the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 22 Dec 2022
| 22 Dec 2022
Chemical ionization mass spectrometry utilizing ammonium ions (NH4+ CIMS) for measurements of organic compounds in the atmosphere
Matthew M. Coggon
Chelsea E. Stockwell
Jessica B. Gilman
Michael A. Robinson
Martin Breitenlechner
Aaron Lamplugh
John D. Crounse
Paul O. Wennberg
J. Andrew Neuman
Gordon A. Novak
Patrick R. Veres
Steven S. Brown
Carsten Warneke
We describe the characterization and field deployment of chemical ionization mass spectrometry (CIMS) using a recently developed focusing ion-molecule reactor (FIMR) and ammonium–water cluster () as the reagent ion (denoted as CIMS). We show that is a highly versatile reagent ion for measurements of a wide range of oxygenated organic compounds. The major product ion is the cluster with produced via ligand-switching reactions. Other product ions (e.g., protonated ion, cluster ion with , with H3O+, and with ) are also produced, but with minor fractions for most of the oxygenated compounds studied here. The instrument sensitivities (ion counts per second per part per billion by volume, cps ppbv−1) and product distributions are strongly dependent on the instrument operating conditions, including the ratio of ammonia (NH3) and H2O flows and the drift voltages, which should be carefully selected to ensure as the predominant reagent ion and to optimize sensitivities. For monofunctional analytes, the chemistry exhibits high sensitivity (i.e., >1000 cps ppbv−1) to ketones, moderate sensitivity (i.e., between 100 and 1000 cps ppbv−1) to aldehydes, alcohols, organic acids, and monoterpenes, low sensitivity (i.e., between 10 and 100 cps ppbv−1) to isoprene and C1 and C2 organics, and negligible sensitivity (i.e., <10 cps ppbv−1) to reduced aromatics. The instrumental sensitivities of analytes depend on the binding energy of the analyte– cluster, which can be estimated using voltage scanning. This offers the possibility to constrain the sensitivity of analytes for which no calibration standards exist. This instrument was deployed in the RECAP campaign (Re-Evaluating the Chemistry of Air Pollutants in California) in Pasadena, California, during summer 2021. Measurement comparisons against co-located mass spectrometers show that the CIMS is capable of detecting compounds from a wide range of chemical classes. The CIMS is valuable for quantification of oxygenated volatile organic compounds (VOCs) and is complementary to existing chemical ionization schemes.
- Article
(5729 KB) - Full-text XML
-
Supplement
(2698 KB) - BibTeX
- EndNote





Quantifying atmospheric volatile organic compounds (VOCs) and their oxidation products is critical for understanding the formation of ozone (O3) and organic aerosol (OA). However, this objective has been a long-standing challenge because of the sheer number and significant chemical complexity of organic compounds in the atmosphere (Goldstein and Galbally, 2007). Chemical ionization mass spectrometry (CIMS) is a widely used and rapidly developing technique to characterize atmospheric trace gases. The advantages of CIMS include fast time response, high selectivity and sensitivity, and detection linearity over a wide range of analyte mixing ratios. In CIMS, the analytes are ionized via ion-molecule reactions with a reagent ion, which is soft and largely preserves the identity of the analytes. The detection capability of CIMS depends on the selection of reagent ions, which are sensitive to different classes of organics. The commonly employed reagent ions include H3O+ to detect reduced and small functionalized VOCs (de Gouw and Warneke, 2007), I− to detect inorganics and polar and acidic organics (B. H. Lee et al., 2014; Robinson et al., 2022), CF3O− to detect organic peroxides and other multifunctional organics (Crounse et al., 2006; Xu et al., 2020), to detect organic acids (Nah et al., 2018), to detect highly oxygenated molecules (Ehn et al., 2014), and protonated amines to detect reactive radicals (Berndt et al., 2018). Exploring novel reagent ions is an active research area to expand the detection capability of CIMS and to provide precise measurements of atmospheric species with high sensitivity. These efforts enable a comprehensive description of the complex mixture of atmospheric organic compounds.
One ionization scheme under active development utilizes the ammonium ion () chemistry. Several recent studies have demonstrated its capability to detect a range of oxygenated organic compounds, including alcohols, aldehydes, ketones, and even the short-lived peroxy radicals (RO2) (Blake et al., 2006; Lindinger et al., 1998; Canaval et al., 2019; Hansel et al., 2018; Müller et al., 2020; Zaytsev et al., 2019; Berndt et al., 2018; Khare et al., 2022). One reason chemistry is attractive is that it detects oxygenated organic compounds in the positive mass spectrometer mode, in contrast to existing reagent ions (i.e., I−, CF3O−, and ) which are operated in negative mode. This offers the potential to rapidly switch between and H3O+ within the same instrument to detect both oxygenated and reduced organic compounds, respectively, without substantial alteration of the electric fields in the mass spectrometer. Zaytsev et al. (2019) and Müller et al. (2020) demonstrated the feasibility of such rapid switching in laboratory conditions. The application of CIMS in recent studies has largely focused on laboratory studies (Berndt et al., 2018; Zaytsev et al., 2019), but its deployment in field measurements and intercomparison with other analytical instruments are scarce (Khare et al., 2022).
The instrument design, including the ion source and the ion-molecule reactor (IMR), differs between studies. Hansel et al. (2018) applied ion chemistry in a PTR3 instrument (Breitenlechner et al., 2017) (i.e., –PTR3) and detected peroxy radicals and other products from cyclohexene ozonolysis with sensitivities up to 2.8×104 cps ppbv−1 in a free-jet flow system. Using a similar instrument, Zaytsev et al. (2019) calibrated 16 compounds, with a maximum sensitivity of 8.9×104 cps ppbv−1 for decanone. In both studies, the major reagent ion is , generated in a corona discharge ion source from a mixture of NH3 and H2O gas. Later, Müller et al. (2020) developed a method to produce using a mixture of water vapor and nitrogen in a hollow cathode glow discharge ion source, which is used in PTR-MS instruments with a traditional drift tube design that includes extraction plates between the hollow cathode ion source and drift tube. Canaval et al. (2019) used a selective reagent ionization time-of-flight mass spectrometer (SRI-ToF-MS) to produce via reaction of He+ and gas NH3. Different instrument designs affect the distribution of reagent ions (i.e., vs. vs. ), detection efficiency, and sensitivity.
In this study, we describe the performance of CIMS using a Tofwerk Vocus long time-of-flight mass spectrometer (Krechmer et al., 2018). We investigate the impacts of instrument conditions on the distribution of reagent ions and the instrumental sensitivities of 60 analytes from several chemical functional classes. Building upon extensive calibrations, we explore the dependence of sensitivity on the ion-molecule reaction rate constant and the binding energy of the analyte– cluster, aiming to derive a relationship to approximate the sensitivity of analytes for which no calibration standards exist. Further, this instrument was deployed during the RECAP campaign (Re-Evaluating the Chemistry of Air Pollutants in California) in Pasadena, California, during the summer of 2021. The instrument performance is further evaluated by comparison to several co-located mass spectrometers.
2.1 Instrument description
The instrument in this work is based on the Tofwerk Vocus, which utilizes a new ion source, a focusing ion-molecule reactor (FIMR), and a long time-of-flight mass spectrometer (LToF). A detailed description of the Vocus can be found in Krechmer et al. (2018). Here we briefly summarize the generation of reagent ions and instrument operation conditions.
The chemical ionization gas entering the ion source is produced by mixing NH3 and H2O from two streams: a 20 sccm flow of water vapor from the headspace of a liquid water reservoir (denoted as H2O flow) and an additional 1 sccm from the headspace of a reservoir containing 0.5 % (vol %) ammonium hydroxide water solution (denoted as NH3 flow, which contains both NH3 and H2O). The ion source consists of two conical surfaces with a voltage gradient. A plasma is produced between the conical surfaces, which primarily ionizes water molecules, producing H3O+. The discharge current is regulated at 2.0 mA. Because NH3 has a larger proton affinity than H2O, the proton transfer reaction (Reaction R1) produces , which then readily clusters with abundant H2O to produce the targeted reagent ion (Reaction R2). Besides H2O, can also cluster with NH3 to produce (Reaction R3). The abundance of H2O in the ion source also leads to formation of cluster ions (Reaction R4). Overall, several ions, (ligand X =NH3 and H2O, ) and , are generated from the ion source and can potentially serve as reagent ions.
The reagent gas flow pushes the ions into the FIMR where they subsequently react with analytes. Sample air enters the FIMR through a 25 mm long PEEK capillary (ID 0.18 mm). The sample flow rate is ∼100 sccm at FIMR pressure of 3 mbar in this study. The FIMR is a 100 mm long glass tube with an inner diameter of 10 mm. A quadrupole radio frequency (RF) field is applied to the FIMR to collimate ions into a narrow beam, significantly enhancing the sensitivity (Krechmer et al., 2018). The FIMR conditions, including temperature, pressure, drift voltage, and the ratio of NH3 to H2O into the ion source, all control the degree of cluster-ion formation, the distribution of reagent ions, and ultimately the sensitivity, as will be discussed in Sect. 3.2 and 3.3. Ions from the FIMR travel through a big segmented quadrupole (BSQ). The BSQ serves as a high-pass band filter to reduce the signal intensity of reagent ions while simultaneously guiding ions into the time-of-flight mass spectrometer. As a result of this filtering, the observed distribution of reagent ions is not the same as the actual distribution in the FIMR (Krechmer et al., 2018). After the BSQ, the ions travel through the primary beam region and are eventually detected by the long time-of-flight mass spectrometer with a mass resolution (full width at half maximum, FWHM) up to 8000 at 100. The extraction frequency of the ToF is set at 17.5 kHz.
2.2 Laboratory characterization
We calibrate the instrumental sensitivities (cps ppbv−1) of 60 organic compounds (Table 1) using two methods, standard gas cylinders (SGC) and a home-built liquid calibration unit using either water or hexane as solvent (LCU-W and LCU-H), as described in Sect. S1.1 in the Supplement. We find minimal dependence of sensitivity on sample relative humidity (RH) as shown in Fig. S1 in the Supplement, consistent with Khare et al. (2022) and observations made when running the Vocus in H3O+ mode (Krechmer et al., 2018). This is mainly because a large amount of water vapor (20 sccm) is deliberately added to the FIMR. As an example, the water amount in a 100 sccm ambient sample under 25 ∘C and 100 % RH is only 15 % of the added 20 sccm water vapor to the FIMR, assuming no water vapor loss in both processes. The instrument background is determined by passing ambient air through a platinum catalytic converter heated to 400 ∘C. The detection limit is defined as 3 standard deviations of measurement background for 1 s integration time.
Table 1Sensitivities (cps ppbv−1), background (cps), and detection limits (pptv for a 1 s integration time) of CIMS.
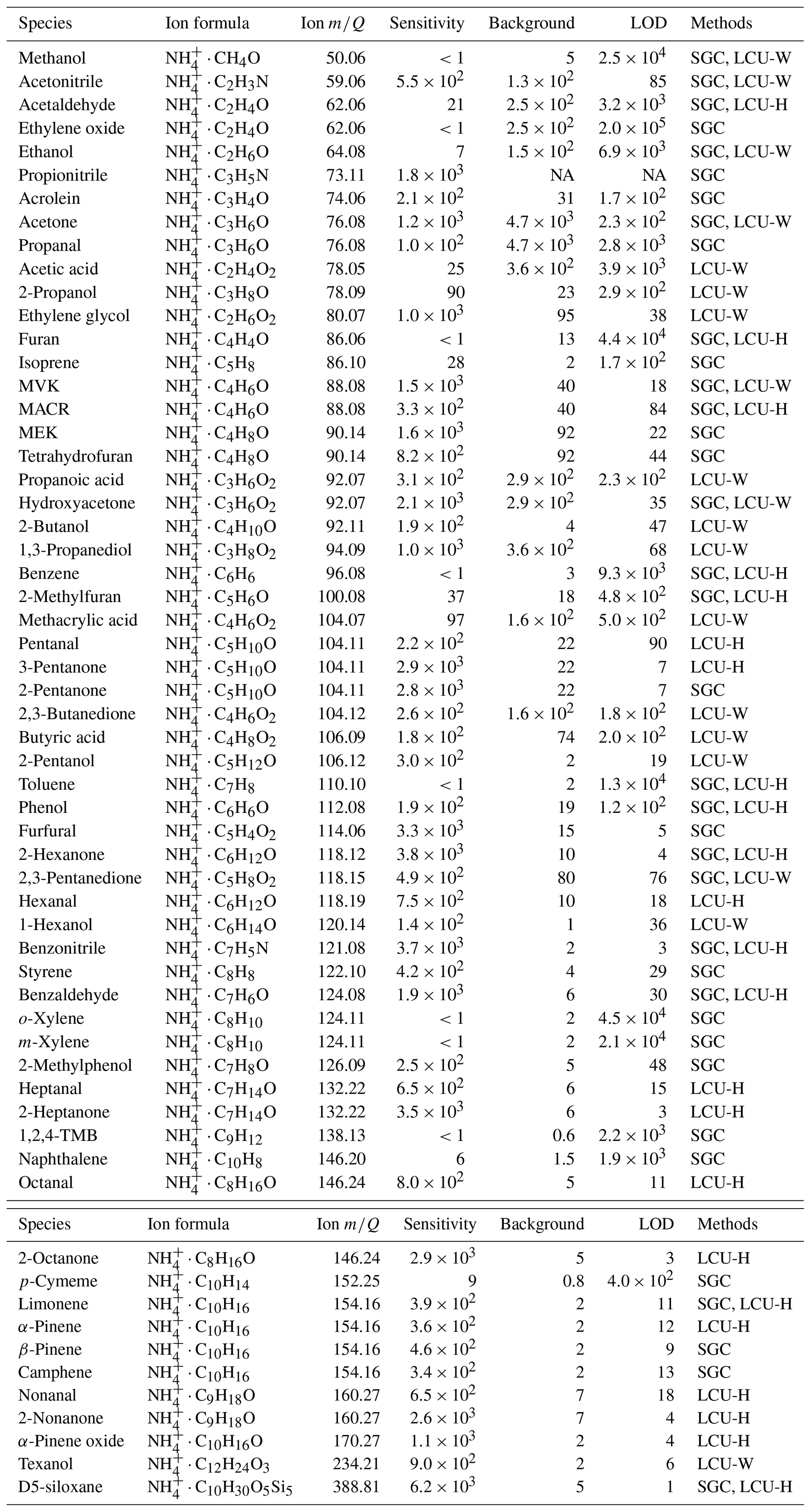
NA: not available.
