determine the cluster formation enthalpies of the ion–molecule clusters and to obtain their most stable geometries. The calculated enthalpies agreed well with the experimental dV50, except for nitric acid, where water molecules accommodated their excess energy and thus enhanced their sensitivities, and 4-nitrophenol, which is charged at the collision limit. Organic compounds formed hydrogen bonding with Br− to generate ion–molecule clusters similar to the conditions in I-CIMS reported by Iyer et al. (2016). Both hydroxyl and carboxyl groups can form hydrogen bonds, while the carbonyl group can enhance molecular polarity and thereby strengthen adjacent hydrogen bonding.
MR-CIMS was deployed in the SAPHIR-CHANEL campaign during the summer of 2024, which conducted a series of oxidation experiments in the outdoor SAPHIR chamber using a mix of VOCs at atmospheric concentrations, including volatile chemical products (VCPs) (Coggon et al., 2021; Gkatzelis et al., 2021; McDonald et al., 2018), cooking (Coggon et al., 2024), gasoline, diesel, and biogenic VOCs. Voltage scanning was conducted and utilized to estimate the sensitivity of MR-CIMS to the gaseous compounds in the chamber. The sensitivities of a total of 290 organic compounds clustered with bromide and 264 organic compounds clustered with iodide were estimated. Their dV50 distributions in relation to the oxygen content were determined. Furthermore, the abundance range of compounds to which the voltage scanning method was applicable in MR-CIMS was also investigated. Based on the nitrous acid (HONO) sensitivity derived from the voltage scanning, we calculated the HONO concentrations measured by MR-CIMS. The results matched those from another calibrated instrument running concurrently, validating the reliability of the voltage scanning. Additionally, we quantified several typical organic compounds originating from various sources in a typical multi-source chamber experiment, whose measurement uncertainties ranged from 8.9 % to 39.0 %, with values dependent on compound sensitivities and concentrations, thereby demonstrating the broad applicability of the voltage scanning technique. Furthermore, halogen compounds were examined with voltage scanning, but the similarity between their intramolecular binding energies and the halogen binding energies formed with reagent ions made it difficult to obtain meaningful dV50 values.
2.1 Instrumentation
MR-CIMS is a newly designed instrument equipped with a bipolar time-of-flight (TOF) mass spectrometer, which can simultaneously measure cations and anions without inner interference.
The design and geometric structure of the IMR of MR-CIMS, i.e., Vocus AIM (Adduct Ionization Mechanism) IMR, has been discussed in detail elsewhere (Riva et al., 2024). The IMR and short quadrupole (SQ) were operated at pressures of around 52 and 1.50 mbar, respectively. In addition, the IMR was heated to 45 °C by a high-precision electronic heater. The time series and histograms of IMR pressure, temperature, and SQ pressure during a representative experiment are provided in Fig. S1 in the Supplement to demonstrate stability. MR-CIMS was operated with four different reagent ions: bromide (Br−), acetone dimers (C6H13O+), iodide (I−), and benzene cations (C6H). These reagent ions were generated via the charging of dibromomethane (CH2Br2, Sigma Aldrich, ≥ 99 %), acetone (C3H6O, Sigma Aldrich, ≥ 99.9 %), methyl iodide (CH3I, Sigma Aldrich, 99.8 %), and benzene (C6H6, Sigma Aldrich, ≥ 99.9 %), respectively, by two Vacuum Ultra-Violet sources (VUV) (Ji et al., 2020). These reagents, emitted from the permeation tubes (Towerk AG, Switzerland) heated to 80 °C, were continuously delivered to the VUV sources via two independent nitrogen flows at 250 sccm (standard cubic centimeters per minute) each and were around 1 : 100 by volume. In each VUV source, a pair of reagents was introduced: one to produce positive ions and the other to generate negative ions. Iodide and benzene cations were produced in one source, and bromide and acetone dimers in the other. Equipped with a fast-switch high voltage mode, the two VUV sources were alternately operated at a frequency of 1 Hz to generate one or other of the two pairs of positive and negative reagent ions. Humidity of samples is well known to have a significant influence on the sensitivity of CIMS (Huey, 2007; Lee et al., 2014). To avoid variations in relative humidity (RH), a dopant flow ranging from 0 to 30 sccm was directed to introduce water vapor from a heated deionized water bottle to the IMR. The flow rate was automatically adjusted at a frequency of 0.5 Hz based on the H2OI− I− ratio (hereafter water ratio) measured in the iodide channel by the TofdaqViewer2 (Tofwerk AG, Switzerland). The target water ratio was set as 0.60, which was around the maximum ratio measured in the SAPHIR chamber, corresponding to a ratio of 0.65 for H2OBr− Br− in the bromide channel. Due to this dopant flow, the RH in the IMR was constant throughout the campaign. Humidity regulation was paused during voltage scanning because the dissociation of adducts changes the water ratio. The time series and histograms of the water ratio in the iodide and bromide channels are provided in Fig. S2 as evidence of stability.
MR-CIMS can identify analytes based on exact molecular weight at high precision. The mass resolution, , for cations and anions are around 5000 and 5300, respectively, at a mass-to-charge ratio () of 200 Th. Data consist of four segments, each corresponding to a different reagent ion, and were recorded with a time resolution of 0.5 Hz. For data processing, each segment was averaged by a factor of 2and thus had an integration time of 1 s. During measurement and voltage scanning, measured analyte signals in counts per second (cps) were ratioed to the total reagent ion signal in cps (determined as the sum of the reagent ion and hydrated reagent ions) and then multiplied by the standard total reagent ion signal value of 1 million cps to yield a sensitivity in units of normalized counts per sec (ncps).
2.2 Voltage scanning method
Voltage scanning is a method developed to experimentally estimate the binding energy of ion–molecule adducts formed by chemical ionization in the IMR. A schematic of the main elements of MR-CIMS including the IMR and quadrupoles, highlighting the region where the voltage was scanned, and the transfer of molecular ions to the TOF MS are shown in Fig. 1. Three electrodes/voltages were adjusted in upstream of big-segmented quadruple (BSQ), including Reactor back, Quad DC, and Skimmer. Reactor back is the direct currency (DC) potential applied at pinhole between IMR and SQ, which is set as 0 V in the routine operation. Quad DC refers to the DC potential applied to the four quadrupole rods that carry the radio frequency (RF) field of SQ. Skimmer voltage corresponds to the DC potential applied to the skimmer electrode, which controls ion extraction and transmission efficiency across the SQ-BSQ interface. An increase in the voltage difference between the BSQ and region upstream of it induces dissociation of the adduct. The binding energy of each adduct can be represented by the voltage gap at which its signal decreases to half of that observed at the start of voltage scanning (dV50). Compounds reacting with the reagent ion at the collision limit typically possess the highest dV50 as their ion–molecule adducts are likely to have strongest binding energy.
The Vocus AIM IMR is configured to be electrically grounded, which differs from earlier CIMS designs (Lopez-Hilfiker et al., 2016). For anions, an increase in the voltage gap between SQ and BSQ will lower the potential of SQ relative to the grounded IMR and thus can generate a reing electric field at the reactor exit. This will reflect ions back toward the IMR because anions are decelerated when moving toward regions of lower electric potential. A similar argument applies to cations in positive channels. This effect results in ion repulsion, i.e., reduced ion transmission, and can result in a decrease in ion signals. Consequently, the measured signal decay rate reflects not only the declustering processes occurring after Skimmer, which indicates the cluster binding energy, but also variations in ion transmission across the IMR–SQ interface. Since voltage scanning is used to derive the cluster binding energy based on the signal decay, we have to minimize the above issue and ensure that signal decay rate is primarily determined by cluster binding energy.
Total ion counts (TIC) serve as a useful indicator of changes in ion transmission efficiency (Fig. S3) (Lopez-Hilfiker et al., 2016). A pinhole bias of −4 V in negative channels and +4 V in positive channels is implemented across the IMR–SQ interface, i.e., Reactor Back, at the start of scanning. This approach results in a fixed ion transmission offset from IMR to pinhole, but it ensures that the subsequent signal decay is dominated by variations in relative binding energies rather than by transmission artefacts from pinhole to SQ. This behavior arises because the strong gas expansion from IMR (∼ 52 mbar) into SQ (∼ 1.5 mbar) can efficiently drive the initial transport of ions, allowing anions to be transmitted with comparable efficiency from pinhole to SQ. It makes ion transmission relatively insensitive to variations in the SQ potential as long as the retarding electric field across the pinhole-SQ region remains moderate. Although ion transmission eventually decreases as the field becomes sufficiently strong, most clusters have already reached their dV50 by that stage.
The increased voltage gap between SQ and BSQ accelerates the velocity of ions. Clusters collided with buffer gas at the entrance of BSQ, and those with higher binding energies required larger kinetic energies to dissociate. Thus, their strengths were inferred from the voltage gaps needed for efficient dissociation. The detailed voltage settings during the scanning are listed in Table S1. The duration for each step of a scan was 20 s and the whole scan lasted for no more than 7 min. For a certain compound, the decay of the adduct signal during a single voltage scan was fitted with a sigmoid function. Based on this, the dV50 was determined as the bias voltage that was applied to decrease the signal to half its original value. The humidity of samples can influence the dV50 of analytes. We measured the dV50 of 3-nitrophenols, acetic acid, propylene glycol, and butyric acid at different dopant flow rates. A slight shift of ±0.5V in dV50 can be observed for all of these compounds when the water ratio increases from 0 (at 0 sccm dopant flow) to 0.75 (at 50 sccm dopant flow) (Fig. S4). Thus, the comparability of dV50 is ensured only when the RH within IMR is constant between sampling and calibration.
Benzene reagent ions mainly charge organic compounds through charge transfer rather than adduct formation, whereas acetone dimers charge them with a combination of both mechanisms. This complicates the interpretation of voltage scanning results in these two positive channels. Therefore, this paper focuses on the results obtained from the bromide and iodide channels.
2.3 Instrument Calibration
A series of species were calibrated with different methods to determine the relationship between dV50 and sensitivity of MR-CIMS to the corresponding compound. Nitric acid and formic acid were calibrated with gravimetrically calibrated permeation tubes (VICI, USA). Chlorine was calibrated with a standard gas cylinder (BOC Limited, UK). Oxygenated volatile organic compounds were calibrated with a liquid calibration system (LCS, Tofwerk AG., Switzerland). The lists of 14 calibrants, methods, and calibration results are given in the Supplement (Table S2).
2.4 Quantum chemistry calculations
Theoretical calculations were performed to confirm the experimentally determined binding enthalpies and to gain a deeper understanding of the formation of ion–molecule clusters. The conformers of compounds and clusters were initially generated using Avogadro software (version 1.101.0) and pre-optimized using the Merck molecular force field (MMFF94) (Halgren, 1996) or, when iodine or iodide was present in the clusters, with Universal Force Field (UFF) (Rappé et al., 1992). Geometry optimizations and vibrational frequencies were performed using the long-range corrected hybrid density function theory methods at the ωB97X-D3/def2-TZVPPD level with TightSCF convergence, employing the RIJCOSX approximation and def2/J auxiliary basis, as implemented in ORCA software (version 6.0.0). Single point electronic energy corrections were computed on the lowest energy geometry at the DLPNO-CCSD(T)/def2-QZVPPD level with the RIJCOSX approximation, AutoAux basis generation, and TightSCF convergence in ORCA. We generally followed the computation theory utilized in the previous investigation (Wang et al., 2021), with additional diffuse functions employed in the single point electronic energy calculations to improve accuracy for the anions. The cluster formation or dissociation enthalpy was computed at 298.15 K, consistent with previous studies (Iyer et al., 2016; Wang et al., 2021). This is because, though the IMR was heated to 45 °C (343.15 K), the quadrupoles and ion optics, where the primary fragmentation reactions occurred, remained near room temperature. Meanwhile, enthalpy values are considerably less sensitive to temperature variations than Gibbs free energies. The cluster formation enthalpy in the bromide channel was calculated for the majority of the calibrated compounds. However, only one aromatic compound, 4-nitrophenol, was included owing to the high computational cost associated with aromatic systems. In the iodide channel, the cluster formation enthalpy of C2H4O2•I−, C2H4O3•I−, C2H2O3•I−, and C2H6O2•I− were computed to validate the modeling results reported by Iyer et al., which were obtained by geometry optimization at the B3LYP/6-31G* level and single-point energy calculations at the DLPNO-CCSD(T)/def2-QZVPP level (Iyer et al., 2016).