During transport, ions get lost in the BSQ, in the ion guides, and in the extraction region of the ToF. We quantify the mass-dependent transmission efficiency relative to the reagent ion by introducing a series of compounds spanning a range of molecular weight (32–370 ) in a large enough quantity to deplete the fraction of reagent ions by ∼ 20 %–30 % (Huey et al., 1995; Heinritzi et al., 2016). The ratio of the increase in the product ions to the decrease in the reagent ion indicates the relative transmission efficiency between these two masses. A detailed derivation can be found in Sect. S1.2.
We have performed laboratory tests and measured the product distribution of 60 organic compounds. The product ions are identified by sampling the headspace of a small vial containing pure analyte. Distance is kept between the instrument inlet and the vial to keep analyte concentration low. Ions correlating with the parent ion () with r2 larger than 0.95 and accounting for larger than 1 % of the parent ion signal are considered to be product ions from the analyte. The distribution of product ions depends on the distribution of reagent ions. In this test, we maintain the ratio between 5 and 20. Under this condition, the ion chemistry of is negligible.
To probe the stability of product ions, we performed voltage scanning tests following the procedure outlined in Lopez-Hilfiker et al. (2016) and Zaytsev et al. (2019). In brief, we vary the voltage gradient (ΔV) between FIMR back and skimmer while keeping the voltage gradient between FIMR front and back constant. A larger ΔV increases the collisional energy, causes stronger collision-induced dissociation of product ions, and tends to decrease the signal of product ions. We define ΔV50 as the voltage gradient at which the parent ion signal drops to half of the maximum signal. This ΔV50 represents the electric field required to break each and is therefore related to the binding energy of . Further, ΔV50 is converted to the kinetic energy of in the center of mass (i.e., KEcm,50) using a parameterization of mass-dependent ion mobility (Zaytsev et al., 2019, and details in the Sect. S1.3). KEcm,50 is a measure of the stability.
2.3 Field deployment
The CIMS was deployed during the RECAP campaign (Re-Evaluating the Chemistry of Air Pollutants in California) in Pasadena, California, from August–September, 2021. The ground sampling site is located on the campus of the California Institute of Technology, which is only one block away from the original sampling site during the 2010 CalNex study (Ryerson et al., 2013). The instrument inlet was set up on a tower 10 m above the ground. The instrument was operated to sample gas phase from 10 to 19 August. Later, the instrument was coupled to a Vocus Inlet for Aerosol (VIA) to automatically switch sampling between gas and particle phases. This study will focus on the gas-phase sampling period. Co-located instruments of relevance to this study include a proton-transfer-reaction mass spectrometer (PTR-MS) (Yuan et al., 2016; de Gouw and Warneke, 2007) and a gas chromatography mass spectrometer (GC-MS) (Lerner et al., 2017). A CF3O− chemical ionization mass spectrometer (CF3O− CIMS) (Crounse et al., 2006; Allen et al., 2022) was deployed at a different site on campus, which is ∼800 m away from the CIMS.
3.1 Overview of ion chemistry
The target primary reagent ion is , which ionizes analytes (A) primarily via ligand-switching reactions (Reaction R5) to form product ion . As analogous to proton affinity, we define affinity as the negative of the enthalpy change in the reaction between and an analyte. If an analyte has a larger affinity than H2O, Reaction (R5) is exothermic and will occur at a rate close to the collision limit when the difference in affinity is sufficiently large (Adams et al., 2003). Otherwise, the ligand-switching reaction is endothermic. The energy imparted via the drift voltage could aid the endothermic reaction to overcome the energy barrier, but the instrument sensitivity in these instances is expected to be low. Besides the target primary ion , ions (X =NH3 and H2O) and () are observed because the chemical ionization gas supply is a mixture of NH3 and H2O. These ions can also serve as reagent ions. Compared to , ionization is softer because the H2O acts as a third body which dissipates some reaction energy. The reactivities of and are also expected to be different, as the NH3 has a larger affinity than H2O (i.e., 108 vs. 86 kJ mol−1, NIST Chemistry WebBook). Therefore, the presence of multiple reagent ions will complicate the ionization chemistry and the interpretation of the mass spectra. To avoid such complication, the instrument conditions need to be carefully optimized to ensure exists as the dominant ion reacting with analytes.
3.2 Modeling the distribution of reagent ions
The distribution of the reagent ions is controlled by several factors, including the FIMR reduced electric field (), temperature (T), pressure (P), the H2O mixing ratio (), and the ratio of NH3 to H2O (). Many of these factors are interdependent – e.g., the depends on pressure and temperature. To unravel the influences of these factors on the distribution of reagent ions, we develop a kinetic model. The model includes a series of reactions between two ions ( and H3O+) and two neutral molecules (NH3 and H2O). Clusters containing up to three molecules are considered, which leads to a total of 14 different ion clusters (Fig. S4). The ion-molecule cluster reaction rate constant (i.e., forward reaction with kforward) is calculated using the parameterization in Su (1994), assuming the reaction proceeds at the collision limit. The reaction rate constant of the declustering reaction (i.e., reverse reaction with kreverse) is calculated using kforward and the equilibrium constant Keq. kreverse for Reaction (R6), for example, is expressed by Eq. (1), where M0 represents the number density (cm−3) under standard condition and Keq represents the reaction equilibrium constant. Keq is calculated using Eq. (2), where ΔH0 and ΔS0 represent the enthalpy and entropy changes of the reaction at standard condition, respectively (Table S1 in the Supplement), and Teff represents the effective temperature of the ions in the FIMR. Teff is calculated using Eq. (3) (de Gouw et al., 2003), where kB is the Boltzmann constant, , mA, and mbuffer are the masses of the ion I+, the neutral analyte A, and the buffer gas, respectively, and the νd is the drift velocity of ion IA+. νd is calculated using Eq. (4), where μ0 is the reduced mobility of IA+ calculated based on the parameterization in Steiner et al. (2014), P and T are the FIMR pressure and temperature, respectively, and E is the electric field strength across the FIMR.
The influences of different FIMR conditions (i.e., , T, P, , and ) on the distribution of reagent ions are intertwined. To visualize their impacts, we first conduct simulations covering wide ranges of all five factors to locate the condition yielding the largest fraction of in total ions (denoted as ). The optimized condition is Td (Townsend), T=330 K, P=5 mbar, , and %. Then, we conduct simulations using the optimal condition as a start point and vary one factor at a time while holding the other four constant to investigate the impact of each factor on the distribution.
The simulation results are shown in Fig. 1. Figure 1a shows that the reduced electric field () strongly impacts the distribution of reagent ions. When the is below 40 Td, is the dominant ion because the electric field is too weak to decluster. When the is above 80 Td, is dominant because the electric field results in strong declustering and because NH3 has higher proton affinity than H2O. Only within a narrow window (50–65 Td) is the target reagent ion the most abundant ion. Within this window, several other ions also exist, including , , and . The FIMR P and T impact the distribution (Fig. 1b and c) through a similar mechanism as , as smaller P and larger T result in larger . As a result, also exhibits a non-monotonic dependence on the FIMR P and T. The impact of H2O mixing ratio in the FIMR () on the distribution is shown in Fig. 1d. The initially increases with the , reaches a maximum when is roughly 0.16–0.18, and then decreases with increasing . This trend is because low limits the supply of H2O to cluster with and high favors the formation of larger clusters. To illustrate, Fig. 1d shows that as increases, the fraction of smaller clusters (i.e., ) decreases, but the fraction of larger clusters (i.e., and ) increases. Lastly, the ratio has a strong impact on the cluster ion distribution (Fig. 1e). A low ratio (<0.2 %) results in an insufficient supply of and therefore ions dominate. A high ratio (>0.55 %) causes to mainly cluster with NH3, producing large amounts of .
Evaluation of the kinetic simulation results by experimental observations is desirable but challenging. One challenge is that the distribution of reagent ions cannot be measured because the BSQ serves as a high-pass band filter which reduces the signal intensity of reagent ions. Another challenge is that voltages in the ion transfer region between the drift tube and the mass analyzer can change the distribution of reagent ions, which causes the measured distribution to be different from that in the FIMR (Krechmer et al., 2018; Breitenlechner et al., 2022; Yuan et al., 2016). Overall, the simulation results illustrate the controlling effects of FIMR conditions on the distribution of reagent ions. The determination of FIMR conditions is eventually based on experimental calibration of instrumental sensitivity, which can be guided by the modeled distribution of reagent ions, as discussed in the next section.
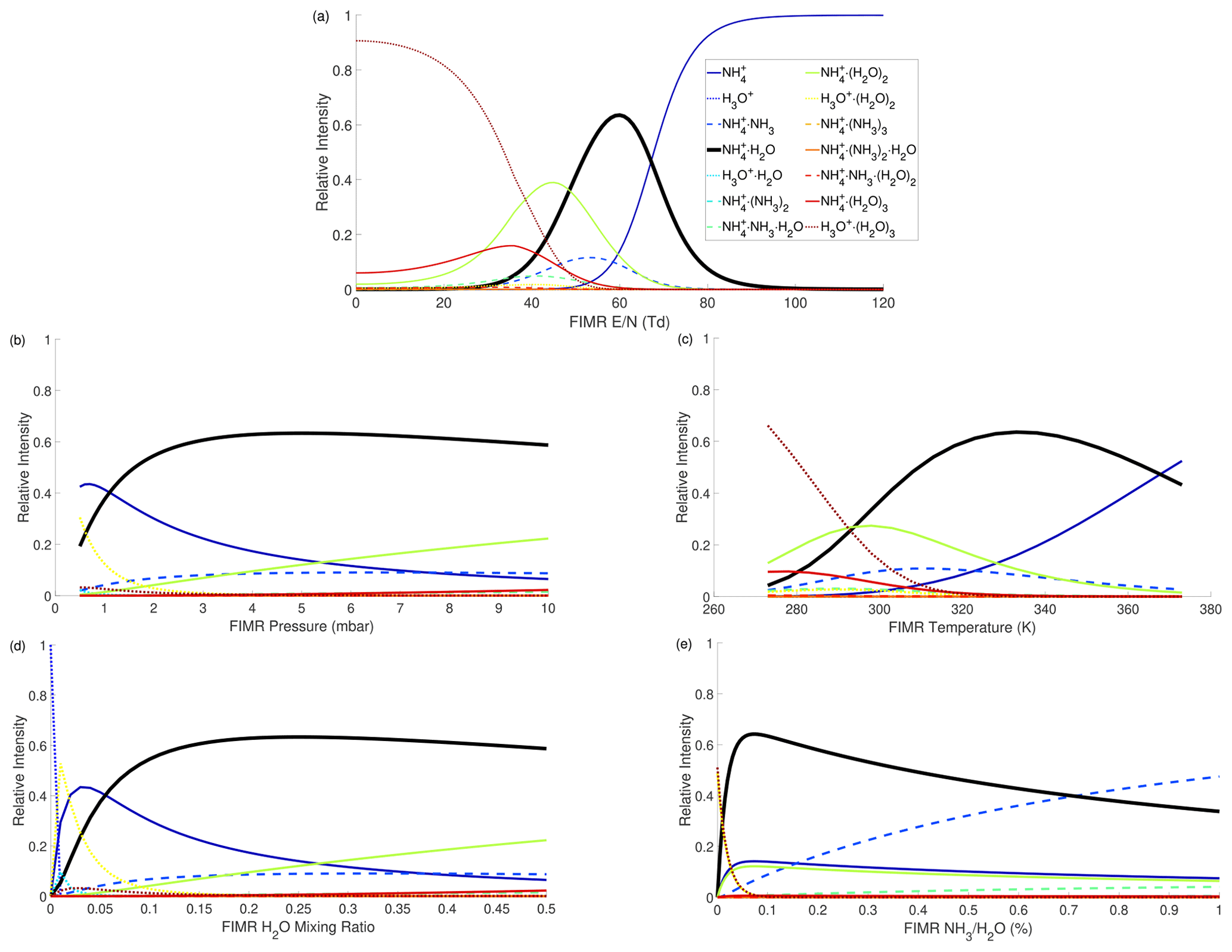
Figure 1The dependence of modeled distribution of reagent ions on FIMR conditions. (a) ; (b) P; (c) T; (d) H2O mixing ratio; (e) . In each panel, the other four factors are held constant at the following conditions: Td, P=5 mbar, T=330 K, H2O mixing ratio =0.25, %. Because the impacts of these factors are intertwined, each panel will change if the other four factors are at different values, as shown in an example in Fig. S5.
3.3 Dependence of sensitivities on FIMR conditions
While the above section modeled the dependence of the distribution of reagent ions on FIMR conditions, in this section we experimentally evaluate the dependence of analyte sensitivities on FIMR conditions. The analyte sensitivity depends not only on the distribution of reagent ions, but also other factors, including the number density of analytes in the FIMR, ion-molecular reaction time, stability of the product ion, and the transmission efficiency of product ions, as discussed below. Similar to the analysis in kinetic modeling, we experimentally vary one factor while holding the others constant.
Figure 2a shows the impacts of on sensitivities of representative analytes. The is varied by ramping the FIMR front voltage from 100 to 600 V, while holding the FIMR back voltage at 5 V. Under FIMR pressure and temperature of 3 mbar and 313 K, respectively, the ranges from 13 to 83 Td. The dependence of sensitivities on follows a similar trend of the modeled distribution of (Fig. 1a). The sensitivities initially increase with increasing , partly because of more reagent ion . As keeps increasing, declusters into , so less causes a decrease in sensitivities. Besides changing the distribution of the reagent ions, changing influences the sensitivity via other mechanisms, including the extent of declustering of and the focusing effect of ions in the FIMR. Krechmer et al. (2018) show that the higher better focuses ions to the central axis of the reactor and increases the sensitivity. This may explain the uptick in sensitivities when increases from 80 to 90 Td, which is not observed in the modeled . Overall, the observed dependence of sensitivities on is a superposition of at least three effects: focusing effects and the extent of declustering of both reagent ions and product ions.
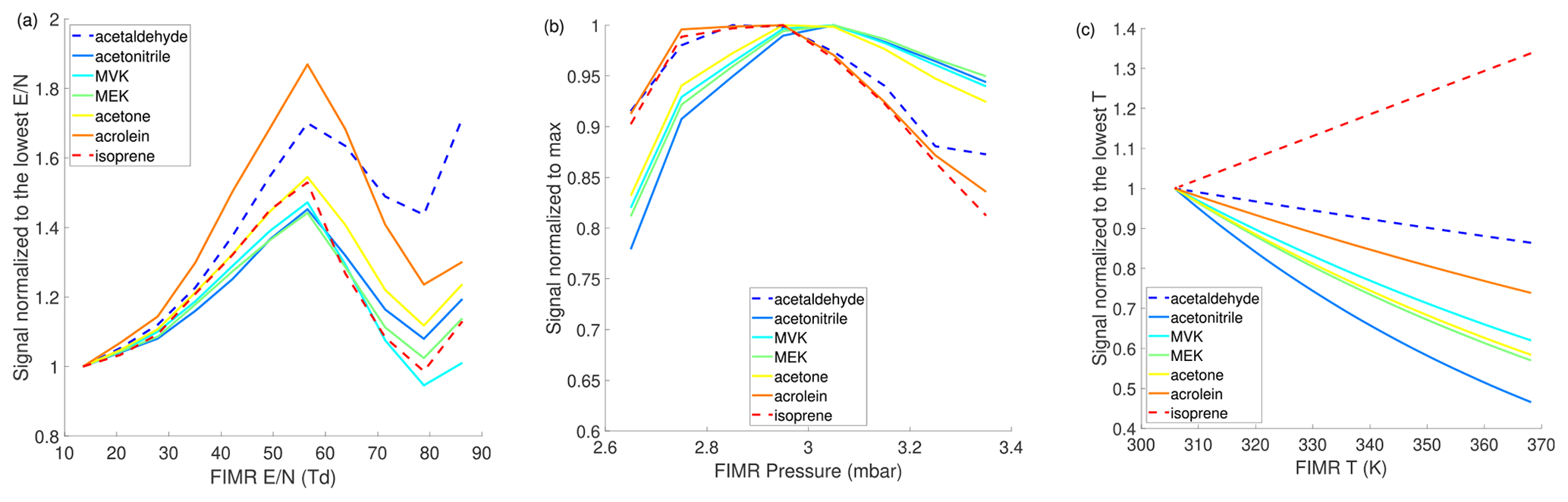
Figure 2Dependence of instrument sensitivities of representative species on FIMR conditions. (a) ; (b) P; (c) T. The range of in panel (a) is obtained by varying the drift voltage while maintaining the P and T at 3 mbar and 313 K, respectively. Analytes with sensitivities lower than 50 cps ppbv−1 are shown in dashed lines. The parent ion is used to quantify the sensitivity.
The effects of FIMR pressure on sensitivities are shown in Fig. 2b. The sensitivities exhibit a non-monotonic dependence on FIMR pressure, in a similar manner as the reagent ion does (Fig. 1b), suggesting the pressure-dependent sensitivities are related to the pressure-dependent distribution of reagent ions. In addition, higher pressure increases the number density of analyte molecules in the FIMR, which tends to increase the sensitivity. However, this effect is smaller than the effect of changing reagent ion on sensitivities, as Fig. 2b shows that the sensitivities decrease with increasing pressure beyond 3 mbar.
The effects of FIMR temperature on sensitivities are shown in Fig. 2c. Among the seven compounds tested here, the sensitivities of six oxygenated compounds exhibit a negative dependence on the temperature between 310 and 370 K. The reduced VOC, isoprene, exhibits a positive dependence. Similar to isoprene, α-pinene sensitivity also increases with temperature in the 303–350 K window as recently reported in Khare et al. (2022). Here we examine the opposite trends of temperature-dependent sensitivity between acetone and α-pinene because their affinities are available in the literature (Sect. S4). α-Pinene has an affinity smaller than that of H2O (i.e., 75 vs. 86 kJ mol−1 from Canaval et al., 2019), resulting in the ligand-switching reaction between α-pinene and being endothermic. Therefore, the reaction is promoted under higher temperature, which enhances the sensitivity. In contrast, the ligand-switching reaction between acetone and is exothermic because acetone has a larger affinity than H2O (i.e., 110 vs. 86 kJ mol−1 from Canaval et al., 2019). For exothermic reactions (ΔH is negative), higher temperature leads to smaller Keq (Eq. 2), a smaller ratio, and hence lower sensitivity. To better understand the temperature-dependent sensitivities, we add the reversible reactions of acetone and α-pinene with to the kinetic model depicted in Fig. S4 and simulate the dependence of their sensitivities on temperature. As shown in Fig. S6, the model can reproduce the observed dependence of their sensitivities on temperature. The affinity of isoprene is not available, but it is expected to be even smaller than α-pinene, given that the isoprene sensitivity is 10 times smaller than that of α-pinene. Thus, the reaction between isoprene and is likely also endothermic, causing the increasing sensitivity with higher temperature as shown in Fig. 2c.
The effects of the ratio on sensitivities are experimentally tested by simultaneously varying the flow rates of NH3 and H2O, while keeping the total flow rate constant. Because the NH3 flow is a mixture of NH3 and H2O, the accurate flow rate of NH3 is unknown. To approximate the ratio, we use the observed ratio of and because these three ions have similar transmission efficiency and their relative abundance directly depends on the ratio. Figure 3 shows the dependence of sensitivities of nearly 50 analytes on the . For the majority of compounds, their sensitivities initially increase with and then show a decreasing trend. This trend is caused by the fact that the initial increase in favors the formation of and hence higher sensitivity, but high produces more clusters, leading to reduced sensitivity (Fig. 1e). Taking acetone as an example, its affinity (110 kJ mol−1) is higher than that of H2O (86 kJ mol−1) but close to that of NH3 (108 kJ mol−1). As a result, the ligand-switching reaction between acetone and is less favorable than that between acetone and . The sensitivities of several compounds, including D5-siloxane, texanol, and several monoterpenes, exhibit a monotonic increase with the ratio within the tested range but will likely decrease at a higher ratio. The maximum sensitivity occurs at different ratios for different compounds, likely because they have different reactivities towards and .
Unlike the other four factors (i.e., , T, P, and ) which can be accurately controlled, the ratio and the resultant ratio change over time owing to the aging effects within the solution that supplies NH3. In the current approach to supply the chemical ionization gas, the ratio is controlled by the combination of the concentration of ammonium hydroxide aqueous solution and flow rates from the water and ammonium hydroxide reservoirs. Because NH3 is more volatile than H2O, the concentration of the ammonium hydroxide water solution decrease over time, resulting in a decreasing trend of over a timescale of weeks. In addition, the temperature variation of the ammonium hydroxide water solution changes the partitioning of NH3 and hence the ratio. One approach to compensate for the NH3 loss is to adjust the flow rate from the ammonium hydroxide reservoir to maintain a relatively constant ratio. Future studies exploring approaches to reliably supply chemical ionization gas are warranted. In summary, the instrument sensitivities should be calibrated as a function of the ratio. Then, the optimal range can be selected based on the analytes of interest.
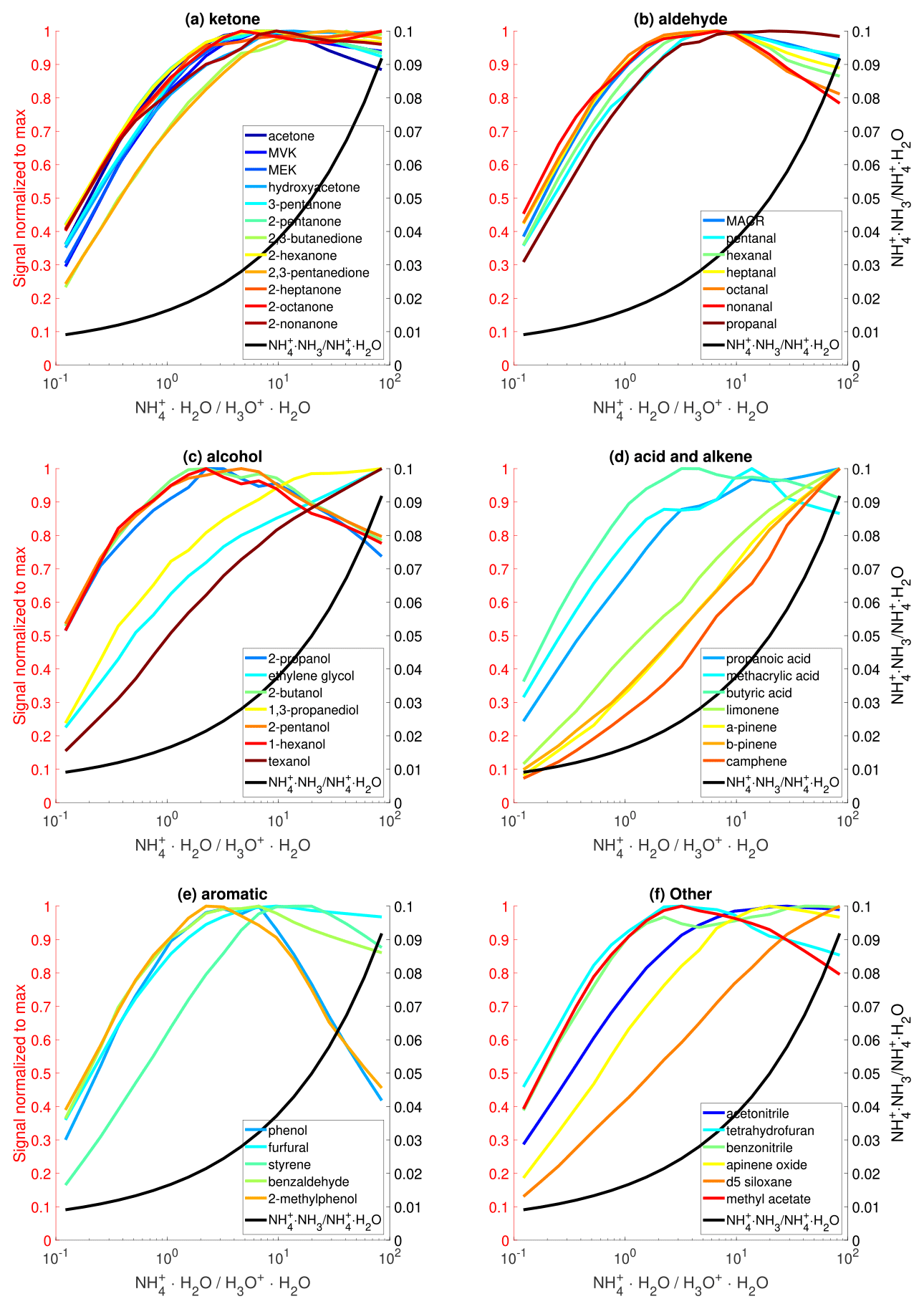
Figure 3The effects of reagent ion distribution on sensitivities of various organic species. The sensitivity of each analyte is normalized to the corresponding maximum value. Only analytes with sensitivity larger than 50 cps ppbv−1 are shown here.