2.5 SAPHIR-CHANEL campaign
During the summer of 2024, a series of VOC oxidation experiments were conducted in the SAPHIR-CHANEL campaign at Forschungszentrum Jülich, Germany, which aims to mimic the atmospheric evolution of VOCs from a wide range of sources under different conditions. The SAPHIR chamber is a large outdoor chamber and has been well described in the previous literature (Fuchs et al., 2013).
In the SAPHIR-CHANEL experiments, different VOC mixes were used as analogues of important compounds emitted from different sources in inhabited areas, including volatile chemical products (VCPs), cooking, gasoline, diesel, and biogenic VOCs. Both single-source and mixed-source oxidation experiments were conducted during the campaign. To maximize the atmospheric relevance in the mixed-source experiments, the VOC categories were combined in the same ratio as those measured in Los Angeles (Coggon et al., 2021; Pfannerstill et al., 2024; Stockwell et al., 2025). Gaseous compounds were sampled from the SAPHIR chamber to the MR-CIMS through a Teflon tube with an outer diameter of 0.5 inches, an inner diameter of 0.36 in., and a length of 0.68 m, with a flow rate of ca. 1.8 L min−1. The tube extended 0.38 m outside the chamber and 0.30 m inside. Detailed experiment descriptions, including other instruments and the chemical regimes, will be provided in a separate manuscript in preparation.
In the chamber experiment results, only the sigmoid fits of signal decays during voltage scanning with a coefficient of determination (R2) greater than 0.5 were retained. A total of 36 voltage scans were conducted at different experiment stages during the multi-source chamber experiments in the campaign in order to cover the widest range of compounds and the median dV50 for each adduct was selected to represent its binding energy. Kernel density estimation (KDE) was applied to estimate the probability density functions of dV50 in both channels, in order to provide better visualization of the effects of oxygen levels on dV50 (Silverman, 2018). KDE was performed using the kdeplot function from the Seaborn package (version 0.13.2) in Python (Waskom, 2021). We utilized the Gaussian kernel and applied a factor of 0.7 to the bandwidth estimated from Scott's Rule to show more details of distributions (Scott, 1979). The detailed calculation methods are provided in Sect. S1.
Since voltage scanning essentially determines cluster stability through cluster dissociation, which significantly reduces the signals of the analytes, it is only suitable for analytes with a sufficient signal-to-noise ratio (SNR) in the mass spectrum. The relationship between SNR and the applicability of voltage scanning was studied here. We followed the calculation principle of noise in previous research (Yan et al., 2016), and the detailed calculation methods of SNR were provided in Sect. S2.
3.1 Determination of collision limit
The collision limit sensitivities of analytes in bromide and iodide adduct channels are difficult to determine, because the required standards are either unavailable, expensive or exhibit significant wall losses during calibration. Aggarwal et al. (2025) determined the collision limit of a total of 39 CIMS and statistically demonstrated that the collision-limited sensitivity relative to the reagent ion remains nearly unchanged across different ionization mechanisms, if the reactor geometry and other key parameters, including reactor temperature, pressure, and water content remain constant (Aggarwal et al., 2025). It should be noted that although different reagent ions share the same collision limit under identical IMR conditions, the exact value must still be experimentally determined for each individual instrument. This is because factors after the IMR, e.g., ion optics transmission efficiency and detector settings, can vary across instruments. We adopted their approach to determine the collision limits of the bromide and iodide channels by measuring that of the benzene channel, as α-pinene, which is readily available, was shown to be ionized at the collision-limit sensitivity by benzene cations in the previous study (Aggarwal et al., 2025). In our instrument, the sensitivity of α-pinene was calibrated with a standard VOC cylinder and was determined to be 13.87 ± 0.69 ncps pptv−1. This value was used as the upper limit with an uncertainty of 5 % in fitting the relationship between dV50 and sensitivity. Aggarwal et al. (2025) have already demonstrated statistically that the collision-limit sensitivities of the benzene, bromide and iodide channels are very similar (Aggarwal et al., 2025). In addition, we further examine the binding enthalpy and sensitivities of the calibrants in the bromide channel, thereby confirming the validity of this choice.
3.2 Relationship between sensitivity and dV50
Measured during the liquid calibration in the laboratory, the variation of some typical ion–molecule adducts' signals in the bromide channel during the voltage scanning are shown in Fig. S5. Most observed signals corresponding to ion–molecule adducts showed a sigmoidal decrease with an increasing voltage gap. A similar trend was observed in the iodide channel and will not be elaborated here to avoid redundancy. The water-reagent ion clusters are known to be among the weakest adducts in I-CIMS and Br-CIMS (Iyer et al., 2016). Our results support this observation, and their dV50 may serve as practical lower limits.
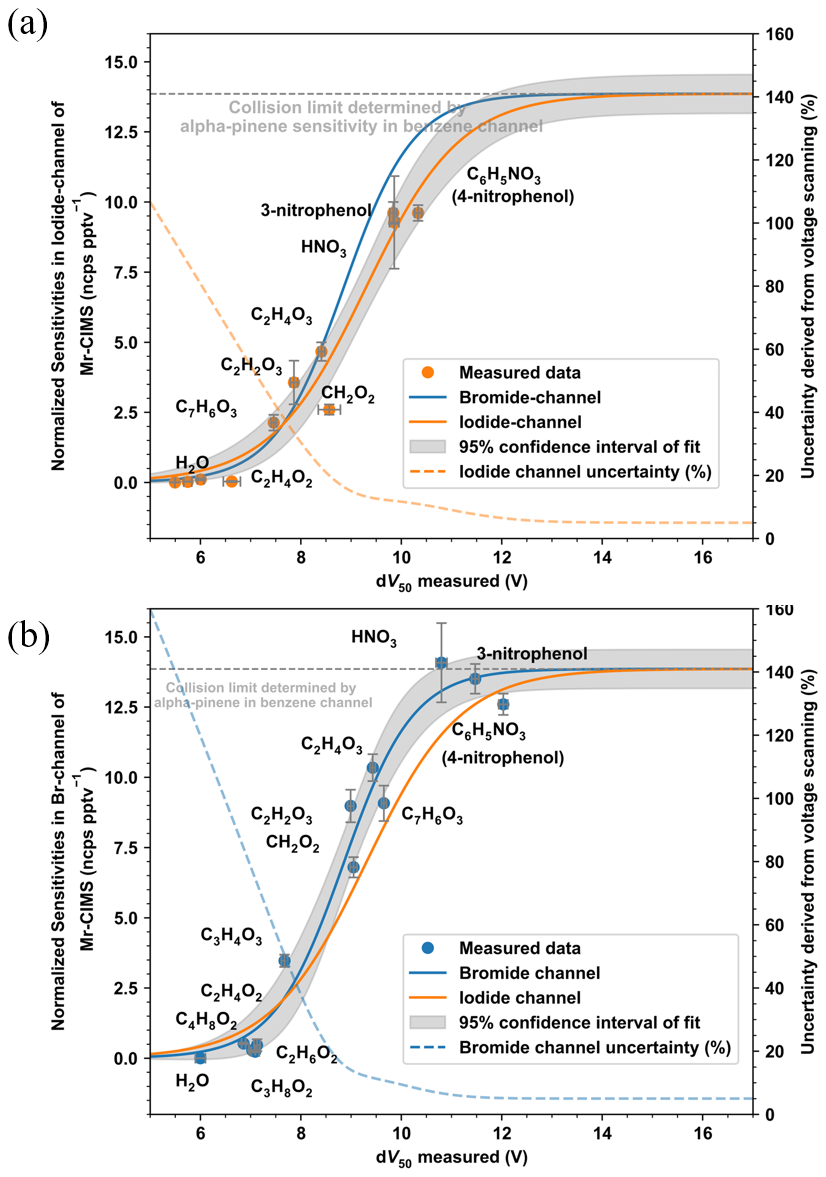
Figure 2Relationships between dV50 of adducts and sensitivities to the corresponding species in the (a) bromide channel and (b) iodide channel of MR-CIMS. Fitting curves and 95 % confidence intervals of dV50 and sensitivity are shown. For comparison, the sigmoidal fit line from the iodide channel is also plotted in panel (a) and that from the bromide channel is also plotted in panel (b). The systematic uncertainty derived from voltage scanning is the sum of squares of 5 % uncertainty from collision limit determination and half the confidence interval of sigmoid fit divided by the corresponding fitted value.
A series of gaseous compounds were calibrated in MR-CIMS and their measured sensitivities show a clear correlation with dV50 (Fig. 2), Generally, iodide shows a higher selectivity than bromide in chemical ionization. The maximum sensitivity experimentally determined from the benzene cation channel is close to the maximum sensitivity determined from the bromide channel, which supports the use of a single value for the collision limit between different channels as recommended by Aggarwal et al. (2025). This offers a promising approach for the calibration of the collision limit, with the potential to significantly reduce time and cost. We did not find suitable calibrants that have collision-limit sensitivity in the iodide channel and can be generated at ignorable loss by our LCS. Therefore, we used the maximum sensitivity in other channels when fitting the empirical relationship for the iodide channel. The uncertainty in the estimation was defined as half the width of the 95 % confidence interval of the sigmoid fit, divided by the corresponding fitted value, and was also treated as a systematic error associated with the use of this method. The uncertainty was considerable for compounds with low dV50, but generally remained below 20 % for those with dV50 greater than 8.60 and 8.52 V in the bromide and iodide channel, respectively, corresponding to sensitivities of 5.69 and 5.30 ncps pptv−1.
Glyoxylic acid (C2H2O3, SMILES: C(=O)C(=O)O) and glycolic acid (C2H4O3, SMILES: OC(=O)CO) exhibit higher sensitivity than acetic acid (C2H4O2, SMILES: CC(=O)O) in both channels. The effect of the benzene ring on sensitivity differs between the two channels, which is evident from the sensitivity of salicylic acid (C7H6O3, SMILES: O=C(O)c1ccccc1O). It effectively reduces the sensitivity in the iodide channel while having minimal impact in the bromide channel.
The carbon chain structure also needs to be considered when estimating the sensitivity of the compound. Glyoxylic acid has a much higher sensitivity than pyruvic acid (C3H4O3, SMILES: CC(=O)C(=O)O) in both channels, and formic acid (CH2O2, SMILES: C(=O)O) is also much more sensitive than acetic acid. However, such a reduction in sensitivity with the increasing carbon chain is not obvious for other compounds. Acetic acid has a very close sensitivity to butyric acid (C4H8O2, SMILES: CCCC(=O)O), whilst ethylene glycol (C2H6O2, SMILES: OCCO) is as sensitive as propylene glycol (C3H8O2, SMILES: CC(O)CO) in both channels, though higher uncertainty is an issue in the calibration of these low sensitivity compounds. Therefore, the influence of the carbon chain cannot be linearly correlated with its size and requires further clarification through theoretical calculations.
Additionally, we also calibrated the three isomers of nitrophenol (2-nitrophenol, 3-nitrophenol, and 4-nitrophenol), to investigate the difference in sensitivity caused by the structures of analytes. 3-nitrophenol and 4-nitrophenol have high and similar sensitivity, which are confirmed by their dV50 results. No meaningful signal corresponding to 2-nitrophenol associated with the bromide or iodide ion was detected. However, a charged anion (C6H4NO) with a modest signal was observed, likely resulting from proton abstraction from 2-nitrophenol.
A test experiment was conducted to confirm that fast-switching settings of MR-CIMS do not influence the determination of dV50 for the analytes. We measured the dV50 of lactic acid under routine operation and under conditions where the VUV lamps were operated without switching. The results of both negative channels are shown in Fig. S6, demonstrating that the differences were within 3 %.
This voltage scanning method incorporating a voltage bias is also applicable to other instruments utilizing Vocus AIM IMR. We employed it on our Vocus chemical ionization time-of-flight mass spectrometer (Vocus CI-TOF, Vocus 2R, Tofwerk AG.). We followed the default operating pressure of Vocus CI-TOF, with its IMR and SQ maintained at 60 and 2.0 mbar, respectively. We set the voltage bias at its electrode “Vocus/Reactor back” and adjusted the potential before BSQ with its electrodes “RF DC” and “Skimmer” by the same increment. The results obtained are comparable to those from MR-CIMS, though the absolute sensitivities of same compounds differed between the two instruments (Fig. S7). This is likely due to factors such as instrument tuning, microchannel plate (MCP) settings, and pressures. In addition, because we did not have an extra dopant flow heater, the water ratio of the Vocus CI-TOF in iodide channel and bromide channel were 0.09 and 0.3, respectively. However, the relationship between dV50 and sensitivities was preserved.
3.3 Theoretical calculations
Quantum chemistry calculations were performed to better understand the dependence of sensitivity on structure, especially that of Br-CIMS. The calculated formation enthalpies were listed in Table 1. The sensitivity to lactic acid (C3H6O3, SMILES: CC(C(=O)O)O) was not determined due to its high background in the calibration system, but its dV50 was obtained. Ethanol (C2H6O, SMILES: CCO) was not detectable by either the iodide channel and bromide channel, and its formation enthalpy with Br− was calculated here for comparison.
Table 1Cluster formation enthalpies and experimentally determined dV50 of different species with bromide or iodide. The cluster geometries were optimized at the ωB97X-D3/def2-TZVPPD level at 298.15 K. The enthalpies were calculated at the DLPNO-CCSD(T)/def2-QZVPPD level at 298.15 K. The error of dV50 represents the standard error of the sigmoid fit.
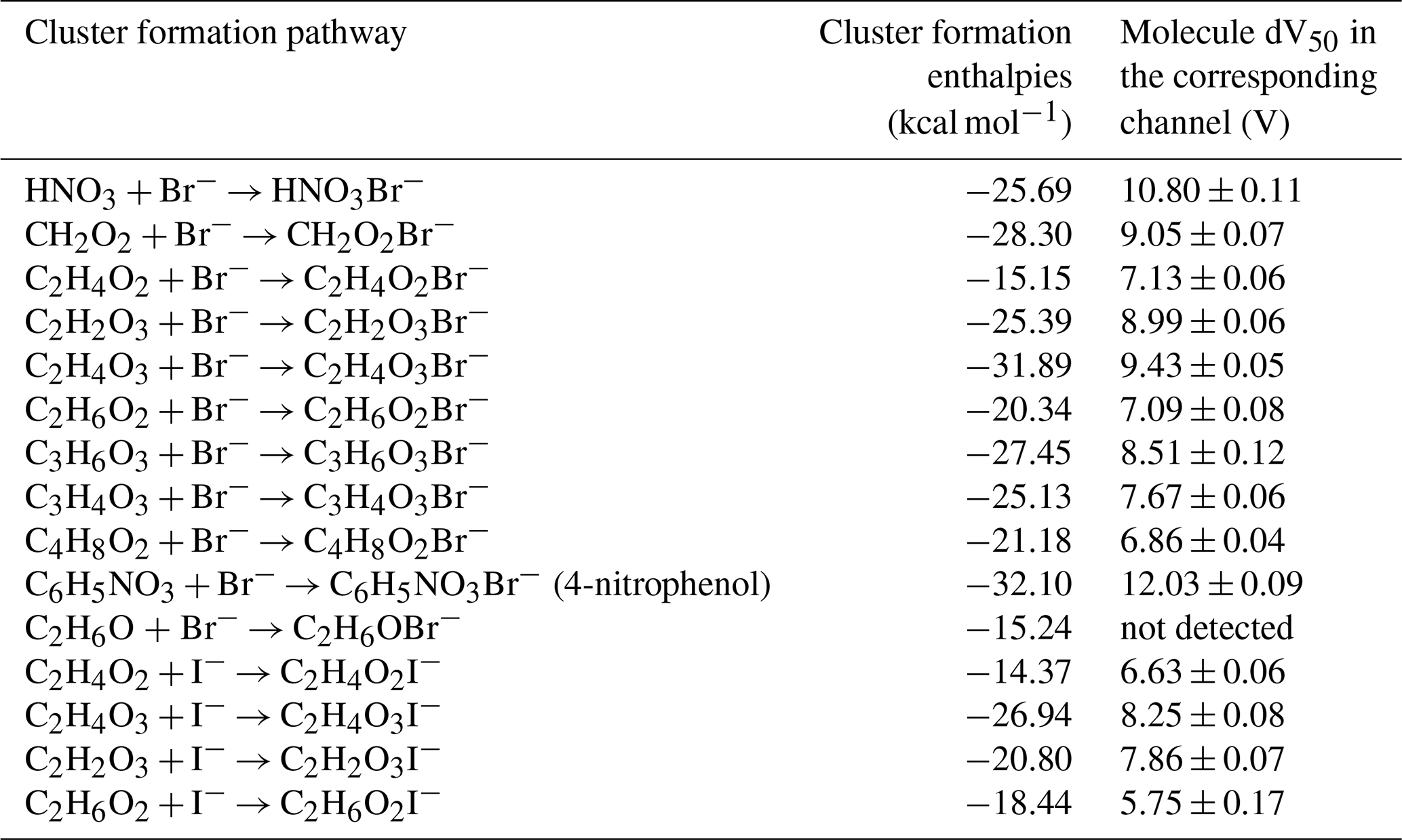
The theoretical formation enthalpies for Br− showed good linear relationship (R2 = 0.744) with experimentally determined binding energies (Fig. 3), except for 4-nitrophenol and HNO3. 4-nitrophenol exhibited a high dV50 and a sensitivity close to that of α-pinene in benzene channel, which is consistent with its large cluster formation enthalpy. Previous investigations showed that iodine can be clustered by bromide at collision-limit sensitivity (Wang et al., 2021), with a calculated cluster formation enthalpy to be −32.15 kcal mol−1 at our calculation level. The cluster formation enthalpy of 4-nitrophenol and bromide was calculated to be −32.10 kcal mol−1, which was nearly identical to that of iodine.
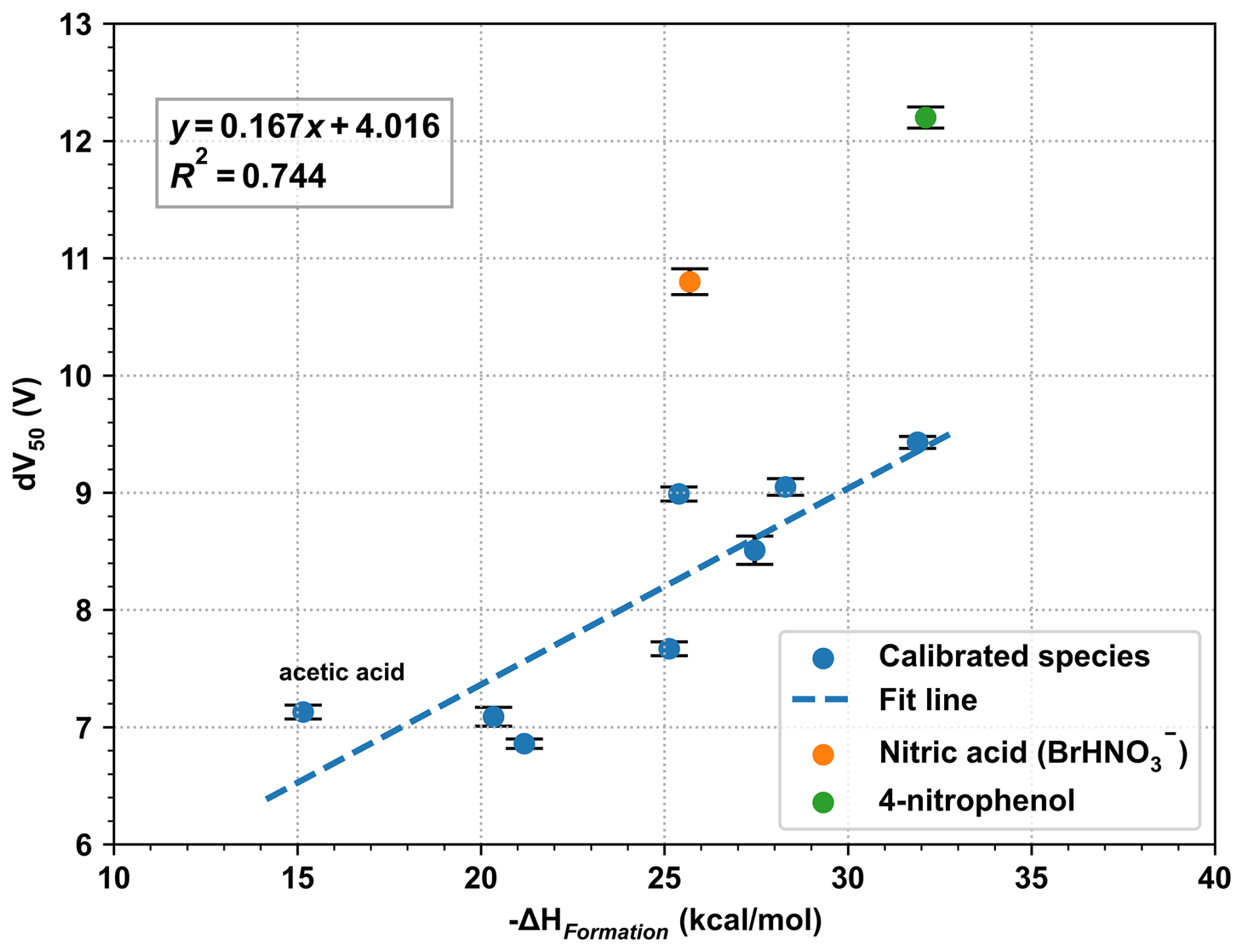
Figure 3Comparison between the cluster formation enthalpy calculated at the DLPNO-CCSD(T)/def2-QZVPPD level at 298.15 K and experimentally determined dV50. The error of dV50 represents the standard error of the sigmoid fit. Nitric acid was an obvious outlier due to the stabilization effects of water clusters. 4-nitrophenol was clustered at the collision limit, causing its dV50 to deviate substantially from the fitted trend of the other data points.