The impacts of various FIMR conditions on instrument sensitivities are highly intertwined. The relationship between instrument sensitivity and individual FIMR condition shown in Fig. 2 could change when other FIMR conditions change. The optimal FIMR conditions should be explored collectively and systematically. The optimal condition for our instrument is FIMR drift voltage 55 Td, 3 mbar, 40 ∘C, 1 sccm from 0.5 % ammonium hydroxide aqueous solution, and 20 sccm water vapor. A temperature value that is slightly higher than ambient temperature is chosen for control purposes.
3.4 Product distributions from the ion-molecule reactions
The desired reagent ion is and the desired ion-molecule reaction is the ligand-switching reaction between and analyte A, which produces cluster as the parent ion (Reaction R5). However, the presence of several reagent ions in the FIMR and the declustering of in the electric field induce a variety of reactions and cause complex product distributions. Besides the target parent ion , we observe the protonated product (AH+), analyte clusters (, , and ), and fragmentation products. The potential ion-molecule reactions and product ions can be generally expressed by Reactions (R7) and (R8).
Figure 4 shows the product distributions for all tested analytes grouped by their chemical class. The analyte sensitivities are represented by the circle size in the figure. Among all classes, acids, ketones, and nitriles have the most desirable product distribution, in which the fraction of parent ion in all product ions (denoted as ) is more than 90 %, with the exception of acetic acid. For 2-octanone and 2-nonanone, is the sole product ion. For the alcohols, the product distribution is diverse. 2-Propanol and 2-butanol have fragmentation products (), which account for ∼5 % of the total products, but the fragmentation mechanism is unclear. For the aldehydes, the generally accounts for more than 80 % of total product ions. The tends to increase with larger molecules, for example when comparing a homologous series of aldehydes (pentanal, hexanal, heptanal, octanal, and nonanal). Four monoterpenes studied here produce a significant amount of protonated product (AH+), which is comparable to that of . The causes of the product distributions of four monoterpenes are possibly explained by their proton affinity and affinity. Three monoterpenes including α-pinene, β-pinene, and camphene have smaller affinities than H2O (Table S2). Thus, their ligand-switching reactions with are endothermic and the production of is likely aided by the energetic collision energy imparted by the drift voltage. These three monoterpenes have higher proton affinity than NH3 (Table S2) so that can undergo internal proton transfer to produce , which breaks in the electric field and produces AH+. In contrast to the above three monoterpenes, limonene has larger affinity than H2O and smaller proton affinity than NH3 (Table S2). Thus, the ligand-switching reaction with is exothermic and the internal proton transfer reaction is thermodynamically unfavorable. For limonene, the C10H16H+ is likely produced from the declustering of in the electric fields. The energy released from the exothermic reaction together with that imparted via the drift voltage could even break into fragments , , and C6H12N+. As a result, limonene produces ∼20 % of fragmentation products. For reduced aromatics (toluene, o-xylene, m-xylene, 1,2,4-TMB, and p-cymene), AH+ is the dominant product and is negligible. The product distributions of reduced aromatics are puzzling because these analytes have lower proton affinity than NH3. Since their sensitivities are <2 cps ppbv−1, it is not recommended to use to quantify reduced aromatics. Compared to reduced aromatics, oxygenated aromatics have higher sensitivities and larger values of . For example, benzaldehyde, 2-methylphenol, and furfural have greater than 90 %.
For a number of analytes in this study, the production of is evident. This product complicates the interpretation of the mass spectra and introduces uncertainties in quantification because the same ion is produced from an analyte (A) clustering with and an analyte with chemical formula A+H2O clustering with . For example, the ion can be produced from either acetone (C3H6O) clustering with or 1,3-propanediol (C3H8O2) clustering with . Cluster ions with are not observed for any compound. Overall, the product distribution is complicated and caution is required in quantification.
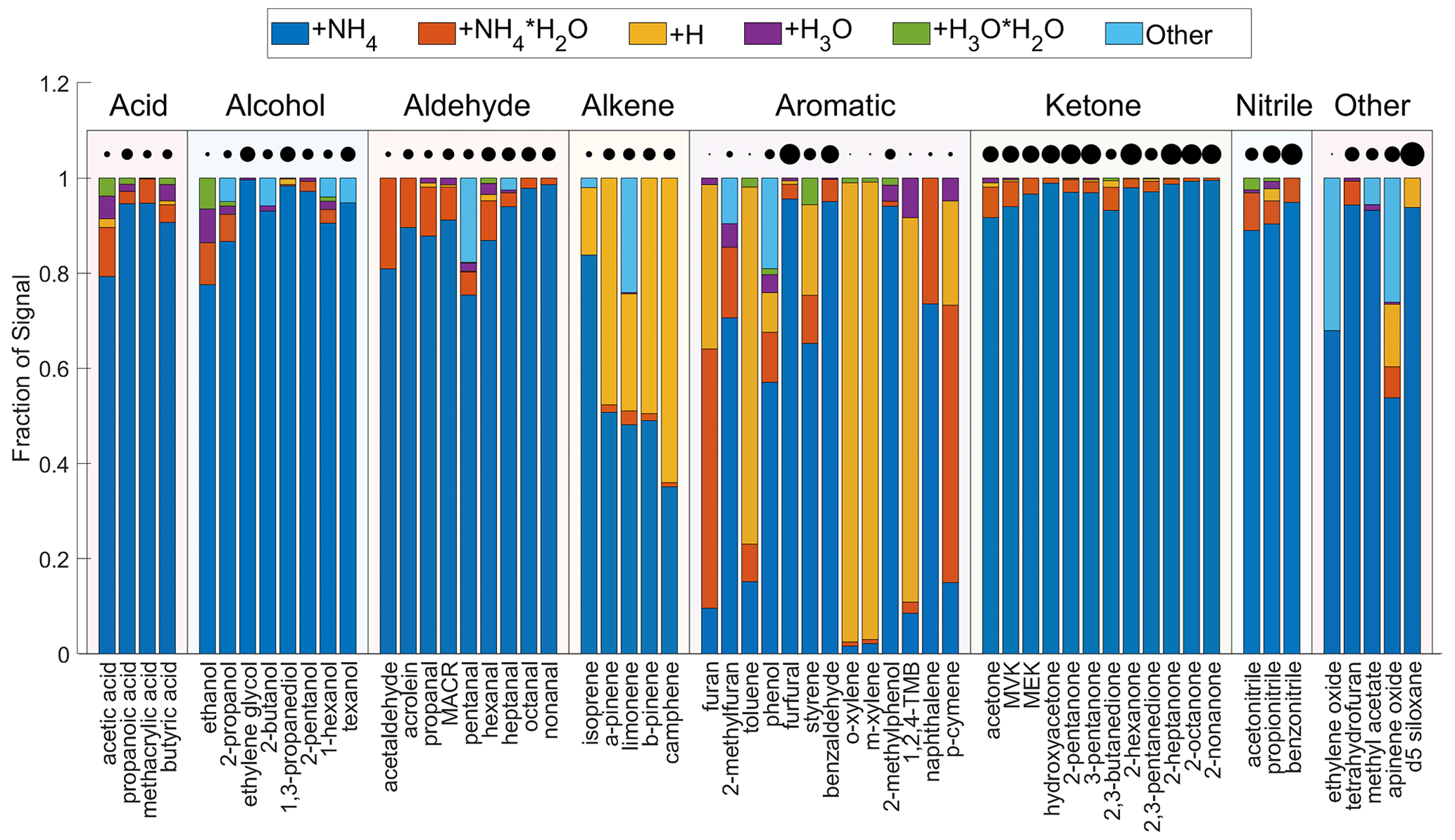
Figure 4The product distributions of analytes in the CIMS. The analytes are grouped by chemical class. Within each class, the analytes are sorted by increasing molecular weight. The distributions are obtained under the condition that the ratio of to is between 5 and 20. The product ion labeled “other” includes charge transfer products (e.g., C6H6O+ for phenol) and fragmentation products (e.g., C5H12N+ for pentanal). The product distribution of benzene is not shown because the signals of its product ions are too low to be reliably fitted. The circles are scaled to the square root of the analyte sensitivity.
3.5 Constraining the sensitivity
Because of a lack of calibration standards, the CIMS sensitivities to the majority of routinely detected multifunctional organic compounds in the atmosphere are not quantifiable. We attempt to constrain the sensitivity by building upon the extensive calibration of organic compounds from various chemical classes in this study. The observed instrument sensitivity (S, cps ppbv−1) is defined as the detected analyte signal (i.e., , cps) at a volume mixing ratio of 1 ppbv. Fundamentally, S depends on the product ion formation and the transmission efficiency of product ions, as expressed by Eq. (5) (Lopez-Hilfiker et al., 2016), where the integral represents the formation of product ions via the ion-molecule reactions in the IMR, represents the fraction of parent ion in all product ions, and TE represents the transmission efficiency of parent ion, which is dependent on the mass-to-charge ratio () and the binding energy (B) of the parent ion. In the integral, [] represents the concentration in the IMR, and k and t represent the reaction rate constant and reaction time between reagent ion and analyte (A) in the IMR, respectively. Using this integral to represent the product ion formation is only valid when the ion-molecule reaction is in the kinetic-limited regime. In the thermodynamic regime, both forward and reverse ion-molecule reactions need to be considered.
Under a constant instrumental condition, the and reaction time are fixed. The sensitivity of an analyte is determined by , k, and TE. Among these three factors, and k are more uncertain than TE. The value of k for exothermic ligand-switching reactions is close to the collisional limit (Adams et al., 2003), which can be calculated according to Su (1994) using the dipole moment and polarizability of the analyte (Table S3). can be experimentally measured, and it is close to 1 for multifunctional organic compounds, as discussed in Sect. 3.4. The TE, which represents the survival chance of ions through ion optics, is difficult to quantify. We assume the overall TE is represented by the product of -dependent TE (denoted asTS) and binding-energy-dependent TE (denoted as TEB) (Eq. 6). TS represents the transmission efficiency through BSQ, the extraction region of the ToF, and other processes that are dependent on . TS is experimentally quantified as described in Sect. 2.2. TEB accounts for the ion loss via collision-induced dissociation caused by energy imparted by electric fields. TEB depends on the binding energy of the parent ion, as the parent ion with stronger bonds between analyte and have a larger chance to survive the electric fields and hence a larger TEB. The binding energy of is experimentally probed from the voltage scanning tests and is represented by the kinetic energy of in the center of mass (i.e., KEcm,50) (Sect. 2.2). In this way, TEB is related to a measurable parameter KEcm,50. The mathematical relationship between TEB and KEcm,50, TE, is the final component to constrain the sensitivity.
We utilize the extensive calibration of 60 compounds from diverse chemical classes to derive the relationship between TEB and KEcm,50. By rearranging Eqs. (5) and (6) and representing as a constant C, TEB can be expressed as Eq. (7), where Scorr represents the sensitivity corrected for , k, and TE. Using Eq. (7), the relationship between TEB and KEcm,50 can be obtained through plotting Scorr against KEcm,50. As shown in Fig. 5, Scorr exhibits a positive dependence on KEcm,50. The relationship between Scorr and KEcm,50 of the majority of compounds can be reasonably described using a Hill equation. Analytes with small KEcm,50 (i.e., <0.15 eV) have very low sensitivity because of declustering of in the electric fields. As KEcm,50 increases, the sensitivity increases. This is because with a stronger bond between A and is more likely to survive the imparted energy from electric fields and hence more likely to be detected. When KEcm,50 exceeds a threshold (i.e., 0.35 eV), does not decluster in the electric field and is detected with maximum Scorr. The maximum Scorr is constrained using 2-hexanone here, but calibrations of analytes with KEcm,50 larger than 0.35 eV are warranted to constrain the maximum Scorr. Such analytes tend to be large oxygenated organic compounds with low volatility, making their calibrations challenging.
A similar relationship between Scorr and KEcm,50 has been reported in Zaytsev et al. (2019), who used –PTR3 and explored the relationship between the sensitivity and KEcm,50 for 16 compounds, 9 of which are ketones. Unlike this study, Zaytsev et al. (2019) did not normalize the sensitivity to the ion-molecule collision rate constant k. This is reasonable as the ion-molecule reaction time in –PTR is ∼3 ms, about 15 times longer than that in our instrument. The long reaction time results in an equilibrium between cluster formation and fragmentation in the IMR for many analytes. In this thermodynamic regime, the product ion formation is proportional to the equilibrium constant of Reaction (R6) (Iyer et al., 2016; Robinson et al., 2022) so that normalizing the sensitivity in –PTR3 by the equilibrium constant may improve the relationship between sensitivity and KEcm,50 in Zaytsev et al. (2019).
In this study, the relationship between Scorr and KEcm,50 is largely defined by monofunctional organic compounds, but we anticipate that this relationship applies to organic compounds containing at least one functional group that binds strongly with , such as C=O, -OH, and nitrile. For example, five multifunctional compounds studied here (i.e., ethylene glycol, 1,3-propanediol, hydroxyacetone, 2,3-butanedione, and 2,3-pentanedione) are well described by the fitted Hill equation. Because the fitted Hill equation does not apply to monocarboxylic acids, for reasons discussed later, the applicability of the relationship to multifunctional organic acids is uncertain and warrants future investigation. Moreover, we compare several structural isomers with monofunctional groups, including acetone vs. propanal, MACR vs. MVK, C5–C9 mono-aldehyde vs. mono-ketones. Despite the difference in Scorr between isomers, their Scorr and KEcm,50 follow the same relationship.
The relationship between Scorr and KEcm,50 depicted in Fig. 5 provides an effective approach to estimate the sensitivity of the CIMS to a suite of oxygenated organic compounds. The KEcm,50 can be calculated from the voltage scan tests. TE can be experimentally quantified following the procedure in Sect. 2.2. is unknown, but it is close to 1 for multifunctional organic compounds, as discussed in Sect. 3.4. The k is also unknown, but it can either be calculated (Su, 1994) or reasonably estimated based on the molecular mass, elemental composition, and functional group (Sekimoto et al., 2017). k is generally on the order of 10−9 cm3 molec.−1 s−1 (Fig. S7). Finally, based on the four abovementioned parameters, the sensitivity can be estimated.
The observed relationship between Scorr and KEcm,50 in Fig. 5 has limitations. First, it is only applicable to analytes for which the ligand-switching reaction with is exothermic. This arises from approximating the ion-molecule reaction rate constant (k) in Scorr using the collisional limiting rate constant. This approximation is not valid for endothermic reactions, which occur at a slower rate. This likely explains why several compounds, including monocarboxylic acids, some monoterpenes, reduced aromatics, isoprene, and 2-methylfuran, are outliers in Fig. 5. For example, the affinity of acetic acid is estimated to be lower than H2O (Sect. S5). Two monoterpenes, limonene and α-pinene, do not follow the fitted line, but the behaviors of monoterpenes are more complicated. The calculated affinities of β-pinene and camphene are smaller than that of H2O (Table S2), causing their ligand-switching reactions to be endothermic, but they fall on the fitted Hill equation. In contrast, limonene has larger affinity than H2O, but it is lower than the fitted line. The reason for such different behavior is unknown but might be related to their structural difference. For example, β-pinene and camphene have an external C=C bond connected to the six-member ring, but α-pinene and limonene do not. Another limitation is that KEcm,50, which is calculated from ΔV50 based on voltage scan, may not be a proper proxy for affinity for some analytes. For example, α-pinene has similar affinity as β-pinene and camphene (Canaval et al., 2019), but the voltage scan test shows that α-pinene has a larger KEcm,50 than the other two (Fig. 5). Another exception is that isoprene and 2-methylfuran are expected to have small affinity considering their low sensitivities, but their KEcm,50 is the highest among all analytes studied here. Similar “false positive” behavior (i.e., large KEcm,50 or binding energy, but low sensitivity) is also observed in the I− CIMS (Iyer et al., 2016). We suspect the voltage scanning affects not only the collisional energy of the , but also the ion-molecule chemistry or ion transmission via some unknown mechanisms. In the voltage scan, the FIMR front voltage is increased simultaneously with FIMR back voltage to keep the upstream voltage gradient constant. It is generally assumed that the absolute voltages do not affect the ion-molecule chemistry and transmission, as long as the voltage gradient is constant, but this assumption may not be valid. For example, in the voltage scan, we observe that the signal of reagent ion becomes noisy when the FIMR front voltage (450 V) is close to the ion source voltage (440 V), suggesting that the FIMR front voltage affects the ion transmission from the ion source into the FIMR.

Figure 5Relationship between Scorr and KEcm,50. Scorr represents the sensitivity (cps ppbv−1) corrected for the fraction of parent ion in all product ions (), -dependent transmission efficiency (TE), and the ion-molecule reaction rate constant (k, 10−9 cm3 molec. s−1), as defined in Eq. (7). The solid line represents a fitting of analytes using a Hill equation. . The dashed line represents a linear fitting for analytes with KEcm between 0.2 and 0.3 eV in a similar fashion done in Zaytsev et al. (2019). Organic acids, naphthalene, isoprene, 2-methyl furan, limonene, α-pinene, and styrene are excluded from both fittings. The ellipses represent the uncertainty range.
3.6 Comparison of sensitivities between instruments
In this section, we compare the sensitivities of our CIMS (denoted as NOAA CIMS) to two other CIMSs and an H3O+ CIMS. The other two CIMSs include a PTR3 instrument with a different IMR design from our Vocus (Zaytsev et al., 2019) (denoted as PTR3 CIMS) and a Vocus instrument with the same IMR design as ours but operated under different conditions (Khare et al., 2022) (denoted as Khare CIMS). The H3O+ CIMS is from our lab (denoted NOAA H3O+ CIMS), which replaced the traditional drift tube with the same FIMR as used in the CIMS. The NOAA H3O+ CIMS was calibrated along with the CIMS using the same calibration methods. The sensitivities of PTR3 CIMS and Khare CIMS are obtained from the corresponding references.
Figure 6 shows the sensitivity ratio of a selected instrument (Si) to the NOAA CIMS () for a number of analytes grouped by their chemical class. Khare CIMS used the same ion source and IMR as NOAA CIMS, but the sensitivities are generally lower than NOAA CIMS by a factor of 5. In particular, the ethylene glycol sensitivity is lower by a factor of 100. The lower sensitivity in Khare et al. (2022) is likely because they used a higher ratio than this study. Khare et al. (2022) used 1 sccm vapor from a 1 % ammonium hydroxide solution, while this study used 1 sccm vapor from a 0.5 % solution. As discussed in Sect. 3.2, a larger ratio leads to a larger fraction of in the total reagent ions and hence reduced sensitivity for most analytes (Fig. 3). The sensitivities in Khare et al. (2022) can be reproduced in NOAA CIMS by using a larger NH3 flow rate. The comparison between NOAA CIMS and Khare CIMS further emphasizes the importance of FIMR conditions for the instrument performance.
The sensitivity ratio of NOAA H3O+ CIMS to NOAA CIMS spans a wide range from 1 to 104. In general, the sensitivity ratio anti-correlates with the sensitivity of NOAA CIMS within each chemical class. This trend is the most evident for aromatics. For example, for reduced aromatics, the sensitivities of which are smaller than 2 cps ppbv−1 in the NOAA CIMS, their sensitivities are 103 higher in the NOAA H3O+ CIMS. However, for oxygenated aromatics, such as benzaldehyde and furfural, the sensitivities of which are on the order of 103 cps ppbv−1 in NOAA CIMS, two instruments have similar sensitivities. Therefore, H3O+ chemistry is more suitable to quantify reduced VOCs and small oxygenated VOCs (e.g., acetic acid, methanol, acetaldehyde) than chemistry. chemistry is better for quantifying larger oxygenated VOCs because it causes less fragmentation than the H3O+ chemistry (Pagonis et al., 2019; Yuan et al., 2017; Sekimoto et al., 2017), which simplifies the interpretation of the mass spectra.
Using the same chemistry, the PTR3 sensitivities are overall 20 times higher than those of NOAA CIMS. This difference is mainly due to different designs of the IMR and ion source. The PTR3 utilized a tripole electrode as the IMR (Breitenlechner et al., 2017). This design enables the IMR to be operated at 60 mbar and 3 ms reaction time (Zaytsev et al., 2019), which are much higher than 3 mbar and 0.2 ms in NOAA CIMS, and leads to enhanced sensitivities. The NOAA CIMS utilizes a low-pressure discharge ion source, which generates more ions than the corona discharge ion source in the PTR3. This compensates for the effects of the lower IMR pressure and short reaction on sensitivity to some extent. The combined influences of ion source, IMR pressure, and reaction time result in the difference in sensitivities between NOAA CIMS and PTR3 CIMS. Despite lower sensitivities, one advantage of the NOAA CIMS is that its sensitivities have a much smaller dependence on the sample relative humidity that the PTR3 CIMS (Zaytsev et al., 2019).
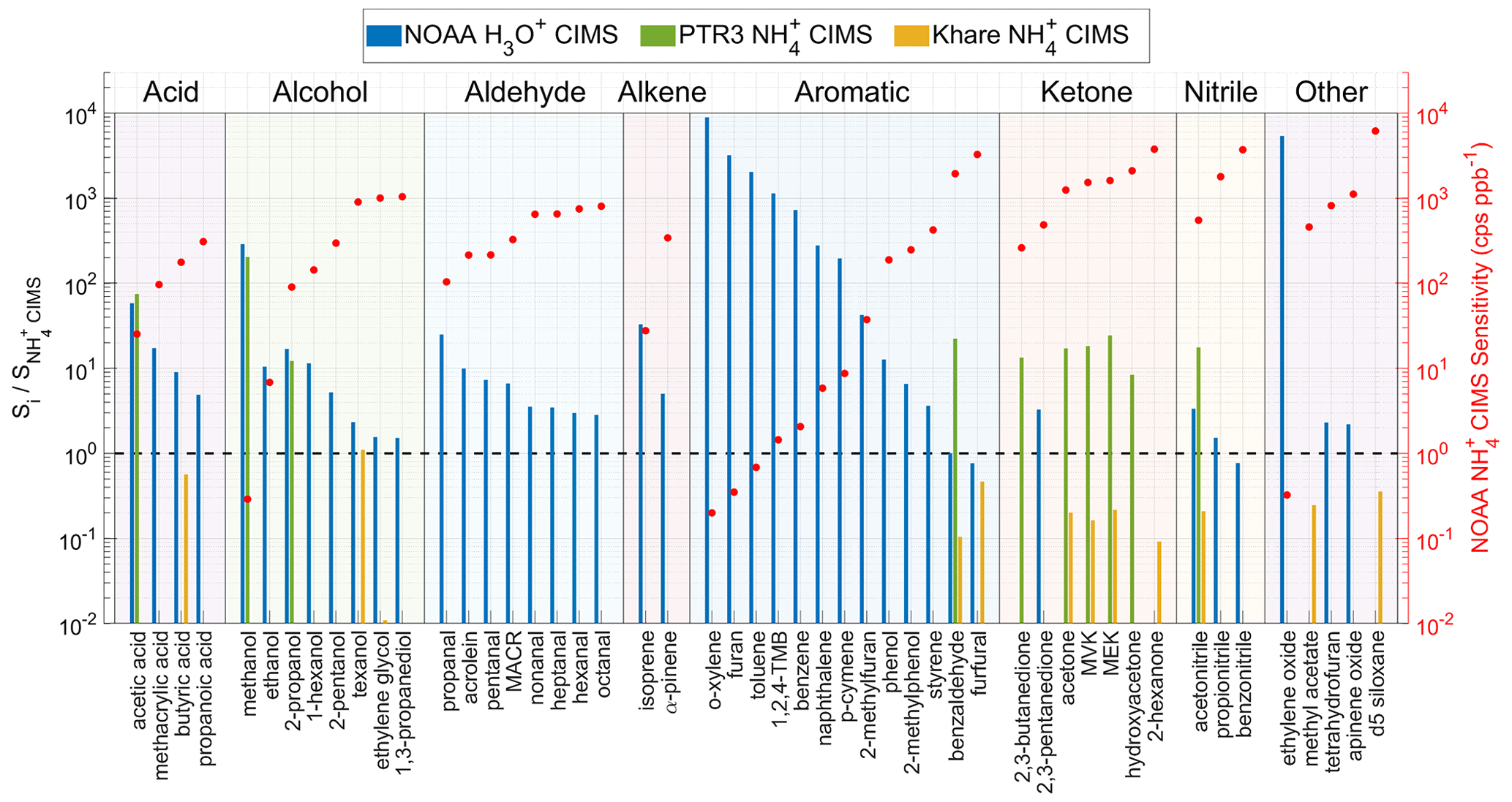
Figure 6The sensitivity ratio of a selected instrument (Si) to the NOAA CIMS (). The selected instrument i includes PTR3 CIMS, Khare CIMS, and NOAA H3O+ CIMS. The analytes are grouped by their chemical class. Within each chemical class, the analytes are sorted by their sensitivity.
The CIMS was deployed during the RECAP campaign in Pasadena, California, in August–September, 2021. Measurements presented in this section were made from 10 to 19 August when the instrument continuously sampled the gas phase.