Although the sensitivity to HNO3 in Br-CIMS approaches the collision limit, the calculated cluster formation enthalpy of BrHNO was only around −25.69 kcal mol−1. This discrepancy can be explained by its interactions with water molecules in the IMR. The cluster formation enthalpy for BrH2O•HNO was calculated to be −24.26 kcal mol−1, only slightly lower than those of the corresponding BrHNO clusters. However, the addition of water molecule can effectively increase number of vibrational modes of small molecules and result in the vibrational relaxation of excess energy, which can stabilize the BrH2O•HNO. Such energy accommodation can efficiently compensate for the slight reduction in cluster formation enthalpy and enhance the sensitivity to HNO3. This is evidenced by the rapid increase in their sensitivity and the concomitant decrease in its fragmentation with increasing RH (Fig. S8). This mechanism was previously reported for compounds with no more than 8 atoms in the IMR of I-CIMS (Lee et al., 2014), and was investigated through quantum Rice-Ramsperger-Kassel (QRRK) calculations by Iyer et al. (2016). It is likewise applicable here, since both instruments employ adduct formation as the basis for ionization, albeit with different reagent ions. Despite BrH2O•HNO being the dominant form of cluster formed from HNO3 in the IMR, the mass spectrum was characterized by BrHNO as the major peak. This can be explained by the low fragmentation enthalpies: BrH2O•HNO dissociating to BrHNO and H2O (10.26 kcal mol−1). Therefore, the weak binding between H2O and Br− can be easily dissociated during the transmission through the quadrupoles and ion lens.
Meanwhile, the stabilization effect from water clustering might also influence small organic compounds in the bromide channel. As QRRK simulations exceed our computational resources, we restrict our analysis to a qualitative comparison. The cluster formation enthalpy of C2H4O2•Br− (−15.15 kcal mol−1) is comparable to that of C2H6O•Br− (−15.24 kcal mol−1), however only C2H4O2•Br− produced a detectable signal of in the MS and its dV50 was even larger than C2H6O2•Br−, whose formation enthalpy was −20.34 kcal mol−1. This suggested that the water cluster stabilization effect was effective for acetic acid (8 atoms) but limited for ethanol (9 atoms) and ethylene glycol (10 atoms). Despite its cluster formation enthalpy with Br− being 4.35 kcal mol−1 more negative than that with BrH2O−, the probability of forming BrH2O− clusters substantially increased owing to the humidified sample conditions. These stabilization effects were inherently reflected in the experimentally determined dV50, which implied the effective binding energy under real ionization conditions. This was supported by the observed sigmoid relationship between dV50 and sensitivities.
Computational results indicated that all of the detectable organic molecules are bound to Br− via hydrogen bonding, forming stable adducts, similar to the conditions modelled in I-CIMS (Iyer et al., 2016). The optimized geometries of representative clusters, including CH2O2•Br−, C2H4O2•Br−, C2H6O2•Br−, C2H4O3•Br−, C3H6O3•Br−, and C2H2O3•Br−, are shown in Fig. 4.
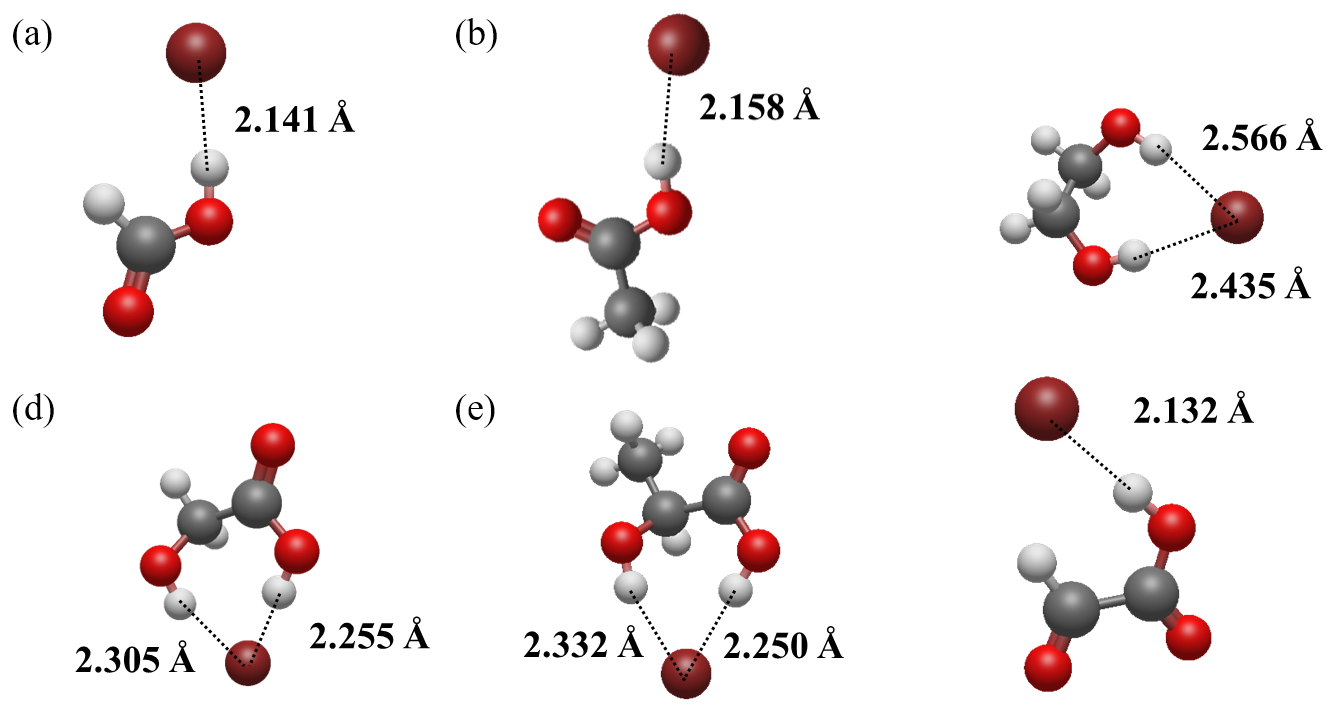
Figure 4Geometries of clusters between Br− and (a) formic acid, (b) acetic acid, (c) ethylene glycol, (d) glycolic acid, (e) lactic acid, and (f) glyoxylic acid. Color coding: oxygen, red; carbon, gray; hydrogen, white; bromine, brown. Hydrogen bond lengths were labeled in the plot.
Carboxyl groups (Fig. 4a, b) and hydroxyl groups (Fig. 4c) can both form a hydrogen bond with Br−. In addition, adjacent hydroxyl and carboxyl groups can jointly form hydrogen bonds with Br−, as observed in glycolic acid (Fig. 4d) and lactic acid (Fig. 4e). Neither carbonyl group nor aldehyde group can form hydrogen bonding with halogen anions because they are all hydrogen bond acceptors. Consequently, ketone and aldehydes cannot be effectively detected in Br-CIMS. However, the sensitivity of glyoxylic acid is much higher than acetic acid. It is because the aldehyde group as a whole is an electron-withdrawing functional group, which can increase the acidity of the adjacent carboxyl group and enhances the partial positive (δ+) character of the carboxylic proton. This promotes hydrogen bonding formation between the carboxylic proton and Br−. This is corroborated by the more negative cluster formation enthalpy of −25.39 kcal mol−1 of C2H2O3•Br− cluster (Fig. 4f) compared to that of C2H4O2•Br− (−15.15 kcal mol−1).
A comparable effect was also observed for alkyl substitution attached to the carboxyl group. The sensitivity of formic acid is larger than that of acetic acid, as the electron-donating methyl group in acetic acid decreases the δ+ character of the carboxylic proton. Therefore, the cluster formation enthalpy of CH2O2•Br− (−28.30 kcal mol−1) is more negative than that of C2H4O2•Br−, which agrees well with the experimental results.
The binding mechanism also accounted for the inability of bromide channel to detect 2-nitrophenol. It possessed a well-known intramolecular hydrogen bond (Borisenko et al., 1994), which effectively inhibited the hydroxyl group from forming a strong hydrogen bond with Br−. In contrast, such intramolecular hydrogen bonding was absent in 3- and 4-nitrophenol (Boll et al., 1958), enabling these isomers to be detected with substantially higher sensitivity. This explanation was also applicable for I-CIMS due to their subtly different but similar binding methods.
The calculated geometries and the order of the formation enthalpies in the iodide channel are generally consistent with those reported by Iyer et al(2016). The only difference is that glycolic acid was previously assumed to be charged by iodide through hydrogen bonding with its hydroxyl group (Iyer et al., 2016). However, our calculations suggest that the hydrogen bond involving the carboxyl group is energetically more stable (Fig. S9), similar to other acids. This difference arises from the higher level of theory employed in our calculations. We also calculated the structure in which glycolic acid forms two hydrogen bonds with iodide, which had a cluster formation enthalpy of −26.16 kcal mol−1, but it is less stable than the structure where iodide is bonded to the carboxyl group (−26.94 kcal mol−1). Such a difference in the adduct formation mechanisms of α-hydroxy acid in the bromide and iodide channel might be attributed to the greater steric effect of iodide than the bromide.
3.4 Applications in chamber experiments
The voltage scanning conducted during the SAPHIR-CHANEL campaign facilitated the quantification of a range of gaseous species. As mentioned above, only sigmoid fits of adduct signal decay with a R2 larger than 0.5 were considered as successful voltage scanning results. Certain signals exhibited insufficient intensity to reveal a distinct sigmoid decay, whereas others showed no decay during voltage scanning and were likely to be fragment ions. We will also investigate the abundance of signal intensities required to be applicable for voltage scanning later.
The distribution density of dV50 for nitrogen-free organic compounds is shown in Fig. 5, which illustrates that estimating sensitivity for CIMS based solely on molecular formula would lead to significant errors without additional voltage scanning information. For clarity, only compounds with at least 2 oxygen atoms (CxHyOz, z ≥ 2) are presented. In the bromide channel, an increase of up to four oxygen atoms generally resulted in a rise of approximately 1 V in dV50 per additional oxygen atom for CxHyO2–4. However, estimating the dV50 of a particular molecule solely based on the number of oxygen atoms resulted in significant errors. The trend of increasing dV50 with oxygen atoms was only observed for CxHyO2–4 in the iodide channel, but some CxHyO2 showed up with the highest dV50. For nitrogen-free compounds with at least 5 oxygen atoms, the distributions of dV50 were broader in both channels. This might be attributed to the increase in both the number and types of oxygen-containing functional groups, leading to greater structural diversity. The distributions of dV50 for organonitrates were more complex. No obvious relationship with effective oxygen atoms and dV50 was observed in both channels (Fig. S10), which highlighted the complicated effect of the nitrate group on the sensitivity of compounds.