4.1 Measurement capability
Figure S8 uses a mass defect plot to illustrate the measurement capability of CIMS. In the RECAP campaign, a total of 288 ions have signals above the detection limit. Half of the ions have the formula CxHyN1Oz (reagent ion included in the formula). These ions mostly represent the non-nitrogen-containing oxygenated organics cluster with or . A total of 70 ions have the formula CxHyN2Oz, which likely represent nitrogen-containing compounds. This assignment is supported by the analysis of product distribution (Sect. 3.4), which shows the product ion contains at most one nitrogen from the reagent ion. A total of 40 out of 288 ions have the formula CxHyOz, which likely represent analytes clustering with ().
4.2 Instrument intercomparison
The co-located instruments in the RECAP campaign enable the evaluation of the field performance of CIMS. In this section, we compare the measurements of several important atmospheric species from different chemical classes by three NOAA mass spectrometers (i.e., CIMS, H3O+ CIMS, GC-MS) and the Caltech CF3O− CIMS. For compounds that are commercially available, we calibrate the instrumental sensitivity and compare the mixing ratio. For multifunctional oxygenated organics that do not have calibration standards, raw signals are compared. If multiple isomers exist for a parent ion and if these isomers are quantified by GC-MS, we apply the GC-MS resolved isomer ratio and the sensitivities of individual isomers to convert the raw cps of the parent ion to the summed mixing ratio of all isomers for CIMS (Sect. S6).
To account for instrument variability, the ion signals are typically normalized to the changing reagent ion signals. However, previous studies using Vocus in H3O+ and chemistry did not normalize the signals to reagent ions (Krechmer et al., 2018; Khare et al., 2022) because the BSQ serves as a high-pass band filter and substantially reduces the signal intensity of reagent ions. In this study, we find that without normalization, the comparisons between CIMS and GC-MS exhibit a significant difference between day and night (Fig. S9a and b), which is consistent with the diurnal trend of reagent ion (Fig. S9c). Normalization to the reagent ion signal largely eliminates this difference. In light of this observation, we normalize the ion signals to that of and then apply the normalized sensitivity to convert the signal (ncps) to mixing ratio (ppbv).
4.2.1 Reduced VOCs
We compare the measurements of isoprene and monoterpenes between CIMS, H3O+ CIMS, and GC-MS (Fig. 7a and b). CIMS has a relatively low sensitivity to isoprene (i.e., 28 cps ppbv−1), but the high mass resolution of the instrument enables a clear separation of isoprene (detected as ) from other isobars. Overall, isoprene measured by CIMS is ∼20 % higher than H3O+ CIMS and GC-MS (Fig. S10). At night, both CIMS and H3O+ CIMS observe a significantly higher isoprene concentration than GC-MS (Fig. 7a). This is likely because isoprene measured by CIMS and H3O+ CIMS has interference from fragments of other species. We found that several aldehydes, including octanal and nonanal, fragment in the H3O+ CIMS and produce C5H8H+. Correcting such interference results in a lower isoprene concentration measured by the H3O+ CIMS, particularly at night, and better agreement between H3O+ CIMS and GC-MS (Fig. 7a). Similarly, pentanal in the CIMS produces , which is the parent ion of isoprene. Because the isoprene sensitivity in CIMS is so low, the production of from an analyte with high sensitivity would lead to large interference in the isoprene concentration. Thus, CIMS is not recommended for quantifying isoprene.
For monoterpenes, GC-MS shows that α-pinene and β-pinene are the dominant monoterpene isomers at the sampling site. The ratio of α-pinene and β-pinene measured by GC-MS is used to convert the signal measured by CIMS to the mixing ratio of total monoterpenes (the Sect. S6). Three instruments show a large difference in measuring monoterpenes (Fig. 7b). The correlation between H3O+ CIMS and CIMS is strong, but H3O+ CIMS observes 3 times more monoterpenes than CIMS (Fig. S10b). CIMS and GC-MS agree well at night, but CIMS detects more monoterpenes in the afternoon than GC-MS (Fig. 7b). The monoterpene concentrations measured by both the CIMS and the H3O+ CIMS are very spiky in the afternoon, and the afternoon peak in the diurnal trend coincides with that of isoprene (Fig. S11). Both observations suggest that the monoterpenes are primary emissions from a local source, which is likely the trees a few meters away from the sampling site. The difference between three measurements may be related to fragmentation interference in the monoterpene signals measured by CIMS (i.e., ) and H3O+ CIMS (). It is also possible that there are shorter-lived monoterpene isomers, other than α-pinene and β-pinene, which are not reported by the GC-MS, leading to the absence of an afternoon peak of monoterpenes in GC-MS.
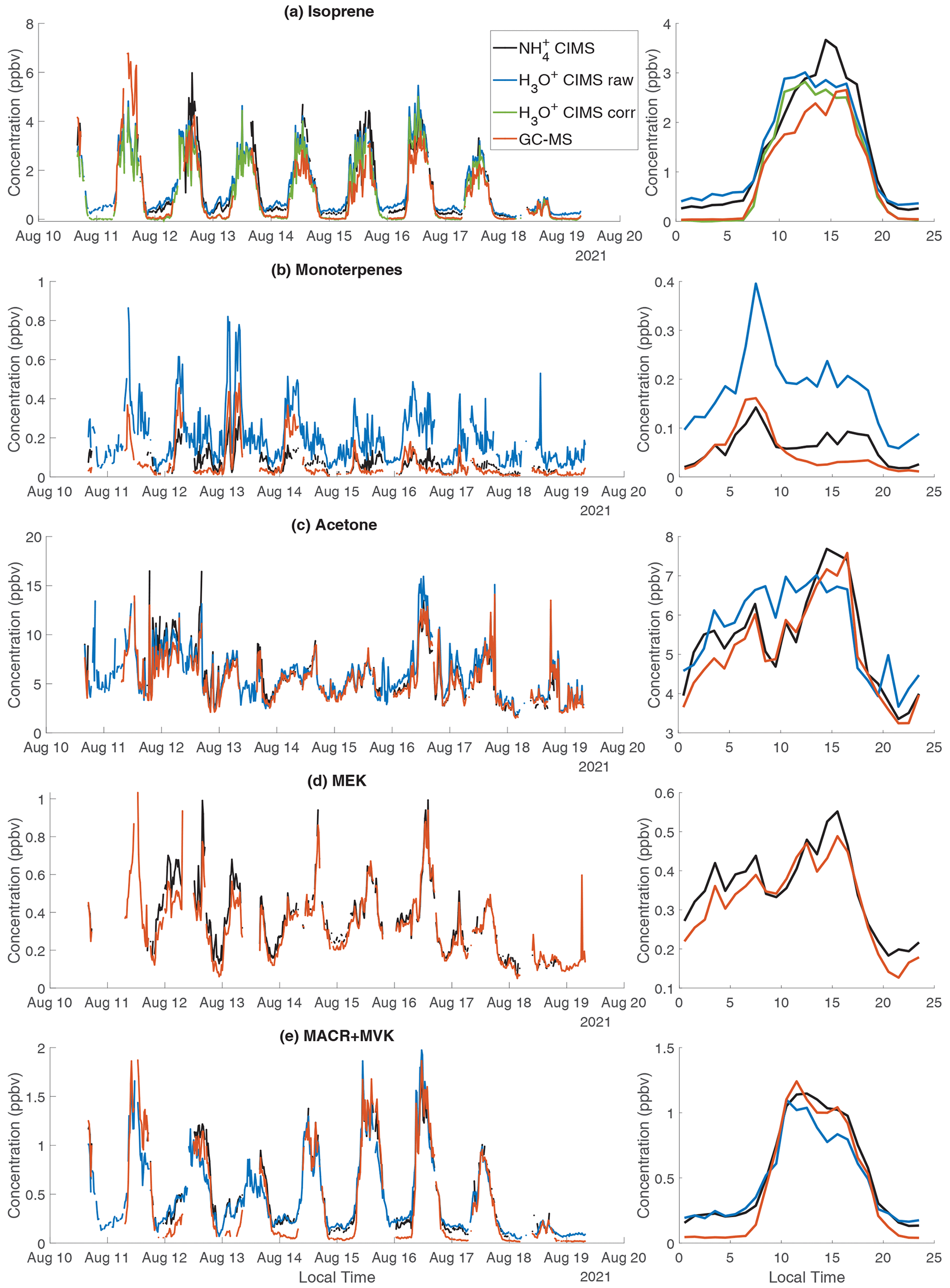
Figure 7The time series and diurnal trend of selected species measured by CIMS, H3O+ CIMS, and GC-MS. (a) Isoprene; (b) monoterpenes; (c) acetone; (d) methyl ethyl ketone (MEK); (e) methacrolein (MACR) + methyl vinyl ketone (MVK). In panel (a), H3O+ CIMS corr represents the isoprene measurement by the H3O+ CIMS after correcting the interference from octanal and nonanal (Sect. S6). In panel (d), MEK measured by the NOAA H3O+ CIMS is not included because its peak fitting (C4H9O+) is degraded by the nearby large signal of .
4.2.2 Carbonyls
Figure 7c shows the time series of acetone measured by CIMS, H3O+ CIMS, and GC-MS. In CIMS, we attribute the solely to acetone and ignore the contribution from its structural isomer propanal because GC-MS shows that the propanal concentration is much lower than acetone and because the CIMS sensitivity to acetone is 10 times larger than propanal (1247 vs. 103 cps ppbv−1). Acetone concentrations measured by the three instruments agree within 15 %, which is within the combined calibration uncertainties. Similar to acetone, the MEK measurement agrees between CIMS and GC-MS within 10 % and an r2 of 0.93 (Fig. 7d).
For MACR+MVK, three instruments agree well in the day, but CIMS and H3O+ CIMS observe a higher concentration of MACR+MVK at night than GC-MS (Fig. 7e). We suspect the nighttime signal measured by CIMS and H3O+ CIMS is due to 2-butenal and 3-butenal from cooking emissions.
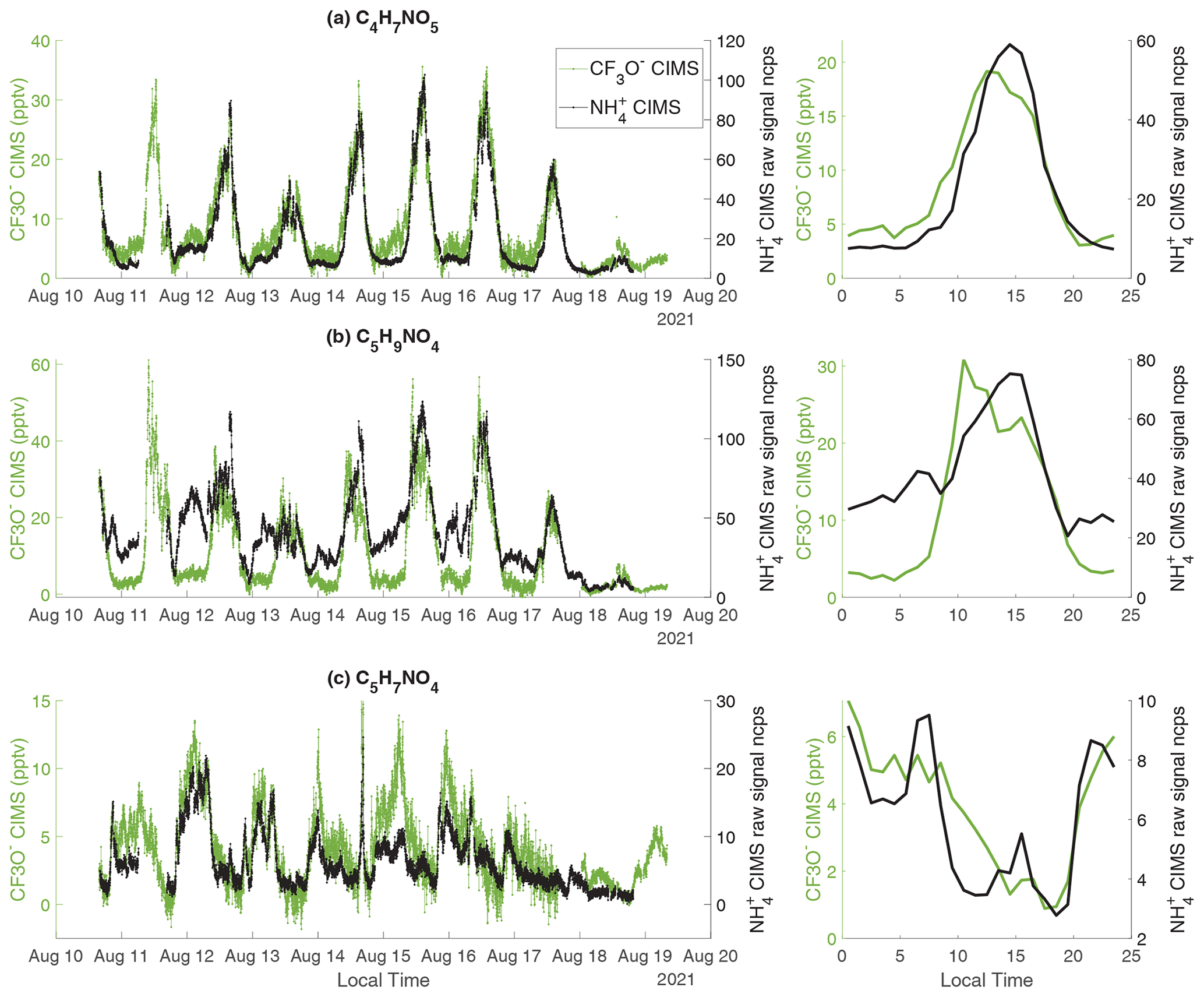
Figure 8The time series and diurnal trend of three nitrogen-containing species measured by CIMS and CF3O− CIMS. (a) C4H7NO5; (b) C5H9NO4; (c) C5H7NO4. Because of a lack of calibration standards, the raw signals (ncps) of CIMS are shown.