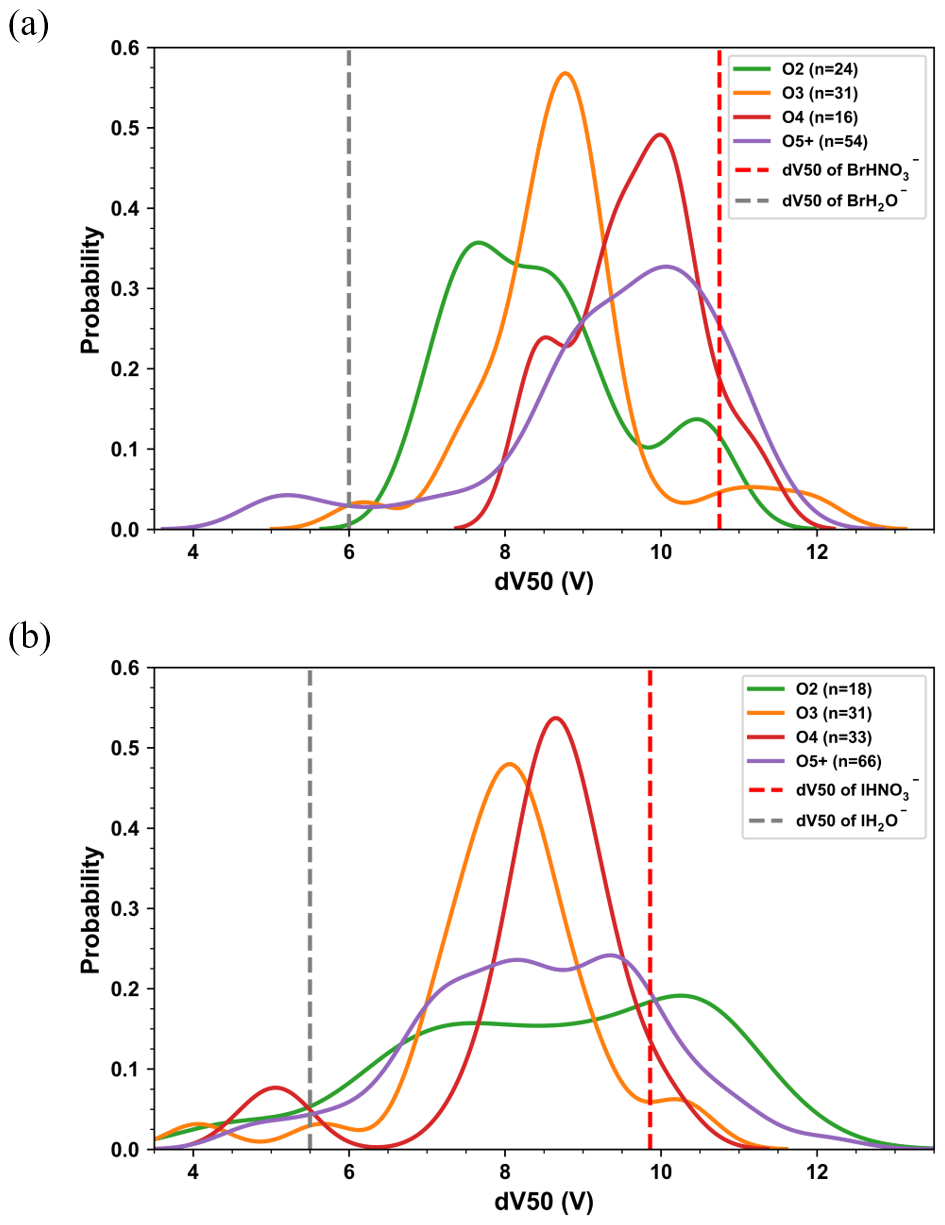
Figure 5Probability density distributions of dV50 for compounds CxHyOz (z ≥ 2) detected in the (a) bromide channel and (b) iodide channel of MR-CIMS. dV50 of water and nitric acid in each channel are shown for comparison. The number of compounds in each category are shown in the label.
In the SAPHIR chamber experiments, the dV50 of a total of 290 and 264 organic compounds were determined in the bromide and iodide channels respectively and thus their sensitivities can be derived. Box and whisker plots of the distributions of 1-second integration SNRs for compounds characterized with successful voltage scanning results from these experiments in Fig. S11. The 5th and 25th percentile of the SNRs are 1.9 and 4.0 in the bromide channel, and 1.6 and 3.7 in the iodide channels, respectively. The signals that yielded successful voltage scanning were evaluated relative to the widely used limit-of-detection threshold of SNR = 3 (Yuan et al., 2017), in order to understand the signal strength necessary for a successful scanning. 84.7 % of compounds with successful voltage scanning results in the bromide channel and 82.3 % in the iodide channel exhibited SNRs large than 3. Therefore, an SNR of 3 was assumed as the thresholds for the abundance of adduct signals applicable to voltage scanning, if their dissociations were observed during the increasing of electric field strength.
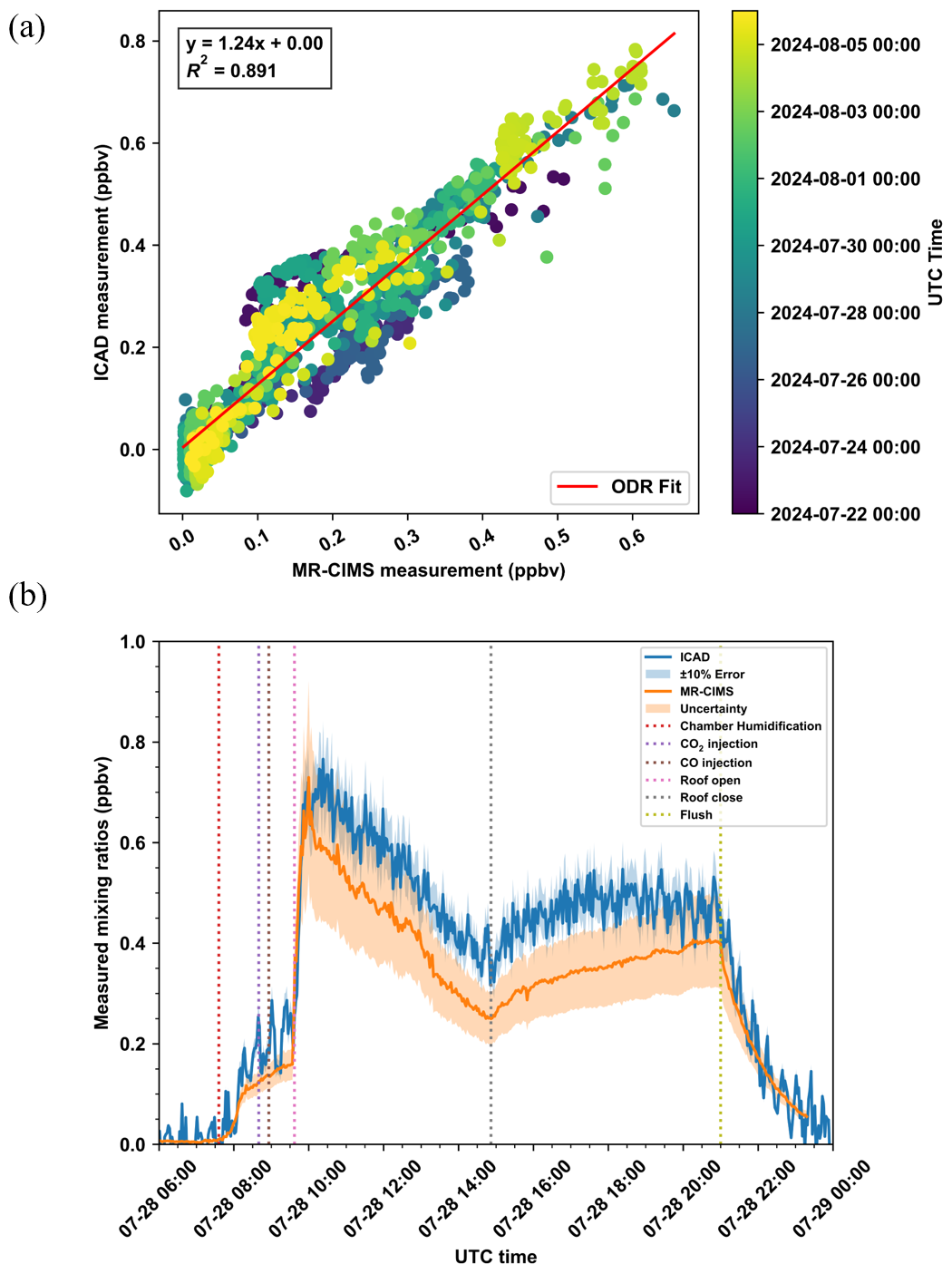
Figure 6(a) Comparison between HONO concentrations measured by the bromide channel of MR-CIMS against ICAD during the SAPHIR-CHANEL campaign averaged at a time resolution of 10 min. The points are colored by their measured time. Fitted using an orthogonal distance regression (ODR). (b) An example comparison of measurements in a background experiment at a time resolution of 2 min. The shaded region indicates the measurement uncertainty. The measurement uncertainty of ICAD was estimated to be 10 %. The measurement uncertainty of MR-CIMS for HONO was estimated to be the RSS of systematic uncertainty and random uncertainty.
The reliability of this method is demonstrated by comparing signals of independently calibrated techniques to those of MR-CIMS using voltage scanning. The HONO concentrations measured by the bromide channel of MR-CIMS and a directly calibrated ICAD (iterative cavity enhanced differential optical absorption spectroscopy) were compared in the chamber experiments during which the voltage scanning approach was applied, shown in Fig. 6a. We estimated HONO sensitivity in the bromide channel to be around 6.53 ± 1.10 ncps pptv−1 based on its dV50, and modified it based on the weekly VOCs calibration results on the benzene channel. The concentrations measured by the two instruments were very similar, as indicated by an orthogonal distance regression (ODR) slope of 1.24 and an R2 of 0.891. Periods of chamber flushing or pre-humidification were excluded from fitting. The measurement uncertainty of ICAD was estimated to be 10 % at 1 min averaging interval (1σ). The total uncertainty of MR-CIMS on the estimated concentration was calculated with the root square sum (RSS) of systematic uncertainty and random uncertainty, which was around 16.8 %. Systematic uncertainty was derived from the sensitivity estimation based on voltage scanning as illustrated above, while random uncertainty was the noise at 1 min averaging interval (1σ) calculated following the method in Sect. S3. A background experiment, conducted without any precursor injection, was used to illustrate the measurement results from the two instruments (Fig. 6b). The discrepancies generally fell within the propagated uncertainties.