4.2.3 Hydroxy nitrates
As shown in Fig. S8, CIMS detects a number of organic nitrates. Due to a lack of calibration standards, the sensitivities of organic nitrates in the CIMS have not been quantified. Here, we explore the measurement capability of the CIMS by comparing to three organic nitrates (i.e., C4H7NO5, C5H9NO4, and C5H7NO4) measured by the CF3O− CIMS, which has been calibrated (L. Lee et al., 2014; Nguyen et al., 2014). All three organic nitrates are detected as adducts with the respective reagent ions in both instruments.
C4H7NO5 matches the formula of hydroxynitrates produced from the oxidation of MACR, MVK, and first-generation organic nitrates from isoprene oxidation. The CIMS and CF3O− CIMS show a correlation with an r2 value of 0.91 (Fig. 8a and S12a). C4H7NO5 corresponds to at least two structural isomers, both of which have three functional groups (-OH, -C=O, and -ONO2). The strong correlation between two instruments could be due to dominance of a single isomer or similar sensitivities to both isomers in each instrument.
C5H9NO4 has been attributed to isoprene hydroxy nitrates (IHNs) in the literature (L. Lee et al., 2014; Xiong et al., 2015; Nguyen et al., 2015; Lee et al., 2016; Teng et al., 2017; Vasquez et al., 2020). The correlation coefficient r2 between the two instruments is 0.63 (Fig. S12b). The C5H9NO4 measured by CF3O− CIMS is close to zero at night (Fig. 8b), consistent with previous isomer-resolved measurements of IHNs by the CF3O− CIMS coupled to a GC front end at the same site in 2017 (Vasquez et al., 2020). In contrast, the C5H9NO4 measured by CIMS is persistently high throughout the night. We hypothesize that the C5H9NO4 signal measured by the CIMS has a large contribution from nitrooxy ketones, which are produced from the oxidation of pentenes by nitrate radicals (Fig. S13). Based on the laboratory characterization, is more sensitive to ketones than alcohols (Table 1 and Fig. 5). Thus, it is possible that nitrooxy ketones have a much higher sensitivity than isoprene hydroxy nitrates in the CIMS. This leads to the observed C5H9NO4 signal in the CIMS largely arising from nitrooxy ketones, even though their concentrations are much smaller than isoprene hydroxy nitrates. In contrast, the CF3O− CIMS likely has similar sensitivity to both classes of chemicals (L. Lee et al., 2014). Further, the nighttime signal of C5H9NO4 measured by the CIMS is consistent with the observation that pentene concentrations are higher at night than in the day (Fig. S14).
C5H7NO4 matches the formula of isoprene carbonyl nitrate produced from isoprene oxidation by nitrate radicals followed by RO2+RO2 reaction (Schwantes et al., 2015). Consistent with this product identification, the measurements of C5H7NO4 by both CIMS and CF3O− CIMS peak at night (Fig. 8d). The correlation r2 between CIMS and CF3O− CIMS is 0.67 (Fig. S12d).
In this study, we describe the development and deployment of CIMS using as a reagent ion. is a highly versatile reagent ion for measurements of a wide range of oxygenated organic compounds. The instrument sensitivities and product distributions are strongly dependent on the instrument conditions, including FIMR reduced electric field, temperature, pressure, the H2O mixing ratio, and the ratio of NH3 to H2O. These conditions should be carefully selected to ensure as the predominant reagent ion and to optimize sensitivities. For example, a comparison between this study and another study using the same instrument but under different FIMR conditions shows that the instrument sensitivity can differ by a factor of 5. Besides the desired reagent ion , several other reagent ions exist in the FIMR even at the optimal condition, which complicates the ion-molecule chemistry and the product distribution. The cluster ion is the predominant product ion for acids, ketones, nitriles, and multifunctional oxygenated compounds. More diverse products, including protonated ion AH+ and fragmentation ions, are observed for small alcohols, biogenic VOCs, and reduced aromatics.
For monofunctional analytes, the chemistry exhibits high sensitivity (i.e., >1000 cps ppbv−1) to ketones, moderate sensitivity (i.e., between 100 and 1000 cps ppbv−1) to aldehydes, alcohols, organic acids, and monoterpenes, low sensitivity (i.e., between 10 and 100 cps ppbv−1) to isoprene and C1 and C2 organics, and negligible sensitivity (i.e., <10 cps ppbv−1) to reduced aromatics. The sensitivity of the CIMS to organic nitrates and highly oxygenated compounds requires further investigation. Overall, the CIMS is complementary to existing chemical ionization schemes. Comparing to two commonly used reagent ions H3O+ and I−, is more suitable to quantify moderately oxygenated compounds with one or two functional groups (i.e., C=O, -OH, and nitrile). These types of compounds have relatively low sensitivity in I− CIMS (B. H. Lee et al., 2014). H3O+ and show similar sensitivity to the moderately oxygenated compounds, and one advantage of chemistry is that it causes less fragmentation than H3O+ chemistry (Pagonis et al., 2019), which simplifies the interpretation of the mass spectra. Moreover, we reveal a strong relationship between instrumental sensitivity and the binding energy of the analyte– cluster, which can be estimated using voltage scanning tests. This offers the possibility to constrain the sensitivity of analytes for which no calibration standards exist. Caution is required when applying this method because the observed relationship is only applicable to analytes for which the ligand-switching reaction with is exothermic and because the measured KEcm,50 may not be a proper proxy for affinity for some analytes. The combination of experimental constraints and theoretical calculation of analyte thermodynamic properties could potentially provide more accurate estimates of analyte sensitivities.
The field performance of the CIMS is evaluated based on comparisons with three co-located mass spectrometers in the RECAP campaign during a 10 d period. CIMS and GC-MS show reasonable agreement in measuring carbonyls (i.e., acetone, MEK, MACR+MVK) but not in isoprene and monoterpenes. Isoprene measured by the CIMS has fragmentation interference. The difference in monoterpene measurements is possibly because of fragmentation interference in the CIMS or some monoterpene isomers are not reported by the GC-MS. Future studies are needed to understand the difference. A number of nitrogen-containing species are detected by the CIMS, and three representative ones are compared to CF3O− CIMS. Strong correlations are observed for C4H7NO5 (likely oxidation products of MACR, MVK, and first-generation organic nitrates from isoprene oxidation) but not for C5H9NO4 (including isoprene hydroxy nitrates and nitrooxy ketones from pentene oxidation). The difference in C5H9NO4 measurements is likely because CIMS and CF3O− CIMS have vastly different sensitivities to different structural isomers. Such comparisons illustrate different measurement capabilities of different reagent ions. It is imperative to understand the isomer-specific sensitivity of the instrument in order to obtain a complete and unbiased understanding of the atmospheric composition.
Data from the RECAP campaign are available to the general public at https://csl.noaa.gov/groups/csl7/measurements/2021sunvex/GroundLA/DataDownload/ (last access: 10 December 2022). The NOAA CSL Tropospheric Chemistry Group data archive for the RECAP campaign is created and maintained by Kenneth Aikin.
The supplement related to this article is available online at: https://doi.org/10.5194/amt-15-7353-2022-supplement.
LX and CW designed the research, LX operated the CIMS, MMC and CES operated the H3O+ CIMS, JBG and AL operated the GC-MS, JDC and POW operated the CF3O− CIMS, MAR, JAN, PRV, GAN, MMC, CES, and SSB provided critical support and comments in CIMS operation, and KHM performed theoretical calculations. All authors commented on the paper.
The contact author has declared that none of the authors has any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
We thank the Caltech Facilities for their support in the RECAP campaign. We thank Kristian H. Møller and Henrik G. Kjaergaard for calculation of dipole moments and polarizabilities.
This work was supported by the NOAA cooperative agreement with CIRES (agreement no. NA17OAR4320101). The NOAA Chemical Sciences Laboratory acknowledges support for this work from the California Air Resources Board (agreement no. 20RD002).
This paper was edited by Anna Novelli and reviewed by two anonymous referees.
Adams, N. G., Babcock, L. M., Mostefaoui, T. M., and Kerns, M. S.: Selected ion flow tube study of NH association and of product switching reactions with a series of organic molecules, Int. J. Mass Spectrom., 223–224, 459–471, https://doi.org/10.1016/S1387-3806(02)00932-6, 2003. a, b
Allen, H. M., Crounse, J. D., Kim, M. J., Teng, A. P., Ray, E. A., McKain, K., Sweeney, C., and Wennberg, P. O.: H2O2 and CH3OOH (MHP) in the Remote Atmosphere: 1. Global Distribution and Regional Influences, J. Geophys. Res.-Atmos., 127, e2021JD035701, https://doi.org/10.1029/2021JD035701, 2022. a
Berndt, T., Scholz, W., Mentler, B., Fischer, L., Herrmann, H., Kulmala, M., and Hansel, A.: Accretion Product Formation from Self- and Cross-Reactions of RO2 Radicals in the Atmosphere, Angew. Chem. Int. Edit., 57, 3820–3824, https://doi.org/10.1002/anie.201710989, 2018. a, b, c
Blake, R. S., Wyche, K. P., Ellis, A. M., and Monks, P. S.: Chemical ionization reaction time-of-flight mass spectrometry: Multi-reagent analysis for determination of trace gas composition, Int. J. Mass Spectrom., 254, 85–93, https://doi.org/10.1016/j.ijms.2006.05.021, 2006. a
Breitenlechner, M., Fischer, L., Hainer, M., Heinritzi, M., Curtius, J., and Hansel, A.: PTR3: An Instrument for Studying the Lifecycle of Reactive Organic Carbon in the Atmosphere, Anal. Chem., 89, 5824–5831, https://doi.org/10.1021/acs.analchem.6b05110, 2017. a, b
Breitenlechner, M., Novak, G. A., Neuman, J. A., Rollins, A. W., and Veres, P. R.: A versatile vacuum ultraviolet ion source for reduced pressure bipolar chemical ionization mass spectrometry, Atmos. Meas. Tech., 15, 1159–1169, https://doi.org/10.5194/amt-15-1159-2022, 2022. a
Canaval, E., Hyttinen, N., Schmidbauer, B., Fischer, L., and Hansel, A.: NH Association and Proton Transfer Reactions With a Series of Organic Molecules, Front. Chem., 7, 191, https://doi.org/10.3389/fchem.2019.00191, 2019. a, b, c, d, e
Chemical Sciences Laboratory: SUNVEx/RECAP Data Download, Pasadena Ground Site, https://csl.noaa.gov/groups/csl7/measurements/2021sunvex/GroundLA/DataDownload/, last access: 10 December 2022.