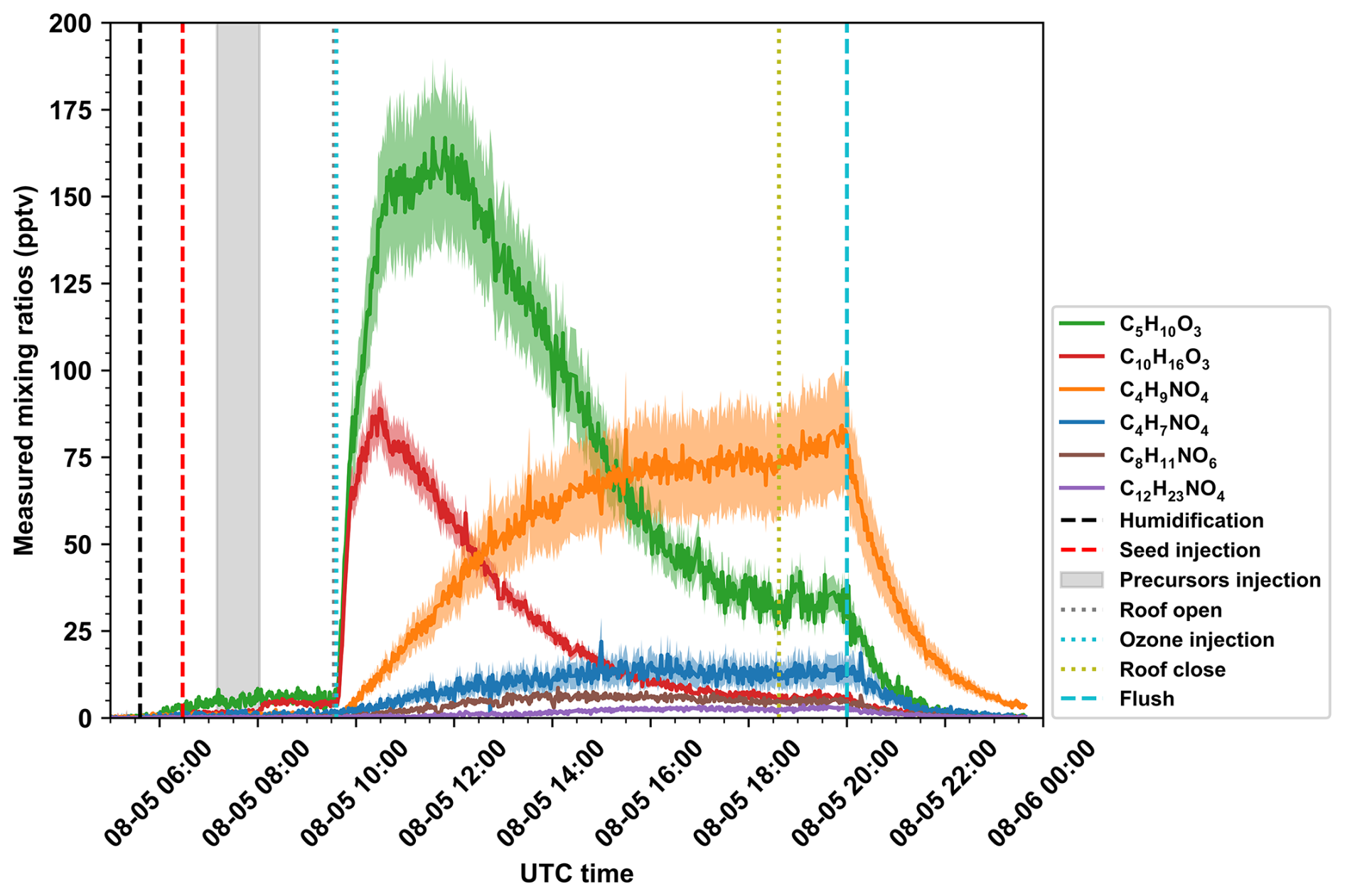
Figure 7Measured by bromide channel, time series of ISOPOOH (C5H10O3), pinonic acid (C10H16O3), butene-derived organonitrates (C4H7NO4 and C4H9NO4), xylene-derived organonitrate (C8H11NO6), and dodecane-derived organonitrate (C12H23NO4) in a multi-source chamber experiment during SAPHIR-CHANEL campaign. The Gray shadow region indicates the periods of precursor injection. Uncertainties were calculated by the RSS of systematic uncertainty and random uncertainty and indicated by the shaded regions around the time series.
Other compounds were also quantified with acceptable total uncertainties. To illustrate the utility of voltage scanning approach, we present data measured by bromide channel and calibrated by the voltage scanning approach from a multi-source chamber experiment designed to simulate daytime oxidation in the Los Angeles atmosphere (Fig. 7). Several precursors were injected, including gasoline, biogenic, cooking, and VCP sources. During the oxidation experiments, a series of compounds that have been previously identified to be formed from certain precursors were detected. These compounds include first-generation intermediate closed-shell products of isoprene oxidation, isoprene hydroxy hydroperoxides (ISOPOOH, C5H10O3) (Wennberg et al., 2018), pinonic acid (C10H16O3) generated from pinene oxidation (Lopez-Hilfiker et al., 2016), two organonitrates generated from the oxidation of butene (C4H7NO4 and C4H9NO4) (Travis et al., 2023), an NO termination product of xylene-derived bicyclic peroxy radical (C8H11NO6) (Bianchi et al., 2019), and an organonitrate generated from dodecane oxidation (C12H23NO4). All of these oxidation products have clearly identifiable precursors as indicated by the master chemical mechanism (MCM) (Jenkin et al., 2003; Saunders et al., 2003). These precursor-product relationships were also confirmed by the mass spectrometry detected in the corresponding single-source chamber experiments.
The compounds mentioned above were detected at varying concentrations, ranging from just a few pptv to as high as 170 pptv. The systematic uncertainties for C5H10O3, C10H16O3, C4H7NO4, C4H9NO4, C8H11NO6, and C12H23NO4 were 13.7 %, 9.3 %, 31.3 %, 20.4 %, 7.9 %, and 13.7 %, respectively. Their random uncertainties varied with signal intensity, with lower values observed at higher SNR. When the chamber roof was open, their 5th to 95th percentile total uncertainties were 13.7 %–13.9 %, 9.4 %–10.4 %, 31.7 %–35.4 %, 20.5 %–21.7 %, 8.9 %–19.9 %, and 16.1 %–39.0 %, respectively. Therefore, they all had acceptable total uncertainties during the daytime oxidation, though characterized by different sensitivities and concentrations.
It deserves notification that the voltage scanning signal might represent a sum of individual responses of structural isomers. If multiple isomers contribute significantly to the ion signal and have different binding energies, the resulting voltage scanning profile cannot be well represented by a single sigmoid fitting. This limitation motivates the use of a sigmoid fitting quality threshold, i.e., R2 > 0.5, below which signals are not retained. In addition, the voltage scanning method can provide insight into compound functionality. As discussed above, different oxygenated functional groups have distinct binding energies, which can be captured in their corresponding dV50 values derived from voltage scanning.
3.5 Halogen compounds
Chlorine was clearly detected in both negative channels, but we only observed the dissociation of the Cl2•Br− cluster when performing voltage scanning. This indicates that the binding energy of Cl2•Br− and Cl2•I− cluster should be quite high. However, this is in contrast with the fact that the sensitivity of Cl2 in the bromide channel and iodide channel were only around 11.33 ± 0.37 and 3.27 ± 0.40 ncps pptv−1, respectively, lower than the collision limit. Cl2, as a small molecule, also benefited from the stabilization effect of water molecules, as evidenced by the concurrent increase in sensitivity and decrease in fragmentation with rising RH (Fig. S12). The cluster formation and fragmentation enthalpy related to Cl2 and I2 with reagent ions and hydrated reagent ions were included in Table 2. It can be shown that the fragmentation enthalpy of the intramolecular bond is close to, or in the iodide channel even lower than, that of the halogen bond formed between halogen compounds and the reagent ions. Therefore, during the voltage scanning, after the dissociation of the halogen–hydrated reagent ion cluster, the dissociation of intramolecular and intermolecular bond was thermodynamically competitive. As a result, the determined dV50 of halogen compounds were likely to be inaccurate because multiple dissociation processes occurred simultaneously. In conclusion, the voltage-scanning method may fail to give meaningful results when the ion–molecule interaction is dominated by a halogen bond. The fragmentation peak Cl− was not included in the discussion of sensitivity here, as both the voltage scanning and quantum chemistry were intended to determine the intermolecular binding energy of the cluster formed between the reagent ions and analytes, i.e., Cl2•Br− and Cl2•I−.
Table 2Fragmentation reaction enthalpies of different species with bromide ions. The cluster geometries are optimized at the ωB97X-D3/def2-TZVPPD level at 298.15 K. The enthalpies are calculated at the DLPNO-CCSD(T)/def2-QZVPPD level at 298.15 K.

In this study, a voltage scanning method was conducted on a multi-reagent ion CIMS instrument (MR-CIMS) to estimate the relative binding energies of ion–molecule adducts in the instrument's bromide and iodide channels. The sigmoid relationship between voltage bias at which the signal reduced by half (dV50) and relative sensitivities in both channels were characterized. These results, together with absolute collision-limit sensitivities obtained via a recently published methodology based on the equivalence of different ionization schemes under identical CIMS operating conditions (Aggarwal et al., 2025), demonstrate that CIMS is capable of quantifying gas-phase mixing ratios for a broad spectrum of atmospheric molecules. A comprehensive understanding of the functionality and structure dependence of bromide-adduct formation chemical ionization was obtained with experimentally determined dV50 and theoretically calculated binding enthalpies. For small molecules, water molecules might effectively accommodate the excess energy of their ion–molecule clusters and enhance their sensitivities, which can also be manifested in their unexpectedly larger dV50.
The dV50 of a series of compounds were determined during a series of experiments conducted at the SAPHIR chamber and the comparison of HONO concentrations validated the reliability of this method. A total of 290 and 264 compounds can be quantified in the bromide channel and iodide channel in the chamber experiments, respectively. The enhancement in sensitivity brought about by oxygenated functionalities was observed, but the specific molecular structures also had a significant influence on sensitivity, which confirmed the calibration results conducted previously. A SNR threshold of 3.0 was suggested to be applied as a threshold for the adduct signal abundance for voltage scanning, provided that dissociation was observed with increasing electric field strength. Measurement uncertainties were discussed for typical compounds generated from different precursors in a daytime oxidation chamber experiment. The comparable intra- and intermolecular bond energies of halogen compounds inherently limit their applicability of this quantification method. This work demonstrates that voltage scanning holds significant potential for the quantification of gaseous compounds and presents substantial opportunities for further development and application.
Data are available from the authors upon request.
The supplement related to this article is available online at https://doi.org/10.5194/amt-19-4583-2026-supplement.
YW, HC, and TB designed and conducted the calibration experiments. YW and FL worked on the method development. YW, HC, and TB wrote the manuscript. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The contact author has declared that none of the authors has any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. The authors bear the ultimate responsibility for providing appropriate place names. Views expressed in the text are those of the authors and do not necessarily reflect the views of the publisher.
YW is supported through NERC grant ConstrAining the RolE of Sulfur in the earth system (CARES), Award number, NE/W009307/1. AV acknowledges funding support from the Natural Environment Research Council (NERC) through the UK National Centre for Atmospheric Science (NCAS). This work was supported by the European Research Council (ERC) under the European Union's Horizon Europe research and innovation program through the Starting Grant CHANEL (grant no. 101076276), which provided the primary funding for the CHANEL campaign. Additional support for SAPHIR chamber operations was provided by the EC ATMO-ACCESS Transnational Access program (grant agreement No 101008004). This project has received funding from the European Research Council under the European Union's Horizon 2020 research and innovation programme under the Consolidator Grant ADAPT (grant No. 101002728). We gratefully acknowledge the entire SAPHIR team at Forschungszentrum Jülich for their technical support and operation of the chamber during the CHANEL campaign. Finally, editorial and coding assistance using artificial intelligence was employed to refine text phrasing and streamline figure generation. The authors retain full responsibility for the scientific content and interpretation of all results. We would like to acknowledge Chelsea Stockwell, Carsten Warneke, Steve Brown and Matthew M. Coggon from National Oceanic and Atmospheric Administration with the preparation and organization of the campaign. We would like to acknowledge Charel Wohl and David Oram from University of East Anglia for the use of the liquid calibration system.
This research has been supported by the Natural Environment Research Council (grant no. NE/W009307/1), the European Research Council, HORIZON EUROPE European Research Council (Starting Grant CHANEL (grant no. 101076276) and Consolidator Grant ADAPT (grant no. 101002728)), and the Integrated Carbon Observation System (ATMO-ACCESS Transnational Access program, grant no. 101008004).
This paper was edited by Bin Yuan and reviewed by two anonymous referees.
Aggarwal, S., Bansal, P., Wang, Y., Jorga, S., Macgregor, G., Rohner, U., Bannan, T., Salter, M., Zieger, P., Mohr, C., and Lopez-Hilfiker, F.: Identifying key parameters that affect sensitivity of flow tube chemical ionization mass spectrometers, Atmos. Meas. Tech., 18, 4227–4247, https://doi.org/10.5194/amt-18-4227-2025, 2025.
Albrecht, S. R., Novelli, A., Hofzumahaus, A., Kang, S., Baker, Y., Mentel, T., Wahner, A., and Fuchs, H.: Measurements of hydroperoxy radicals (HO2) at atmospheric concentrations using bromide chemical ionisation mass spectrometry, Atmos. Meas. Tech., 12, 891–902, https://doi.org/10.5194/amt-12-891-2019, 2019.
Bertram, T. H., Kimmel, J. R., Crisp, T. A., Ryder, O. S., Yatavelli, R. L. N., Thornton, J. A., Cubison, M. J., Gonin, M., and Worsnop, D. R.: A field-deployable, chemical ionization time-of-flight mass spectrometer, Atmos. Meas. Tech., 4, 1471–1479, https://doi.org/10.5194/amt-4-1471-2011, 2011.
Bianchi, F., Kurtén, T., Riva, M., Mohr, C., Rissanen, M. P., Roldin, P., Berndt, T., Crounse, J. D., Wennberg, P. O., Mentel, T. F., Wildt, J., Junninen, H., Jokinen, T., Kulmala, M., Worsnop, D. R., Thornton, J. A., Donahue, N., Kjaergaard, H. G., and Ehn, M.: Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol, Chem. Rev., 119, 3472–3509, https://doi.org/10.1021/acs.chemrev.8b00395, 2019.
Boll, P. M., Hordvik, A., Tvedten, O., Linderot, T., Veige, S., and Diczfalusy, E.: Intra- and Intermolecular Hydrogen Bonds in Nitro- and Nitrosophenols., Acta Chem. Scand., 12, 1777–1781, https://doi.org/10.3891/acta.chem.scand.12-1777, 1958.
Borisenko, K. B., Bock, C. W., and Hargittai, I.: Intramolecular Hydrogen Bonding and Molecular Geometry of 2-Nitrophenol from a Joint Gas-Phase Electron Diffraction and ab Initio Molecular Orbital Investigation, J. Phys. Chem., 98, 1442–1448, https://doi.org/10.1021/j100056a012, 1994.
Chen, Y., Xia, M., Zhang, J., Tsiligiannis, E., Wu, C., Yan, C., Cai, R., Zheng, G., Li, Y., Guo, J., An, Z., Li, Y., Zhao, X., Qu, Q., Hua, C., Wang, Z., Wang, S., Liu, Y., Cao, L., He, K., Kulmala, M., Hallquist, M., Wang, T., Worsnop, D., and Jiang, J.: Chloramine chemistry as a missing link in atmospheric chlorine cycling, Sci. Adv., 11, 1–13, https://doi.org/10.1126/sciadv.adv4298, 2025.
Coggon, M. M., Gkatzelis, G. I., McDonald, B. C., Gilman, J. B., Schwantes, R. H., Abuhassan, N., Aikin, K. C., Arend, M. F., Berkoff, T. A., Brown, S. S., Campos, T. L., Dickerson, R. R., Gronoff, G., Hurley, J. F., Isaacman-VanWertz, G., Koss, A. R., Li, M., McKeen, S. A., Moshary, F., Peischl, J., Pospisilova, V., Ren, X., Wilson, A., Wu, Y., Trainer, M., and Warneke, C.: Volatile chemical product emissions enhance ozone and modulate urban chemistry, P. Natl. Acad. Sci. USA, 118, https://doi.org/10.1073/pnas.2026653118, 2021.
Coggon, M. M., Stockwell, C. E., Xu, L., Peischl, J., Gilman, J. B., Lamplugh, A., Bowman, H. J., Aikin, K., Harkins, C., Zhu, Q., Schwantes, R. H., He, J., Li, M., Seltzer, K., McDonald, B., and Warneke, C.: Contribution of cooking emissions to the urban volatile organic compounds in Las Vegas, NV, Atmos. Chem. Phys., 24, 4289–4304, https://doi.org/10.5194/acp-24-4289-2024, 2024.
Crounse, J. D., McKinney, K. A., Kwan, A. J., and Wennberg, P. O.: Measurement of gas-phase hydroperoxides by chemical ionization mass spectrometry, Anal. Chem., 78, 6726–6732, https://doi.org/10.1021/ac0604235, 2006.
Ehn, M., Thornton, J. A., Kleist, E., Sipilä, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I. H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurtén, T., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., Canagaratna, M., Maso, M. D., Berndt, T., Petäjä, T., Wahner, A., Kerminen, V. M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476–479, https://doi.org/10.1038/nature13032, 2014.
Finkenzeller, H., Iyer, S., He, X. C., Simon, M., Koenig, T. K., Lee, C. F., Valiev, R., Hofbauer, V., Amorim, A., Baalbaki, R., Baccarini, A., Beck, L., Bell, D. M., Caudillo, L., Chen, D., Chiu, R., Chu, B., Dada, L., Duplissy, J., Heinritzi, M., Kemppainen, D., Kim, C., Krechmer, J., Kürten, A., Kvashnin, A., Lamkaddam, H., Lee, C. P., Lehtipalo, K., Li, Z., Makhmutov, V., Manninen, H. E., Marie, G., Marten, R., Mauldin, R. L., Mentler, B., Müller, T., Petäjä, T., Philippov, M., Ranjithkumar, A., Rörup, B., Shen, J., Stolzenburg, D., Tauber, C., Tham, Y. J., Tomé, A., Vazquez-Pufleau, M., Wagner, A. C., Wang, D. S., Wang, M., Wang, Y., Weber, S. K., Nie, W., Wu, Y., Xiao, M., Ye, Q., Zauner-Wieczorek, M., Hansel, A., Baltensperger, U., Brioude, J., Curtius, J., Donahue, N. M., Haddad, I. El, Flagan, R. C., Kulmala, M., Kirkby, J., Sipilä, M., Worsnop, D. R., Kurten, T., Rissanen, M., and Volkamer, R.: The gas-phase formation mechanism of iodic acid as an atmospheric aerosol source, Nat. Chem., 15, 129–135, https://doi.org/10.1038/s41557-022-01067-z, 2023.
Fuchs, H., Hofzumahaus, A., Rohrer, F., Bohn, B., Brauers, T., Dorn, H. P., Häseler, R., Holland, F., Kaminski, M., Li, X., Lu, K., Nehr, S., Tillmann, R., Wegener, R., and Wahner, A.: Experimental evidence for efficient hydroxyl radical regeneration in isoprene oxidation, Nat. Geosci., 6, 1023–1026, https://doi.org/10.1038/ngeo1964, 2013.
Gkatzelis, G. I., Coggon, M. M., McDonald, B. C., Peischl, J., Gilman, J. B., Aikin, K. C., Robinson, M. A., Canonaco, F., Prevot, A. S. H., Trainer, M., and Warneke, C.: Observations Confirm that Volatile Chemical Products Are a Major Source of Petrochemical Emissions in U.S. Cities, Environ. Sci. Technol., 55, 4332–4343, https://doi.org/10.1021/acs.est.0c05471, 2021.
Halgren, T. A.: Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94, J. Comput. Chem., 17, 490–519, https://doi.org/10.1002/(SICI)1096-987X(199604)17:5/6<490::AID-JCC1>3.0.CO;2-P, 1996.
Huey, L. G.: Measurement of trace atmospheric species by chemical ionization mass spectrometry: Speciation of reactive nitrogen and future directions, Mass Spectrom. Rev., 26, 166–184, https://doi.org/10.1002/mas.20118, 2007.
Huey, L. G., Hanson, D. R., and Howard, C. J.: Reactions of SF6- and I- with atmospheric trace gases, J. Phys. Chem., 99, 5001–5008, https://doi.org/10.1021/j100014a021, 1995.
Iyer, S., Lopez-Hilfiker, F., Lee, B. H., Thornton, J. A., and Kurtén, T.: Modeling the Detection of Organic and Inorganic Compounds Using Iodide-Based Chemical Ionization, J. Phys. Chem. A, 120, 576–587, https://doi.org/10.1021/acs.jpca.5b09837, 2016.
Jenkin, M. E., Saunders, S. M., Wagner, V., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part B): tropospheric degradation of aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 181–193, https://doi.org/10.5194/acp-3-181-2003, 2003.
Ji, Y., Huey, L. G., Tanner, D. J., Lee, Y. R., Veres, P. R., Neuman, J. A., Wang, Y., and Wang, X.: A vacuum ultraviolet ion source (VUV-IS) for iodide–chemical ionization mass spectrometry: a substitute for radioactive ion sources, Atmos. Meas. Tech., 13, 3683–3696, https://doi.org/10.5194/amt-13-3683-2020, 2020.
Kercher, J. P., Riedel, T. P., and Thornton, J. A.: Chlorine activation by N2O5: simultaneous, in situ detection of ClNO2 and N2O5 by chemical ionization mass spectrometry, Atmos. Meas. Tech., 2, 193–204, https://doi.org/10.5194/amt-2-193-2009, 2009.
Lee, B. H., Lopez-Hilfiker, F. D., Mohr, C., Kurtén, T., Worsnop, D. R., Thornton, J. A., Kurte, T., Worsnop, D. R., Thornton, J. A., Kurtén, T., Worsnop, D. R., and Thornton, J. A.: An iodide-adduct high-resolution time-of-flight chemical-ionization mass spectrometer: Application to atmospheric inorganic and organic compounds, Environ. Sci. Technol., 48, 6309–6317, https://doi.org/10.1021/es500362a, 2014.
Lee, B. H., Lopez-Hilfiker, F. D., Veres, P. R., McDuffie, E. E., Fibiger, D. L., Sparks, T. L., Ebben, C. J., Green, J. R., Schroder, J. C., Campuzano-Jost, P., Iyer, S., D'Ambro, E. L., Schobesberger, S., Brown, S. S., Wooldridge, P. J., Cohen, R. C., Fiddler, M. N., Bililign, S., Jimenez, J. L., Kurtén, T., Weinheimer, A. J., Jaegle, L., and Thornton, J. A.: Flight Deployment of a High-Resolution Time-of-Flight Chemical Ionization Mass Spectrometer: Observations of Reactive Halogen and Nitrogen Oxide Species, J. Geophys. Res.-Atmos., 123, 7670–7686, https://doi.org/10.1029/2017JD028082, 2018.
Lopez-Hilfiker, F. D., Iyer, S., Mohr, C., Lee, B. H., D'Ambro, E. L., Kurtén, T., and Thornton, J. A.: Constraining the sensitivity of iodide adduct chemical ionization mass spectrometry to multifunctional organic molecules using the collision limit and thermodynamic stability of iodide ion adducts, Atmos. Meas. Tech., 9, 1505–1512, https://doi.org/10.5194/amt-9-1505-2016, 2016.
McDonald, B. C., De Gouw, J. A., Gilman, J. B., Jathar, S. H., Akherati, A., Cappa, C. D., Jimenez, J. L., Lee-Taylor, J., Hayes, P. L., McKeen, S. A., Cui, Y. Y., Kim, S. W., Gentner, D. R., Isaacman-VanWertz, G., Goldstein, A. H., Harley, R. A., Frost, G. J., Roberts, J. M., Ryerson, T. B., and Trainer, M.: Volatile chemical products emerging as largest petrochemical source of urban organic emissions, Science, 359, 760–764, https://doi.org/10.1126/science.aaq0524, 2018.
Mohr, C., Thornton, J. A., Heitto, A., Lopez-Hilfiker, F. D., Lutz, A., Riipinen, I., Hong, J., Donahue, N. M., Hallquist, M., Petäjä, T., Kulmala, M., and Yli-Juuti, T.: Molecular identification of organic vapors driving atmospheric nanoparticle growth, Nat. Commun., 10, 1–7, https://doi.org/10.1038/s41467-019-12473-2, 2019.