Crounse, J. D., McKinney, K. A., Kwan, A. J., and Wennberg, P. O.: Measurement of Gas-Phase Hydroperoxides by Chemical Ionization Mass Spectrometry, Anal. Chem., 78, 6726–6732, https://doi.org/10.1021/ac0604235, 2006. a, b
de Gouw, J. and Warneke, C.: Measurements of volatile organic compounds in the earth's atmosphere using proton-transfer-reaction mass spectrometry, Mass Spectrom. Rev., 26, 223–257, https://doi.org/10.1002/mas.20119, 2007. a, b
de Gouw, J., Warneke, C., Karl, T., Eerdekens, G., van der Veen, C., and Fall, R.: Sensitivity and specificity of atmospheric trace gas detection by proton-transfer-reaction mass spectrometry, Int. J. Mass Spectrom., 223–224, 365–382, https://doi.org/10.1016/S1387-3806(02)00926-0, 2003. a
Ehn, M., Thornton, J. A., Kleist, E., Sipila, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I.-H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurten, T., Nielsen, L. B., Jorgensen, S., Kjaergaard, H. G., Canagaratna, M., Maso, M. D., Berndt, T., Petaja, T., Wahner, A., Kerminen, V.-M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476–479, https://doi.org/10.1038/nature13032, 2014. a
Goldstein, A. H. and Galbally, I. E.: Known and unexplored organic constituents in the earth's atmosphere, Environ. Sci. Technol., 41, 1514–1521, https://doi.org/10.1021/es072476p, 2007. a
Hansel, A., Scholz, W., Mentler, B., Fischer, L., and Berndt, T.: Detection of RO2 radicals and other products from cyclohexene ozonolysis with NH and acetate chemical ionization mass spectrometry, Atmos. Environ., 186, 248–255, https://doi.org/10.1016/j.atmosenv.2018.04.023, 2018. a, b
Heinritzi, M., Simon, M., Steiner, G., Wagner, A. C., Kürten, A., Hansel, A., and Curtius, J.: Characterization of the mass-dependent transmission efficiency of a CIMS, Atmos. Meas. Tech., 9, 1449–1460, https://doi.org/10.5194/amt-9-1449-2016, 2016. a
Huey, L. G., Hanson, D. R., and Howard, C. J.: Reactions of SF6- and I- with Atmospheric Trace Gases, J. Phys. Chem., 99, 5001–5008, https://doi.org/10.1021/j100014a021, 1995. a
Iyer, S., Lopez-Hilfiker, F., Lee, B. H., Thornton, J. A., and Kurtén, T.: Modeling the Detection of Organic and Inorganic Compounds Using Iodide-Based Chemical Ionization, J. Phys. Chem. A, 120, 576–587, https://doi.org/10.1021/acs.jpca.5b09837, 2016. a, b
Khare, P., Krechmer, J. E., Machesky, J. E., Hass-Mitchell, T., Cao, C., Wang, J., Majluf, F., Lopez-Hilfiker, F., Malek, S., Wang, W., Seltzer, K., Pye, H. O. T., Commane, R., McDonald, B. C., Toledo-Crow, R., Mak, J. E., and Gentner, D. R.: Ammonium adduct chemical ionization to investigate anthropogenic oxygenated gas-phase organic compounds in urban air, Atmos. Chem. Phys., 22, 14377–14399, https://doi.org/10.5194/acp-22-14377-2022, 2022. a, b, c, d, e, f, g, h, i
Krechmer, J., Lopez-Hilfiker, F., Koss, A., Hutterli, M., Stoermer, C., Deming, B., Kimmel, J., Warneke, C., Holzinger, R., Jayne, J., Worsnop, D., Fuhrer, K., Gonin, M., and de Gouw, J.: Evaluation of a New Reagent-Ion Source and Focusing Ion–Molecule Reactor for Use in Proton-Transfer-Reaction Mass Spectrometry, Anal. Chem., 90, 12011–12018, https://doi.org/10.1021/acs.analchem.8b02641, 2018. a, b, c, d, e, f, g, h
Lee, B. H., Lopez-Hilfiker, F. D., Mohr, C., Kurtén, T., Worsnop, D. R., and Thornton, J. A.: An Iodide-Adduct High-Resolution Time-of-Flight Chemical-Ionization Mass Spectrometer: Application to Atmospheric Inorganic and Organic Compounds, Environ. Sci. Technol., 48, 6309–6317, https://doi.org/10.1021/es500362a, 2014 a, b
Lee, B. H., Mohr, C., Lopez-Hilfiker, F. D., Lutz, A., Hallquist, M., Lee, L., Romer, P., Cohen, R. C., Iyer, S., Kurtén, T., Hu, W., Day, D. A., Campuzano-Jost, P., Jimenez, J. L., Xu, L., Ng, N. L., Guo, H., Weber, R. J., Wild, R. J., Brown, S. S., Koss, A., de Gouw, J., Olson, K., Goldstein, A. H., Seco, R., Kim, S., McAvey, K., Shepson, P. B., Starn, T., Baumann, K., Edgerton, E. S., Liu, J., Shilling, J. E., Miller, D. O., Brune, W., Schobesberger, S., D'Ambro, E. L., and Thornton, J. A.: Highly functionalized organic nitrates in the southeast United States: Contribution to secondary organic aerosol and reactive nitrogen budgets, P. Natl. Acad. Sci. USA, 113, 1516–1521, https://doi.org/10.1073/pnas.1508108113, 2016. a
Lee, L., Teng, A. P., Wennberg, P. O., Crounse, J. D., and Cohen, R. C.: On Rates and Mechanisms of OH and O3 Reactions with Isoprene-Derived Hydroxy Nitrates, J. Phys. Chem. A, 118, 1622–1637, https://doi.org/10.1021/jp4107603, 2014. a, b, c
Lerner, B. M., Gilman, J. B., Aikin, K. C., Atlas, E. L., Goldan, P. D., Graus, M., Hendershot, R., Isaacman-VanWertz, G. A., Koss, A., Kuster, W. C., Lueb, R. A., McLaughlin, R. J., Peischl, J., Sueper, D., Ryerson, T. B., Tokarek, T. W., Warneke, C., Yuan, B., and de Gouw, J. A.: An improved, automated whole air sampler and gas chromatography mass spectrometry analysis system for volatile organic compounds in the atmosphere, Atmos. Meas. Tech., 10, 291–313, https://doi.org/10.5194/amt-10-291-2017, 2017. a
Lindinger, W., Hansel, A., and Jordan, A.: On-line monitoring of volatile organic compounds at pptv levels by means of proton-transfer-reaction mass spectrometry (PTR-MS) medical applications, food control and environmental research, Int. J. Mass Spectrom., 173, 191–241, https://doi.org/10.1016/S0168-1176(97)00281-4, 1998. a
Lopez-Hilfiker, F. D., Iyer, S., Mohr, C., Lee, B. H., D'Ambro, E. L., Kurtén, T., and Thornton, J. A.: Constraining the sensitivity of iodide adduct chemical ionization mass spectrometry to multifunctional organic molecules using the collision limit and thermodynamic stability of iodide ion adducts, Atmos. Meas. Tech., 9, 1505–1512, https://doi.org/10.5194/amt-9-1505-2016, 2016. a, b
Müller, M., Piel, F., Gutmann, R., Sulzer, P., Hartungen, E., and Wisthaler, A.: A novel method for producing NH4+ reagent ions in the hollow cathode glow discharge ion source of PTR-MS instruments, Int. J. Mass Spectrom., 447, 116254, https://doi.org/10.1016/j.ijms.2019.116254, 2020. a, b, c
Nah, T., Ji, Y., Tanner, D. J., Guo, H., Sullivan, A. P., Ng, N. L., Weber, R. J., and Huey, L. G.: Real-time measurements of gas-phase organic acids using SF chemical ionization mass spectrometry, Atmos. Meas. Tech., 11, 5087–5104, https://doi.org/10.5194/amt-11-5087-2018, 2018. a
Nguyen, T. B., Crounse, J. D., Schwantes, R. H., Teng, A. P., Bates, K. H., Zhang, X., St. Clair, J. M., Brune, W. H., Tyndall, G. S., Keutsch, F. N., Seinfeld, J. H., and Wennberg, P. O.: Overview of the Focused Isoprene eXperiment at the California Institute of Technology (FIXCIT): mechanistic chamber studies on the oxidation of biogenic compounds, Atmos. Chem. Phys., 14, 13531–13549, https://doi.org/10.5194/acp-14-13531-2014, 2014. a
Nguyen, T. B., Crounse, J. D., Teng, A. P., St. Clair, J. M., Paulot, F., Wolfe, G. M., and Wennberg, P. O.: Rapid deposition of oxidized biogenic compounds to a temperate forest, P. Natl. Acad. Sci. USA, 112, E392–E401, https://doi.org/10.1073/pnas.1418702112, 2015. a
Pagonis, D., Sekimoto, K., and de Gouw, J.: A Library of Proton-Transfer Reactions of H3O+ Ions Used for Trace Gas Detection, J. Am. Soc. Mass Spectr., 30, 1330–1335, https://doi.org/10.1007/s13361-019-02209-3, 2019. a, b
Robinson, M. A., Neuman, J. A., Huey, L. G., Roberts, J. M., Brown, S. S., and Veres, P. R.: Temperature dependent sensitivity of iodide chemical ionization mass spectrometers, EGUsphere [preprint], https://doi.org/10.5194/amt-2022-295, 2022. a, b
Ryerson, T. B., Andrews, A. E., Angevine, W. M., Bates, T. S., Brock, C. A., Cairns, B., Cohen, R. C., Cooper, O. R., de Gouw, J. A., Fehsenfeld, F. C., Ferrare, R. A., Fischer, M. L., Flagan, R. C., Goldstein, A. H., Hair, J. W., Hardesty, R. M., Hostetler, C. A., Jimenez, J. L., Langford, A. O., McCauley, E., McKeen, S. A., Molina, L. T., Nenes, A., Oltmans, S. J., Parrish, D. D., Pederson, J. R., Pierce, R. B., Prather, K., Quinn, P. K., Seinfeld, J. H., Senff, C. J., Sorooshian, A., Stutz, J., Surratt, J. D., Trainer, M., Volkamer, R., Williams, E. J., and Wofsy, S. C.: The 2010 California Research at the Nexus of Air Quality and Climate Change (CalNex) field study, J. Geophys. Res.-Atmos., 118, 5830–5866, https://doi.org/10.1002/jgrd.50331, 2013. a
Schwantes, R. H., Teng, A. P., Nguyen, T. B., Coggon, M. M., Crounse, J. D., St. Clair, J. M., Zhang, X., Schilling, K. A., Seinfeld, J. H., and Wennberg, P. O.: Isoprene NO3 Oxidation Products from the RO2+ HO2 Pathway, J. Phys. Chem. A, 119, 10158–10171, https://doi.org/10.1021/acs.jpca.5b06355, 2015. a
Sekimoto, K., Li, S.-M., Yuan, B., Koss, A., Coggon, M., Warneke, C., and de Gouw, J.: Calculation of the sensitivity of proton-transfer-reaction mass spectrometry (PTR-MS) for organic trace gases using molecular properties, Int. J. Mass Spectrom., 421, 71–94, https://doi.org/10.1016/j.ijms.2017.04.006, 2017. a, b
Steiner, G., Jokinen, T., Junninen, H., Sipilä, M., Petäjä, T., Worsnop, D., Reischl, G. P., and Kulmala, M.: High-Resolution Mobility and Mass Spectrometry of Negative Ions Produced in a 241Am Aerosol Charger, Aerosol Sci. Tech., 48, 261–270, https://doi.org/10.1080/02786826.2013.870327, 2014. a
Su, T.: Parametrization of kinetic energy dependences of ion–polar molecule collision rate constants by trajectory calculations, J. Chem. Phys., 100, 4703–4703, https://doi.org/10.1063/1.466255, 1994. a, b, c
Teng, A. P., Crounse, J. D., and Wennberg, P. O.: Isoprene Peroxy Radical Dynamics, J. Am. Chem. Soc., 139, 5367–5377, https://doi.org/10.1021/jacs.6b12838, 2017. a
Vasquez, K. T., Crounse, J. D., Schulze, B. C., Bates, K. H., Teng, A. P., Xu, L., Allen, H. M., and Wennberg, P. O.: Rapid hydrolysis of tertiary isoprene nitrate efficiently removes NOx from the atmosphere, P. Natl. Acad. Sci. USA, 117, 33011–33016, https://doi.org/10.1073/pnas.2017442117, 2020. a, b
Xiong, F., McAvey, K. M., Pratt, K. A., Groff, C. J., Hostetler, M. A., Lipton, M. A., Starn, T. K., Seeley, J. V., Bertman, S. B., Teng, A. P., Crounse, J. D., Nguyen, T. B., Wennberg, P. O., Misztal, P. K., Goldstein, A. H., Guenther, A. B., Koss, A. R., Olson, K. F., de Gouw, J. A., Baumann, K., Edgerton, E. S., Feiner, P. A., Zhang, L., Miller, D. O., Brune, W. H., and Shepson, P. B.: Observation of isoprene hydroxynitrates in the southeastern United States and implications for the fate of NOx, Atmos. Chem. Phys., 15, 11257–11272, https://doi.org/10.5194/acp-15-11257-2015, 2015. a
Xu, L., Møller, K. H., Crounse, J. D., Kjaergaard, H. G., and Wennberg, P. O.: New Insights into the Radical Chemistry and Product Distribution in the OH-Initiated Oxidation of Benzene, Environ. Sci. Technol., 54, 13467–13477, https://doi.org/10.1021/acs.est.0c04780, 2020. a
Yuan, B., Koss, A., Warneke, C., Gilman, J. B., Lerner, B. M., Stark, H., and de Gouw, J. A.: A high-resolution time-of-flight chemical ionization mass spectrometer utilizing hydronium ions (H3O+ ToF-CIMS) for measurements of volatile organic compounds in the atmosphere, Atmos. Meas. Tech., 9, 2735–2752, https://doi.org/10.5194/amt-9-2735-2016, 2016. a, b
Yuan, B., Koss, A. R., Warneke, C., Coggon, M., Sekimoto, K., and de Gouw, J. A.: Proton-Transfer-Reaction Mass Spectrometry: Applications in Atmospheric Sciences, Chem. Rev., 117, 13187–13229, https://doi.org/10.1021/acs.chemrev.7b00325, 2017. a
Zaytsev, A., Breitenlechner, M., Koss, A. R., Lim, C. Y., Rowe, J. C., Kroll, J. H., and Keutsch, F. N.: Using collision-induced dissociation to constrain sensitivity of ammonia chemical ionization mass spectrometry (NH CIMS) to oxygenated volatile organic compounds, Atmos. Meas. Tech., 12, 1861–1870, https://doi.org/10.5194/amt-12-1861-2019, 2019. a, b, c, d, e, f, g, h, i, j, k, l, m