Pfannerstill, E. Y., Arata, C., Zhu, Q., Schulze, B. C., Ward, R., Woods, R., Harkins, C., Schwantes, R. H., Seinfeld, J. H., Bucholtz, A., Cohen, R. C., and Goldstein, A. H.: Temperature-dependent emissions dominate aerosol and ozone formation in Los Angeles, Science, 384, 1324–1329, https://doi.org/10.1126/science.adg8204, 2024.
Rappé, A. K., Casewit, C. J., Colwell, K. S., Goddard, W. A., and Skiff, W. M.: UFF, a Full Periodic Table Force Field for Molecular Mechanics and Molecular Dynamics Simulations, J. Am. Chem. Soc., 114, 10024–10035, https://doi.org/10.1021/ja00051a040, 1992.
Rissanen, M. P., Mikkilä, J., Iyer, S., and Hakala, J.: Multi-scheme chemical ionization inlet (MION) for fast switching of reagent ion chemistry in atmospheric pressure chemical ionization mass spectrometry (CIMS) applications, Atmos. Meas. Tech., 12, 6635–6646, https://doi.org/10.5194/amt-12-6635-2019, 2019.
Riva, M., Rantala, P., Krechmer, J. E., Peräkylä, O., Zhang, Y., Heikkinen, L., Garmash, O., Yan, C., Kulmala, M., Worsnop, D., and Ehn, M.: Evaluating the performance of five different chemical ionization techniques for detecting gaseous oxygenated organic species, Atmos. Meas. Tech., 12, 2403–2421, https://doi.org/10.5194/amt-12-2403-2019, 2019.
Riva, M., Pospisilova, V., Frege, C., Perrier, S., Bansal, P., Jorga, S., Sturm, P., Thornton, J. A., Rohner, U., and Lopez-Hilfiker, F.: Evaluation of a reduced-pressure chemical ion reactor utilizing adduct ionization for the detection of gaseous organic and inorganic species, Atmos. Meas. Tech., 17, 5887–5901, https://doi.org/10.5194/amt-17-5887-2024, 2024.
Robinson, M. A., Roberts, J. M., Neuman, J. A., Jernigan, C. M., Xu, L., Coggon, M. M., Stockwell, C. E., Warneke, C., Peischl, J., Gilman, J. B., Lamplugh, A., Rollins, A. W., Zuraski, K., Rivera-Rios, J. C., Wang, Y., Ng, N. L., Liu, S., Brown, S. S., and Veres, P. R.: Online Calibration of a Chemical Ionization Mass Spectrometer for Multifunctional Biogenic Organic Nitrates, ACS ES&T Air, 1, 1066–1083, https://doi.org/10.1021/acsestair.4c00056, 2024.
Saunders, S. M., Jenkin, M. E., Derwent, R. G., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): tropospheric degradation of non-aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 161–180, https://doi.org/10.5194/acp-3-161-2003, 2003.
Scott, D. W.: On optimal and data-based histograms, Biometrika, 66, 605–610, https://doi.org/10.1093/biomet/66.3.605, 1979.
Silverman, B. W.: Density Estimation for Statistics and Data Analysis, Routledge, 186 pp., https://doi.org/10.1201/9781315140919, 2018.
Slusher, D. L., Huey, L. G., Tanner, D. J., Flocke, F. M., and Roberts, J. M.: A thermal dissociation – Chemical ionization mass spectrometry (TD-CIMS) technique for the simultaneous measurement of peroxyacyl nitrates and dinitrogen pentoxide, J. Geophys. Res.-Atmos., 109, 1–13, https://doi.org/10.1029/2004JD004670, 2004.
Song, M., He, S., Li, X., Liu, Y., Lou, S., Lu, S., Zeng, L., and Zhang, Y.: Optimizing the iodide-adduct chemical ionization mass spectrometry (CIMS) quantitative method for toluene oxidation intermediates: experimental insights into functional-group differences, Atmos. Meas. Tech., 17, 5113–5127, https://doi.org/10.5194/amt-17-5113-2024, 2024.
Stockwell, C. E., Coggon, M. M., Schwantes, R. H., Harkins, C., Verreyken, B., Lyu, C., Zhu, Q., Xu, L., Gilman, J. B., Lamplugh, A., Peischl, J., Robinson, M. A., Veres, P. R., Li, M., Rollins, A. W., Zuraski, K., Baidar, S., Liu, S., Kuwayama, T., Brown, S. S., McDonald, B. C., and Warneke, C.: Urban ozone formation and sensitivities to volatile chemical products, cooking emissions, and NOx upwind of and within two Los Angeles Basin cities, Atmos. Chem. Phys., 25, 1121–1143, https://doi.org/10.5194/acp-25-1121-2025, 2025.
Tham, Y. J., He, X. C., Li, Q., Cuevas, C. A., Shen, J., Kalliokoski, J., Yan, C., Iyer, S., Lehmusjärvi, T., Jang, S., Thakur, R. C., Beck, L., Kemppainen, D., Olin, M., Sarnela, N., Mikkilä, J., Hakala, J., Marbouti, M., Yao, L., Li, H., Huang, W., Wang, Y., Wimmer, D., Zha, Q., Virkanen, J., Gerard Spain, T., O'Doherty, S., Jokinen, T., Bianchi, F., Petäjä, T., Worsnop, D. R., Mauldin, R. L., Ovadnevaite, J., Ceburnis, D., Maier, N. M., Kulmala, M., O'Dowd, C., Maso, M. D., Saiz-Lopez, A., and Sipilä, M.: Direct field evidence of autocatalytic iodine release from atmospheric aerosol, P. Natl. Acad. Sci. USA, 118, 1–8, https://doi.org/10.1073/pnas.2009951118, 2021.
Travis, K. R., Crawford, J. H., Soja, A. J., Gargulinski, E. M., Moore, R. H., Wiggins, E. B., Diskin, G. S., DiGangi, J. P., Nowak, J. B., Halliday, H., Yokelson, R. J., McCarty, J. L., Simpson, I. J., Blake, D. R., Meinardi, S., Hornbrook, R. S., Apel, E. C., Hills, A. J., Warneke, C., Coggon, M. M., Rollins, A. W., Gilman, J. B., Womack, C. C., Robinson, M. A., Katich, J. M., Peischl, J., Gkatzelis, G. I., Bourgeois, I., Rickly, P. S., Lamplugh, A., Dibb, J. E., Jimenez, J. L., Campuzano-Jost, P., Day, D. A., Guo, H., Pagonis, D., Wennberg, P. O., Crounse, J. D., Xu, L., Hanisco, T. F., Wolfe, G. M., Liao, J., St. Clair, J. M., Nault, B. A., Fried, A., and Perring, A. E.: Emission Factors for Crop Residue and Prescribed Fires in the Eastern US During FIREX-AQ, J. Geophys. Res.-Atmos., 128, https://doi.org/10.1029/2023JD039309, 2023.
Vasquez, K. T., Crounse, J. D., Schulze, B. C., Bates, K. H., Teng, A. P., Xu, L., Allen, H. M., and Wennberg, P. O.: Rapid hydrolysis of tertiary isoprene nitrate efficiently removes NOx from the atmosphere, P. Natl. Acad. Sci. USA, 117, 33011–33016, https://doi.org/10.1073/PNAS.2017442117, 2020.
Veres, P. R. and Roberts, J. M.: Development of a photochemical source for the production and calibration of acyl peroxynitrate compounds, Atmos. Meas. Tech., 8, 2225–2231, https://doi.org/10.5194/amt-8-2225-2015, 2015.
Veres, P. R., Roberts, J. M., Wild, R. J., Edwards, P. M., Brown, S. S., Bates, T. S., Quinn, P. K., Johnson, J. E., Zamora, R. J., and de Gouw, J.: Peroxynitric acid (HO2NO2) measurements during the UBWOS 2013 and 2014 studies using iodide ion chemical ionization mass spectrometry, Atmos. Chem. Phys., 15, 8101–8114, https://doi.org/10.5194/acp-15-8101-2015, 2015.
Wang, C., Liggio, J., Wentzell, J. J. B., Jorga, S., Folkerson, A., and Abbatt, J. P. D.: Chloramines as an important photochemical source of chlorine atoms in the urban atmosphere, P. Natl. Acad. Sci. USA, 120, 2017, https://doi.org/10.1073/pnas.2220889120, 2023.
Wang, M., He, X.-C., Finkenzeller, H., Iyer, S., Chen, D., Shen, J., Simon, M., Hofbauer, V., Kirkby, J., Curtius, J., Maier, N., Kurtén, T., Worsnop, D. R., Kulmala, M., Rissanen, M., Volkamer, R., Tham, Y. J., Donahue, N. M., and Sipilä, M.: Measurement of iodine species and sulfuric acid using bromide chemical ionization mass spectrometers, Atmos. Meas. Tech., 14, 4187–4202, https://doi.org/10.5194/amt-14-4187-2021, 2021.
Waskom, M.: Seaborn: Statistical Data Visualization, J. Open Source Softw., 6, 3021, https://doi.org/10.21105/joss.03021, 2021.
Wennberg, P. O., Bates, K. H., Crounse, J. D., Dodson, L. G., McVay, R. C., Mertens, L. A., Nguyen, T. B., Praske, E., Schwantes, R. H., Smarte, M. D., St Clair, J. M., Teng, A. P., Zhang, X., and Seinfeld, J. H.: Gas-Phase Reactions of Isoprene and Its Major Oxidation Products, Chem. Rev., 118, 3337–3390, https://doi.org/10.1021/acs.chemrev.7b00439, 2018.
Xu, L., Coggon, M. M., Stockwell, C. E., Gilman, J. B., Robinson, M. A., Breitenlechner, M., Lamplugh, A., Crounse, J. D., Wennberg, P. O., Neuman, J. A., Novak, G. A., Veres, P. R., Brown, S. S., and Warneke, C.: Chemical ionization mass spectrometry utilizing ammonium ions (NH CIMS) for measurements of organic compounds in the atmosphere, Atmos. Meas. Tech., 15, 7353–7373, https://doi.org/10.5194/amt-15-7353-2022, 2022.
Yan, C., Nie, W., Äijälä, M., Rissanen, M. P., Canagaratna, M. R., Massoli, P., Junninen, H., Jokinen, T., Sarnela, N., Häme, S. A. K., Schobesberger, S., Canonaco, F., Yao, L., Prévôt, A. S. H., Petäjä, T., Kulmala, M., Sipilä, M., Worsnop, D. R., and Ehn, M.: Source characterization of highly oxidized multifunctional compounds in a boreal forest environment using positive matrix factorization, Atmos. Chem. Phys., 16, 12715–12731, https://doi.org/10.5194/acp-16-12715-2016, 2016.
Yuan, B., Koss, A. R., Warneke, C., Coggon, M., Sekimoto, K., and De Gouw, J. A.: Proton-Transfer-Reaction Mass Spectrometry: Applications in Atmospheric Sciences, Chem. Rev., 117, 13187–13229, https://doi.org/10.1021/acs.chemrev.7b00325, 2017.
Zaytsev, A., Breitenlechner, M., Koss, A. R., Lim, C. Y., Rowe, J. C., Kroll, J. H., and Keutsch, F. N.: Using collision-induced dissociation to constrain sensitivity of ammonia chemical ionization mass spectrometry (NH CIMS) to oxygenated volatile organic compounds, Atmos. Meas. Tech., 12, 1861–1870, https://doi.org/10.5194/amt-12-1861-2019, 2019.
Zhang, W., Xu, L., and Zhang, H.: Recent advances in mass spectrometry techniques for atmospheric chemistry research on molecular-level, Mass Spectrom. Rev., 1–44, https://doi.org/10.1002/mas.21857, 2023.